

I. Фізіологічний апоптоз відбувається:
а) у період ембріогенезу (імплантація, органогенез, онтогенетична інволюція, метаморфоз) відповідно до генетичної програми знищення клітин, які вже більше не потрібні;
б) під час гормон-залежної інволюції органів у дорослих (загибель клітин ен-дометрію під час менструального циклу, атрезія фолікулів яєчника під час менопаузи, зменшення маси молочних залоз після припинення лактації);
в) у популяціях клітин з високою інтенсивністю проліферативних процесів для підтримання сталої кількості клітин (наприклад, у криптах слизової тонкої кишки);
г) у клітинах, що вже виконали свою функцію (у нейтрофілах по завершенню гострого запалення, у лімфоцитах у кінці імунної відповіді;
ґ) у потенційно небезпечних для організму лімфоцитах (клітинах так званих "заборонених клонів'1);
д) під час дії цитотоксичних Т-лімфоцитів (Т-кілерів) на уражені вірусами і пухлинні клітини.
II. Апоптоз в патологічних умовах може відбуватися:
а) унаслідок дії патогенних чинників, що спричиняють необоротне ушкодження ДНК (іонізуюча радіація, протипухлинні препарати);
б) в інфікованих вірусами клітинах (наприклад, при вірусному гепатиті);
в) у паренхіматозних органах у процесі розвитку атрофії, що виникає як наслідок обтурації їхніх вивідних проток (підшлункова і слинні залози);
г) у пухлинах — як під час їхньої регресії, так і в процесі активного росту;
ґ) під впливом чинників, що збільшують проникність мітохондріальних мембран.
11.23. Як здійснюється апоптоз?
Розрізняють три послідовні фази апоптозу. І. Фаза ініціювання. Суть її полягає в послідовній активації так званих "суїцид-них" ферментів — каспаз (їх сьогодні налічують понад 10). Існує два шляхи такої активації: зовнішній і внутрішній.
Зовнішній (рецепторопосередкований) шлях пов'язаний з існуванням на поверхні клітинної мембрани так званих "рецепторів смерті" (із сімейства рецепторів до фактора некрозу пухлин - ФНП). Взаємодія цих рецепторів з ФНП та деякими іншими лігандами спричиняється до появи в цитоплазмі (з внутрішнього боку плазматичної мембрани поблизу активованого рецептора) б ілка-п о середника, який активує протеазу- каспазу-8 (у людини- каспазу-10). Активна каспаза-8 започатковує каскад реакцій (у тому числі і аутокаталітичних), під час яких відбувається перетворення неактивних прокаспаз в активні каспази - ферменти, що "вбивають" клітину.
Внутрішній (мітохондріальний) шлях пов'язаний зі збільшенням проникності мітохондріальних мембран (утворенням "мітохондріальної пори") та виходом із мітохондрій у цитоплазму так званих проапоптичних сполук- цитохрому с,
апоптозіндукуючого фактора (АІФ) та ін. Утворення "мітохондріальної пори " відбувається внаслідок заміни антиапоптичних білків в мембрані мітохондрій на проапоптичні білки. Така заміна має місце тоді, коли припиняється дія на клітину факторів росту та інших стимуляторів клітинної активності. Цитохром с, потрапивши в цитоплазму, разом з білком цитозолю - апоптозактивуючим фактором (ААФ) утворює комплекс, який активує каспазу-9. Зазначена протеаза започатковує процес утворення інших активних каспаз (див. вище).
II. Фаза вбивання ("екзекуції"). У результаті наведених вище подій утворюються активні форми каспаз-екзекуторів (каспаза-3, каспаза-6 та ін.). Ці протеази: а) розщеплюють білки цитоскелета; б) руйнують білки матриксу ядра; в) активують цитоплазматичну ДНК-азу через розщеплення її білкового інгібітора. Унаслідок цього змінюються форма і об'єм клітини, ядро розпадається на окремі фрагменти, а сама клітина на так звані апоптичні тільця — утвори, що зовні мають мембрану, а всередині містять спресовані органели і окремі фрагменти ядра.
Ш. Фаза вилучення загиблих клітин. Здійснюється макрофагами шляхом фагоцитозу мертвих клітин і апоптичних тілець. Цьому сприяє вивільнення клітинами ще на ранніх фазах їхнього апоптозу речовин-хемотаксинів, а також поява на поверхні клітин, що гинуть, білків-маркерів, які дають змогу макрофагам розпізнавати мертві клітини серед ще живих. Важливе значення цього етапу полягає в тому, що своєчасний фагоцитоз загиблих клітин запобігає їхньому некрозу, виходу з клітин лізосомних ферментів і розвитку запалення.
Універсальні механізми клітинного пошкодження.
Які зміни на молекулярному рівні мають велике значення в патогенезі ушкодження клітини? *
Можна виділити 6 груп молекулярних механізмів, що відіграють важливу роль у патогенезі ушкодження клітин: ліпідні (пероксидне окиснення ліпідів, активація мембранних фосфоліпаз, детергентна дія вільних жирових кислот), кальцієві, електролітно-осмотичні, ацидотичні, протеїнові і нуклеїнові.
11.6. У чому сутність пероксидного окиснення ліпідів?
Пероксидним окисненням ліпідів (ПОЛ) називають вільнорадикальне окиснення ненасичених жирових кислот, що входять до складу фосфоліпідів клітинних мембран.
Ініціаторами ПОЛ є вільні радикали, серед яких найбільше значення мають: 'О " (Н02") - супероксидний радикал; ОН' - гідроксильний радикал; Н' - водневий радикал; *02 - синглетний (збуджений) кисень.
11. 7. Які реакції лежать в основі ініціювання пероксидного окиснемня ліпідів?
Первинний вільний радикал, що з'явився в клітині (А') взаємодіє з молекулою ненасиченої жирової кислоти (RH), у результаті чого утворюється вільний радикал цієї кислоти (R') і молекулярний продукт реакції (НА):
![]()
Вільний радикал жирової кислоти, що утворився, взаємодіє з молекулярним киснем, який завжди міститься в клітині, у результаті чого з'являється пероксидний радикал цієї кислоти (ROO'):
![]()
Пероксидний радикал, у свою чергу, вступає у взаємодію з новою молекулою ненасиченої жирової кислоти, що міститься поруч. У ході цієї реакції утворюється гідропероксид (ROOH) і новий вільний радикал:
![]()
Слід зазначити дві важливі особливості ПОЛ. Перша полягає в тому, що реакції ПОЛ мають ланцюговий характер. Це означає, що в ході реакцій ПОЛ не відбувається знищення вільних радикалів і в процес утягуються все нові й нові молекули нена-сичених жирових кислот.
Друга особливість- це розгалужений характер ПОЛ. Інакше кажучи, у реакціях ПОЛ у кількості, що наростає, з'являються вільні радикали, джерелом яких є самі проміжні продукти ПОЛ. Прикладом може бути утворення вільних радикалів з гідропероксидів ліпідів при їхній взаємодії з наявними в клітині металами змінної валентності:
![]()
Через те що в багатьох біохімічних реакціях за умов норми утворюється невелика кількість вільних радикалів, існує постійна небезпека активації ПОЛ в клітині. Однак у природних умовах цього не відбувається, оскільки клітина має у своєму розпорядженні механізми антиоксидантного захисту, завдяки яким досягається інактивація вільних радикалів, обмеження й гальмування ПОЛ.
11.8. Які антиоксидантні системи є в клітинах?
І. Ферментні антиоксидантні системи: 1. Супероксгіддисмутазна.
Компоненти: супероксиддисмутаза (СОД), каталаза. Призначення: інактивація супероксидних радикалів (HOj):
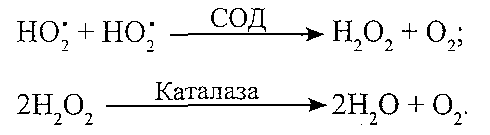
Причини порушень: набуті розлади синтезу ферментів, дефіцит міді і заліза. 2. Глютатіонова.
Компоненти: глютатіон (Г), глютатіонпероксидаза (ГП), глютатіонредуктаза (ГР), НАДФ-Н2.
Призначення: інактивація і руйнування гідропероксидів ліпідів:
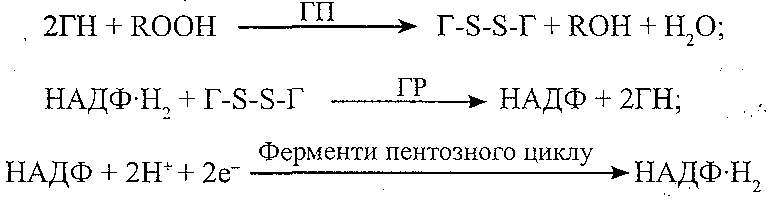
Причини порушень: спадково обумовлені і набуті порушення синтезу ферментів, дефіцит селену, порушення пентозного циклу (зменшення утворення НАДФН2).
II. Неферментні антиоксиданти: 1. "Істинні" антиоксиданти.
Компоненти: токофероли, убіхінони, нафтохінони, флавоноїди, стероїдні гормони, біогенні аміни.
Призначення: інактивація вільних радикалів жирових кислот:
![]()
де In — антиоксидант; In' — вільний радикал цього антиоксиданту, що має низьку реакційну здатність.
Причини порушень: гіповітаміноз Е, порушення регенерації "істинних" антиоксидантів. 2. Допоміжні антиоксиданти.
Компоненти: аскорбінова кислота, сполуки, що містять сірку - глютатіон, цистин, цистеїн.
Призначення: регенерація "істинних" антиоксидантів:
![]()
де DH - відновлена, D — окиснена форма допоміжного антиоксиданту.
Причини порушень: гіповітаміноз С, порушення пентозного циклу, дефіцит сполук, що містять сірку.
11.9. У яких випадках відбувається активація ПОЛ?
Активація ПОЛ відбувається:
1) при надмірному утворенні первинних вільних радикалів (ультрафіолетове й іонізуюче випромінювання, гіпероксІя, отруєння чотирихлористим вуглецем, гіпервітаміноз D та ін.);
2) при порушенні функціонування антиоксидантних систем (недостатність ферментів - супероксиддисмутази, каталази, глютатіон пер оксид ази, глютатіонредуктази; дефіцит міді, заліза, селену; гіповітамінози Е, С; порушення пентозного циклу).
11.10. Які механізми лежать в основі порушень бар'єрних функцій клітинних мембран при активації ПОЛ?
I. Іонофорнш механізм обумовлений появою в клітині речовин, що мають властивості іонофорів, тобто сполук, здатних полегшувати дифузію іонів через мембрану завдяки утворенню комплексів, що проходять через її шари. У процесі активації ПОЛ серед проміжних продуктів його реакцій з'являються речовини-іонофори для іонів кальцію і водню. У результаті цього підвищується проникність клітинних мембран до цих іонів.
II. Механізм електричного пробою пов'язаний з існуванням на багатьох мембранах (плазматичній, внутрішній мітохондріальній) різниці потенціалів. У результаті появи гідрофільних продуктів ПОЛ порушуються електроізоляційні властивості гідрофобного шару клітинних мембран, що призводтггь до електричного пробою мембрани, тобто до електромеханічного її розриву з утворенням нових трансмембранних каналів іонної провідності.
11.11. Як порушується матрична функція мембран у процесі активації ПОЛ?
Сутність матричної функції ліпідного подвійного шару мембран полягає в тому, що в ньому вмонтовані мембранні ферменти та деякі спеціалізовані білки.
У процесі ПОЛ порушується активність мембранних ферментів, оскільки змінюється їх ліпідне мікрооточення, яке багато в чому визначає властивості білкових молекул. Крім того, у ході реакцій ПОЛ відбувається утворення "зшивок" між молекулами білків і фосфоліпідів, а також окиснення сульфгідрильних груп активних центрів, що призводить до необоротної інактивації ферментів.
11.12. Яким чином підвищення активності фосфоліпаз впливає на ушкодження клітинних мембран?
У патогенезі ушкодження клітини велике значення має надмірна активація фос-фоліпази А2 - ферменту, що здійснює гідролітичне відщеплення ненасичених жирових кислот від молекул фосфоліпідів клітинних мембран (рис. 33). У результаті цього утворюються лізофосфоліпіди, в молекулах яких лише один жировокислотний "хвіст", у зв'язку з чим вони мають здатність до міцелоутворення і є дуже сильними
детергентами (рис. 34). З детергентною дією лізофосфоліпідів і пов'язане ушкодження клітинних мембран в умовах надмірної активації фосфоліпази А,, яку, як правило, викликають високі концентрації іонів кальцію в цитоплазмі клітини.
11.13. За яких умов виникає небезпека детергентноїдії вільних жирових кислот на клітинні мембрани?
Детергентна дія вільних жирових кислот (ВЖК) виявляється при збільшенні їхнього вмісту в клітині. Це може бути обумовлене (оис. 35):
а) посиленим надходженням ВЖК у клітину при гіперліпацидемії (наприклад, активація ліполізу в жировій тканині при стресі, цукровому діабеті);
б) вивільненням ВЖК у лізосомах із тригліцеридної частини ліпопротеїдів, що надійшли в клітину із плазми крові (наприклад, гіперліпопротеїнемія при атеросклерозі);
в) вивільненням ВЖК із фосфоліпідів мембран під дією мембранних фосфоліпаз;
г) порушеннями використання ВЖК як джерела енергії (зменшення активності ферментів Р-окиснення і циклу Кребса, наприклад, при гіпоксії).
Для того щоб запобігти ушкоджувальній дії надлишку жирових кислот, клітина має у своєму розпорядженні систему ферментів, які переводять вільні жирові кислоти у форму триглщеридів. При цьому відбувається невластиве для норми відкладення тригліцеридів у вигляді жирових крапель, тобто виникає жирова дистрофія клітини.
11.14. У яких випадках іони кальцію втягуються в патогенез ушкодження клітини? З якими ефектами цих іонів пов 'язана їхня участь в ушкодженні клітинних структур?
Ушкодження клітинних структур може бути обумовлене стійким підвищенням концентрації іонів Са2+ у цитоплазмі клітини. Така ситуація виникає або в результаті надмірного надходження іонів Са2+ у цитоплазму (гіперкальціємія, підвищення проникності плазматичної мембрани), або в результаті порушення механізмів, що забезпечують видалення іонів Са2+ із цитоплазми (порушення Са-насосів, Na-Ca-об-мінного механізму, Са-акумулгоючої функції мітохондрій).
Підвищення концентрації іонів Са2т у цитоплазмі викликає:
а) контрактуру (пер є скорочення) фібрилярних структур клітини (міофібрил, елементів цитоскєлета);
б) активацію фосфоліпази А,;
в) роз'єднання процесів окиснення й фосфорування.
11. 15. Чим можуть бути обумовлені зміни вмісту іонів натрію й калію в клітині і яка роль таких порушень у патогенезі клітинного ушкодження?
Вирівнювання концентрацій іонів Na+ і К + по обидва боки плазматичної мембрани (збільшення вмісту Na+ і зменшення вмісту К+ у цитоплазмі) у своїй основі може мати два механізми: 1) посилену дифузію іонів через плазматичну мембрану за існуючим концентраційним і електричним градієнтом і 2) порушення механізмів активного транспорту Na+ і К+ (Na-K-насоса).
Перший механізм реалізується в умовах загальних порушень водно-електролітного обміну (гіпернатріємія, гіпокаліємія) і порушень бар'єрної функції плазматичної мембрани (підвищення її іонної проникності).
Розлади функції Na-K-насоса можуть бути обумовлені дефіцитом АТФ у клітині, збільшенням вмісту холестеролу в ліпідному бішарі мембрани (наприклад, при атеросклерозі), дією цілої низки специфічних інгібіторів Na-K-АТФ-ази (наприклад, строфантину).
Зміни вмісту іонів Na+ і К+ викликають:
а) втрату клітиною електричного мембранного потенціалу (потенціалу спокою), а отже і збудливості;
б) набряк клітини;
в) осмотичне розтягнення клітинних мембран, що супроводжується підвищенням їх проникності.
11. 16. Чим може бути обумовлений розвиток внутрішньоклітинного ацидозу і які зміни в клітині можуть бути з ним пов 'язані?
До розвитку внутрішньоклітинного ацидозу можуть спричинятися:
1) надмірне надходження іонів Н+ у клітину з позаклітинного середовища (декомпенсований газовий або негазовий ацидоз);
2) надмірне утворення кислих продуктів у самій клітині при активації гліколізу (молочна кислота), порушеннях циклу Кребса (три- і дикарбонові кислоти), гідролітичному розщепленні фосфоліпідів клітинних мембран (вільні жирові кислоти, фосфорна кислота) та ін.;
3) порушення зв'язування вільних іонів Н+ у результаті недостатності буферних систем клітини;
4) порушення виведення іонів Н+ з клітини при розладах Na-H-обмінного механізму, а також в умовах порушеного місцевого кровообігу в тканині.
Внутрішньоклітинний ацидоз викликає: а) зміну конформації білкових молекул з порушенням їх ферментативних, скоротливих та інших властивостей; б) підвищення проникності клітинних мембран; в) активацію лізосомних гідролітичних ферментів.
11.17. Які зміни білкових молекул мають значення в патогенезі ушкодження клітини?
До білкових (протеїнових) механізмів ушкодження клітини можна віднести:
1) інгібуванння ферментів (оборотне і необоротне);
2) денатурацію, тобто порушення нативної будови білкових молекул у результаті обумовлених розривом ковалентних зв'язків змін вторинної й третинної структур білка;
3) протеоліз, що здійснюється під дією лізосомних протеолітичних ферментів (ка-тепсинів) і протеаз, які активуються іонами Са2+, У результаті протеолізу можуть з'являтися пептиди, що мають властивості фізіологічно активних речовин. З виходом останніх з ушкоджених клітин може бути пов'язаний розвиток як місцевих, так і загальних реакцій організму (запалення, гарячка).
11. 18. Які порушення функціонування генетичного апарату клітини можуть призводити до її ушкодження?
Основу ушкодження клітини можуть становити так звані нуклеїнові механізми, обумовлені порушеннями процесів:
1) реплікації ДНК (денатурація ДНК, ушкодження ДНК-репліказної ферментної системи, дефіцит трифосфонуклеотидів - АТФ, ГТФ, ТТФ і ЦТФ);
2) транскрипції (мутаційні дефекти генної матриці, інгібування ДНК-залежної РНК-полімерази антибіотиками й токсинами, порушення посттранскрипційної модифікації інформаційної РНК: неприєднання "кепа" до головного кінця молекули, порушення утворення полі-А-хвоста, розлади сплайсингу тощо);
3) трансляції (дефіцит або якісні зміни інформаційної, рибосомної або транспортної РНК, а також рибосомних ферментів і нєферментних білків; дефіцит вільних амінокислот і АТФ; інгібування процесу антибіотиками й мікробними токсинами).
11.19. Які існують універсальні механізми підвищення проникності клітинних мембран при ушкодженні клітини?
Підвищення проникності клітинних мембран може бути обумовлене:
1) активацією пероксидного окиснення ліпідів;
2) активацією фосфоліпаз;
3) осмотичним розтягненням мембран;
4) адсорбцією білків (поліелектролітів) на мембрані;
5) змінами фазового стану мембранних ліпідів (ацидоз, зміна температури).
Які порушення виникають у клітині в результаті ушкодження окремих її органоїдів (плазматичної мембрани, мітохондрій, ендоплазматичногоретикулуму лізосом)?
Порушення бар'єрної функції плазматичної мембрани призводить до вирівнювання існуючих у нормі концентраційних градієнтів речовин: у клітину надходять іони Na+, Са2+, СІ", а виходять іони К+, Mg2+, неорганічного фосфату, низько- і високо-молекулярні органічні сполуки (АМФ, АДФ, проміжні продукти клітинного обміну, білки-ферменти). З ушкодженнями білків і глікопротеїдних комплексів, вбудованих у плазматичну мембрану, пов'язані порушення систем активного транспорту речовин (Na-K-, Са-насосів; Na-Ca- і Na-H-обмінних механізмів); зміни специфічних іонних каналів (Na-, K-, Са-каналів); порушення клітинних рецепторів, що сприймають зовнішні регуляторні сигнали (а- і )3-адренорецепторів, ш- і n-холінорецепторів та ін.); порушення міжклітинних взаємодій; зміни антигенних властивостей клітини.
Ушкодження мітохондрій супроводжується або пригніченням процесів клітинного дихання, або ефектом роз'єднання процесів окиснення й фосфорування. І в тому, і в другому випадку результатом розладів мітохондріальних функцій буде порушення енергозабезпечення клітини (рис. 36).
Ушкодження шорсткого ендоплазматичного ретикулуму призводить до деза-грегації полісом, унаслідок чого порушуються реакції біосинтезу білка в клітині. У результаті ушкодження гладкого ендоплазматичного ретикулуму і його ферментних систем страждають процеси детоксикації, мікросомного окиснення та ін. У деяких
клітинах, наприклад м'язових, порушується здатність ендоплазматичного (саркоплазма-тичного) ретикулуму депонувати іони Са2+, що сприяє реалізації так званих кальцієвих механізмів ушкодження клітини.
Підвищення проникності лізосомних мембран призводить до виходу в цитоплазму гідролітичних ферментів, активація яких у кінцевому підсумку викликає необоротні зміни клітини — її аутоліз.
Механізми клітинного захисту і адаптації клітин до дії пошкоджуючих факторів.
Які захисно-компенсаторні механізми має ушкоджена клітина?
Все різноманіття захисно-компенсаторних реакцій клітини у відповідь на її ушкодження можна умовно розділити на дві групи.
I. Реакції, спрямовані на відновлення порушеного внутрішньоклітинного гомеостазу:
а) активація механізмів активного транспорту речовин (Na-K-, Са-насосів; Na-Ca-, Na-H-обмінних механізмів, мікровезикулярного транспорту);
б) посилення регенерації антиоксидантів;
в) зв'язування вільних жирових кислот (синтез тригліцеридів);
г) активація синтезу білків, нуклеїнових кислот, фосфоліпідів та ін. Неодмінною умовою реалізації цих механізмів є достатнє енергозабезпечення
клітини. Це досягається підвищенням інтенсивності енергетичного обміну (активація гліколізу, клітинного дихання, пентозного циклу) і перерозподілом наявних у клітин енергетичних ресурсів.
II. Реакції, спрямовані на створення функціонального спокою ушкодженої клітини. їхня мета полягає в тому, щоб усунути можливі додаткові зрушення внутрішньоклітинного гомеостазу при дії фізіологічних збуджувальних факторів (стабілізація ушкодження) і звести до мінімуму енергетичні витрати на виконання специфічних функцій клітини.
До таких реакцій можна віднести:
а) утворення клітиною простагландинів і блокада ними |3-адренорецепторів (рис. 37);
б) інгібування адеі-іілатциклази і підвищення активності фосфодіестерази, що руйнує цАМФ;
в) утворення аденозину - природного блокатора Са-каналів та ін.
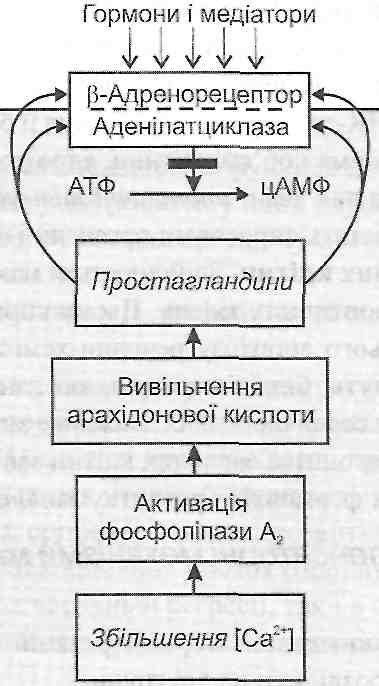
Рис. 37. Захисна роль простагландинів при ушкодженні клітини
Які існують підходи до патогенетичного лікування ушкоджених клітин?
Основні принципи впливу на ушкоджені клітини:
1) обмеження і пригнічення молекулярних механізмів ушкодження (блокада Са-каналів, застосування антиоксидантів, інгібіторів фосфоліпази А2 і протеаз, активація біосинтезу білків та ін.);
2) створення функціонального спокою (щадний режим, дієта, блокада клітинних рецепторів та ін.);
3) енергетичне й пластичне забезпечення гомеостатичних механізмів клітини (вплив на периферичний кровообіг і мікроциркуляцію з метою поліпшення доставки кисню і поживних речовин до ушкоджених клітин, введення енергетичних і пластичних субстратів).
Артеріальна і венозна гіперемії: визначення поняття, прояви, види, причини і механізми розвитку, варіанти завершення і наслідки.
Артеріальна гіперемія - це збільшення кровонаповнення органа або тканини за рахунок надмірного надходження крові по артеріальних судинах.
Які функціональні зміни й клінічні ознаки характеризують артеріальну гіперемію?
При артеріальній гіперемії спостерігають розширення дрібних артерій, артеріол, вен і капілярів; прискорення течії крові в них, пульсацію дрібних артерій і капілярів, збільшення числа видимих оком судин, збільшення тиску в артеріолах, капілярах і венах. У результаті зазначених змін виникає почервоніння, підвищується місцева температура, збільшується об'єм гіперемованої ділянки, підвищується тургор тканини, посилюються обмін речовин і функція органа.
Які фактори можуть бути причиною артеріально)' гіперемії? Що мають на увазі, коли кажуть про фізіологічну й патологічну артеріальну гіперемію?
Причиною артеріальної гіперемії може бути вплив фізичних, хімічних і біологічних факторів зовнішнього середовища; збільшення навантаження на орган або ділянку тканини; психогенні впливи.
ФЬіологічною називають артеріальну гіперемію, що виникає під дією звичайних фізіологічних подразників (збільшення навантаження на орган, психогенні впливи). Основними її різновидами є робоча й реактивна гіперемія.
Робоча гіперемія - це збільшення течії крові в органі під час посилення його функції (збільшення вінцевого кровообігу при посиленні роботи серця, гіперемія слинних залоз під час приймання їжі та ін.).
Реактивна гіперемія являє собою збільшення течії крові після його короткочасного обмеження. Розвивається зазвичай у нирках, головному мозку, шкірі, кишках, м'язах.
Патологічна артеріальна гіперемія виникає під дією незвичайних (патологічних) подразників або в результаті підвищення чутливості судин до звичайних впливів. Вона супроводжує розвиток таких патологічних процесів, як запалення, алергія, опіки, гарячка, її клінічними прикладами можуть бути інфекційний або алергічний висип, почервоніння обличчя при багатьох інфекційних хворобах (кір, скарлатина, висипний тиф), почервоніння половини обличчя при невралгії трійчастого нерва і т. д.
13.5. Назвіть основні механізми розвитку патологічної артеріальної гіперемії.
Розрізняють два механізми: нейрогенний і пов'язаний з дією місцевих хімічних (метаболічних) факторів.
Нейрогенна артеріальна гіперемія залежно від конкретних механізмів її розвитку може бути нейротонічного і нейропаралітичного типів.
Гіперемію, пов'язану з дією місцевих гуморальних факторів, іноді називаютьмі-опаралітичною, підкреслюючи тим самим первинність порушень скоротливих властивостей гладких м'язів судин під впливом хімічних агентів.
13.6. У чому сутність нейротонічного механізму розвитку артеріальної гіперемії?
Нейротонічний тип артеріальної гіперемії розвивається при збільшенні імпуль-сації по судинорозширювальних нервах (вазодилататорах). До останніх належать парасимпатичні нерви і симпатичні холінергічні нерви, медіатором яких є ацетилхолін.
В експерименті на тваринах цей тип артеріальної гіперемії відтворюють шляхом подразнення судинорозширювальних нервів. Так, подразнення chorda tympani (гілка n. facialis) викликає артеріальну гіперемію і посилення секреції піднижньощелепної слинної залози (дослід К. Бернара).
У клініці нейротонічна гіперемія виникає найчастіше рефлекторно у зв'язку з подразненням екстеро- та інтерорецепторів, а також при подразненні судинорозширювальних нервів та їх центрів. Цим, зокрема, пояснюється почервоніння обличчя і шиї при патологічних процесах у внутрішніх органах (яєчниках, серці, печінці).
13.7. Поясніть нейропаралітичний механізм розвитку артеріальної гіперемії.
Нейропаролітична артеріальна гіперемія розвивається при припиненні імпуль-сації по симпатичних адренергічних нервах, що мають судинозвужувальну дію. В експерименті на тваринах її моделюють за допомогою хірургічних втручань і фармакологічних способів. Часто застосовують перетинання симпатичних адренергічних волокон і нервів. Так, при перетинанні симпатичних волокон сідничного нерва спостерігають розширення судин лапки жаби (дослід А. Вальтера), а при видаленні шийного вузла симпатичного стовбура- почервоніння й підвищення температури вуха кроля збоку операційного втручання (дослід К. Бернара).
Серед фармакологічних способів відтворення артеріальної гіперемії нейропаралітичного типу — блокада передачі нервових імпульсів у симпатичних вузлах за допомогою гангліоблокаторів, порушення утворення, депонування й виділення катехоламінів закінченнями симпатичних нервів (застосування симпатолітиків); блокада а-адрено-рецепторів судинних гладких м'язів за допомогою відповідних адреноблокаторів.
13.8. Які гуморальні фактори можуть викликати розвиток артеріальної гіперемії?
Артеріальна гіперемія міопаралітичного типу може розвиватися за умов дії трьох груп гуморальних факторів:
1) неорганічних іонів (калію, водню) і дефіциту кисню;
2) метаболітів (молочної кислоти, органічних кислот циклу Кребса, АДФ, АМФ, аденозину);
3) біологічно активних речовин (гістаміну, серотоніну, кінінів, простагландинів).
Зазначені фактори спричинюють розширення судин, діючи або безпосередньо на гладкі м'язи судинної стінки, або опосередковано, через вплив на ендотелій судин.
13.9. Яка роль ендотелію кровоносних судин у розвитку артеріальної гіперемії?
Під впливом цілого ряду місцевих гуморальних факторів, зокрема, біологічно активних речовин, ендотеліальні клітини судин виділяють речовину, що отримала була назву фактора релаксації ендотеліального походження. Нині відомо, що цією речовиною є оксид азоту (II), який утворюється з амінокислоти аргініну під впливом ферменту NO-синтази. Оксид азоту діє на гладком'язові клітини судинної стінки й викликає їхню гіперполяризацію. Результатом цього є зменшення базального тонусу кровоносних судин та їх розширення під дією тиску крові.
13.10. Назвіть можливі наслідки артеріальної гіперемії.
У більшості випадків артеріальна гіперемія супроводжується збільшенням інтенсивності обміну речовин і посиленням діяльності органа, що є пристосуванням до дії підвищеного функціонального навантаження.
Однак можливі й несприятливі наслідки. При артеріосклерозі, наприклад, різке розширення судини може супроводжуватися розривом її стінки і крововиливом у тканину. Особливо часто подібне явище спостерігається в головному мозку.
13.11. Що таке венозна гіперемія?
Венозна гіперемія - це збільшення кровонаповнення органа або ділянки тканини в результаті утрудненого відтоку крові по венах.
13.12. Які фактори можуть бути причиною венозної гіперемії?
Порушення відтоку крові по венах може бути пов'язане з такими чинниками:
1) внутрішньосудинними (закупорка вен тромбом або емболом);
2) позасудинними (здавлювання вен пухлиною, рубцем, збільшеною маткою, набряковою рідиною);
3) факторами самої судинної стінки (конституціональна слабкість еластичного апарату вен, недостатній розвиток і знижений тонус гладком'язових елементів їхніх стінок);
4) порушеннями загальної гемодинаміки (ослаблення функції правого шлуночка серця, зменшення присмоктувальної дії грудної клітки, утруднення течії крові в малому колі кровообігу).
13.13. Якими ознаками виявляє себе венозна гіперемія?
Для венозної гіперемії характерне збільшення об'єму органа або ділянки тканини, ціаноз, місцеве зниження температури, набряк, підвищення тиску у венах і капіля-
pax застійної ділянки, уповільнення течії крові, вихід еритроцитів за межі судинного русла (діапедез). На завершальному етапі гіперемії можливий маятникоподібний рух крові і стаз. Тривале розширення вен призводить до розтягнення їхньої стінки, що може супроводжуватися гіпертрофією м'язової оболонки, явищами флебосклерозу і варикозного розширення вен.
13.14. Які місцеві й загальні порушення можуть бути наслідком венозної гіперемії?
Місцеві зміни при венозній гіперемії пов'язані в основному з кисневим голодуванням (гіпоксією) тканини. Гіпоксія при цьому спочатку обумовлена обмеженням припливу артеріальної крові, а потім дією на тканинні ферментні системи продуктів порушеного обміну, наслідком чого є порушення утилізації кисню.
Кисневе голодування при венозній гіперемії обумовлює розлади тканинного метаболізму, викликає атрофічні і дистрофічні зміни, надмірне розростання сполучної тканини (наприклад, цироз печінки при венозному застої, викликаному недостатністю функції серця).
Якщо венозна гіперемія має генералізований характер, то можливим є ряд загальних гемодинамічних порушень з важкими наслідками. Найчастіше вони виникають при закупорці великих венозних судин - ворітної, нижньої порожнистої вен. Скупчення крові в зазначених судинних резервуарах (до 90 % всієї крові) супроводжується різким зниженням артеріального тиску, порушенням живлення життєво важливих органів (серця, мозку), що може призвести до смерті
Ішемія: визначення поняття, прояви, види, причини, механізми розвитку, наслідки. Механізми ішемічного пошкодження клітин. Синдром ішемія-реперфузія.
Ішемія - це зменшення кровонаповнення органа або ділянки тканини в результаті обмеження або повного припинення припливу артеріальної крові. Ішемію називають ще місцевим недокрів'ям.
13.16. Які ознаки характерні для ішемії?
Ішемія характеризується зблідненням ділянки органа, зниженням його температури, порушенням чутливості (відчуття оніміння, поколювання, "повзання мурашок"), больовим синдромом, зменшенням швидкості течії крові і об'єму органа, зниженням артеріального тиску на ділянці артерії, розташованій нижче перешкоди; зниженням напруги кисню в ішемізованій ділянці органа або тканини, зменшенням утворення тканинної рідини і зниженням тургору тканини, порушенням функції органа або тканини, дистрофічними змінами.
13.17. Назвіть основні типи ішемії залежно від причини й механізмів її виникнення.
Основними типами ішемії є компресійна, обтураційна і ангіоспастична.
Компресійна ішемія виникає в результаті здавлювання артерій ззовні лігатурою, рубцем, пухлиною, стороннім предметом та ін.
Обтураційна ішемія є наслідком часткового звуження або повного закриття просвіту артерій атеросклеротичною бляшкою, тромбом або емболом.
Ангіоспастична ішемія виникає внаслідок спазму артерій, викликаного емоційним впливом (страх, хвилювання, гнів), фізичними факторами (холод, травма, механічне подразнення), хімічними агентами, біологічними подразниками (токсини бактерій) і т. д. В основі спазму можуть лежати нервові рефлекторні механізми або безпосередня дія подразників на гладкі м'язи судин (вплив вазопресину, ангіотензи-ну II, ендотеліну).
13.18. Чим визначається характер обмінних, функціональних і структурних порушень у тканині при її ішемії?
Характер таких порушень визначається ступенем кисневого голодування. Важкість гіпоксії, у свою чергу, залежить від швидкості розвитку й типу ішемії, її тривалості, локалізації, характеру колатерального кровообігу, функціонального стану органа або тканини.
Ішемія, що виникає на ділянці повної обтурації або компресії артерій, за інших рівних умов викликає важчі зміни, ніж при спазмі. Ішемія, що швидко розвивається, як і тривала, протікає важче порівняно з ішемією, що розвивається повільно, або нетривалою.
Ішемія життєво важливих органів (мозок, серце) має важчі наслідки, ніж ішемія нирок, легень, селезінки, а ішемія останніх - важчі, якщо порівнювати з ішемією скелетної, м'язової, кісткової або хрящової тканини. Мозок і серце характеризуються високим рівнем енергетичного обміну, але, незважаючи на це, їх колатеральні судини функціонально не здатні компенсувати порушення кровообігу. Навпаки, скелетні м'язи й особливо сполучна тканина, завдяки низькому рівню енергетичного обміну в них, більш стійкі в умовах ішемії.
Утруднення припливу артеріальної крові при підвищеній функціональній активності органа або тканини небезпечніше, ніж у стані спокою.
13.19. Якими послідовними стадіями характеризується патогенез порушень в ішемізованій тканині?
Виділяють такі стадії патогенезу
I. Порушення енергетичного обміну. Вони виявляють себе зниженням ефективності циклу Кребса і тканинного дихання, активацією гліколізу, а в кінцевому підсумку - зменшенням вмісту в клітинах макроергічних сполук — креатинфосфату і АТФ. Порушення утворення енергії на ділянці ішемії патогенетично пов'язане з недостатньою доставкою кисню й необхідних для окиснення субстратів, зниженням активності й синтезу ферментів, виходом ферментів з ушкоджених клітин, роз'єднанням окиснення й фосфорування.
II. Порушення енергозалежних процесів у клітинах, викликане зменшенням утворення енергії. При цьому порушуються специфічні функції клітин (скоротлива, секреторна та ін.), механізми активного транспорту речовин, зокрема, робота іонних насосів; знижується біосинтез білків неколагенового типу, що беруть участь у структурній організації клітин і тканин. В остаточному підсумку розвиваються ушкодження клітин і некробіотичні зміни, які в найважчих випадках закінчуються утворенням осередку некрозу - інфаркту.
III. Посилення біосинтезу компонентів сполучної тканини - колагену, глікозамі-ногліканів, глікопротеїнів, що є основою для наступного склерозування ішемізо-ваної ділянки тканини або органа.
Тромбоз: визначення поняття, види тромбів. Причини, механізми, наслідки тромбоутворення.
Тромбоз -це процес прижиттєвого утворення на внутрішній поверхні стінки судин згустків крові, що складаються з її елементів. Ці згустки отримали назву тромбів.
Тромби бувають пристінковими (частково зменшують просвіт судини) і закупорювальними.
Залежно від будови розрізняють білі, червоні й змішані тромби. У першому випадку тромб утворюють тромбоцити, лейкоцити, а також невелика кількість білків плазми; у другому - еритроцити, скріплені нитками фібрину; змішані тромби складаються з білих та червоних шарів, що чергуються між собою.
13.23. Назвіть три основні фактори, що сприяють тромбоутворенню (тріада Вірхова).
1. Ушкодження судинної стінки. Воно може виникати під дією фізичних факторів (механічна травма, електричний струм), хімічних, біологічних (ендотоксини мікроорганізмів), а також у результаті порушення живлення й метаболізму судинної стінки.
2. Порушення активності систем зсідання і протизсідання у крові та судинній стінці. Для утворення тромбів велике значення має як підвищення активності системи зсідання крові за рахунок збільшення в ній концентрації прокоагулянтів, так і зниження активності антикоагулянтної і фібринолітичної систем.
3. Уповільнення течії крові. Цей чинник дає можливість пояснити, чому у венах тромби утворюються частіше, ніж в артеріях, у венах нижніх кінцівок - частіше, ніж у венах верхніх кінцівок; а також високу частоту тромбоутворення при декомпенсації кровообігу, тривалому перебуванні хворої людини в ліжку.
13.24. З яких фаз складається процес утворення тромбу? У чому їх сутність?
Процес тромбоутворення має дві фази: клітинну і плазматичну.
Сутність клітинної фази полягає в адгезії, агрегації й аглютинації тромбоцитів (докладно див. розд. 26).
Плазматична фаза (фаза коагуляції крові) являє собою ланцюг послідовних біохімічних реакцій зсідання крові, кінцевим результатом яких є утворення фібрину -важливого компонента тромбу.
13.25. Які негативні наслідки може мати тромбоутворення в умовах патології?
"При різних захворюваннях утворення тромбів може супроводжуватися важкими наслідками, зумовленими гострим порушенням кровообігу в зоні судини, в якій виник тромб (ішемія при тромбозі артерій і застій крові при тромбозі вен). Кінцевим етапом може бути розвиток некрозу (інфаркту) у басейні тромбованої, позбавленої колатералей судини. Особливо велика роль тромбозу вінцевих артерій у розвитку інфаркту міокарда. Тромбоз артерій може призводити до трофічних порушень із наступним розвитком гангрени кінцівок при атеросклерозі, облітеративному ендартеріїті, цукровому діабеті.
Емболія: визначення поняття, види емболів. Особливості патогенезу емболії великого і малого кіл кровообігу, системи ворітної вени.
Емболія — це закупорка судин тілами, принесеними течією крові або лімфи. Ці тіла називаються емболами.
13.27. Які виділяють види емболії?
Залежно від характеру емболів та їхнього походження розрізняють емболію екзогенну й ендогенну.
За локалізацією виділяють емболію великого і малого кола кровообігу, а також системи ворітної вени.
Дуже рідко буває так звана ретроградна емболія, коли рух ембола відбувається не за гемодинамічними законами, а відповідно до сили тяжіння самого ембола, і парадоксальна емболія, що спостерігається при незарощенні міжпередсердної або між-шлуночкової перегородки, у результаті чого емболи з вен великого кола кровообігу й правої половини серця переходять у ліву, обминаючи мале коло.
13.28. Назвіть основні причини емболії екзогенного походження.
Залежно від природи й характеру емболів, що надходять ззовні, розрізняють такі види екзогенної емболії: повітряну, газову, бактеріальну, паразитарну, емболію твердими сторонніми тілами.
Повітряна емболія виникає при пораненні великих вен голови й шиї, які слабко спадаються й тиск у яких близький до нуля або негативний. У результаті в ушкоджені вени засмоктується повітря, особливо на висоті вдиху, з наступною емболією судин малого кола кровообігу.
Газова емболія розвивається при різкому перепаді атмосферного тиску від підвищеного до нормального (у робочих кесонів і водолазів) або від нормального до зниженого (при швидкому піднятті на висоту або під час розгерметизації кабіни висотного літального апарата). При цьому зменшується розчинність газів у тканинах і крові, відбувається десатурація, тобто перехід газів з розчиненого стану в газоподібний, і закупорка бульбашками цих газів (у першу чергу азоту) капілярів, розташованих головним чином у системі великого кола кровообігу.
13.29. Назвіть основні причини емболії ендогенного походження.
До ендогенної емболії відносять емболію тромбом (тромбоемболію), жирову емболію, тканинну емболію, емболію навколоплідними водами.
Джерелом тромбоемболії є тромб, що відірвався, найчастіше при асептичному або гнійному його розплавленні.
Жирова емболія виникає при потраплянні в кровоносні судини крапель жиру. Причиною цього найчастіше є ушкодження (роздроблення, сильний струс) кісткового мозку, підшкірної або тазової клітковини.
Тканинна емболія може бути обумовлена занесенням у кровоносне русло частинок різних тканин організму при їх ушкодженні. Особливе значення має емболія судин клітинами злоякісних пухлин, оскільки вона є основним механізмом утворення метастазів.
Емболія навколоплідними водами виникає в результаті потрапляння навколоплідних вод під час пологів в ушкоджені судини матки на ділянці плаценти, що відокремилася.
Стаз: визначення поняття, види, причини, патогенез, наслідки.
Стаз - це уповільнення й зупинка течії крові в капілярах, дрібних артеріях і венах.
13.21. Назвіть основні варіанти стазу та їхні причини.
Розрізняють ішемічний, венозний та істинний (капілярний) стаз.
Ішемічний і венозний стаз розвивається як наслідок ішемії та венозної гіперемії, а тому має ті ж причини, що й зазначені місцеві розлади кровообігу.
Причиною істинного стазу можуть бути фізичні (холод, тепло), хімічні (отрути, концентрований розчин натрію хлориду й інших солей, скипидар) і біологічні фактори (токсини мікроорганізмів).
У патогенезі істинного стазу велике значення надають двом факторам:
1) внутрішньокапілярній агрегації еритроцитів, тобто їх склеюванню й утворенню конгломератів, що утрудняють течію крові;
2) уповільненню течії крові в капілярах унаслідок згущення крові. В останньому випадку провідна роль належить підвищенню проникності стінок судин, розташованих у зоні стазу.
Порушення мікроциркуляції, класифікація. Сладж-синдром: визначення поняття, причини і механізми розвитку. Порушення місцевого лімфообігу, види, причини і механізми розвитку.
Мікроциркуляція — це рух крові й лімфи по мікроциркуляторних кровоносному і лімфоносному руслах.
Мікроциркуляторне кровоносне русло складається з судин, діаметр яких не перевищує 100 мкм, тобто артеріол, метартеріол, капілярних судин, венул і артеріоло-ве-нулярних анастомозів
Мікроциркуляторне лімфоносне русло представлене початковим відділом лімфатичної системи, у якому відбувається утворення лімфи і надходження її в лімфатичні капіляри.
Порушення мікроциркуляції поділяють на три типи: внутрішньосудиннї, порушення, пов 'язані зі змінами самих судин; позасудинні.
- Великі успіхи у вивченні закономірностей мікроциркуляції в нормі і при патології пов'язані з іменем О. М. Чернуха.
У чому сутність феномена під назвою "сладж"?
Сладж є внутрішньо судинним порушенням мікроциркуляції, пов'язаним зі зміною реологічних властивостей крові. Основні фактори таких змін — це порушення суспензійної стабільності крові, а також підвищення її в'язкості.
Головними особливостями крові при сладжі є прилипання один до одного еритроцитів, лейкоцитів і тромбоцитів, збільшення в'язкості крові, що утруднює її рух по мі-кросуцинах. При цьому течія крові різко уповільнюється й нагадує переміщення мулу по дну ріки (назва "сладж" походить від англ. sludge - густа грязь, баговиння, мул).
Залежно від розмірів агрегатів клітин крові, характеру їхніх контурів і щільності впакування еритроцитів розрізняють такі типи сладжу: класичний (великі розміри агрегатів, нерівні обриси контурів і щільне впакування еритроцитів), декстриновий (різна величина агрегатів, округлі обриси, щільне впакування еритроцитів) і аморфний гранулоподібний (величезна кількість дрібних агрегатів у вигляді гранул, що складаються всього з декількох еритроцитів).
Що таке недостатність лімфообігу? Назвіть основні її форми.
Недостатність лімфообігу - це стан, при якому лімфатичні судини не виконують свою основну функцію - здійснення постійного й ефективного дренажу інтерстиціального простору.
Розрізняють такі форми недостатності лімфообігу. 1. Механічна недостатність. Виявляє себе утрудненням відтоку лімфи у зв'язку з наявністю органічних (здавлювання пухлиною, рубцем, облітерація лімфатичних
судин при їх запаленні, тромбозі та ін.) або функціональних причин (підвищення тиску в магістральних венозних судинах, спазм лімфатичних судин, припинення м'язових скорочень та ін.).
2. Динамічна недостатність. Виникає тоді, коли об'єм транссудації міжклітинної рідини перевищує можливості лімфатичної системи забезпечувати ефективний дренаж інтерстиціальної тканини,
3. Резорбційна недостатність. Обумовлена структурними змінами інтерстиціальної тканини, накопиченням білків і осадженням їхніх патологічних видів в інтерстиції.
Основними проявами недостатності лімфообігу в гострій стадії є набряк, накопичення білків і продуктів їхнього розпаду в інтерстиціальній тканині, а в хронічній стадії - розвиток фіброзу й склерозу.
Запалення: визначення поняття, принципи класифікації. Характеристика гострого та хронічного запалення. Загальні прояви та місцеві ознаки запалення. Етіологія запалення.
Запалення - це типовий патологічний процес, що виникає в результаті ушкодження тканини й виявляє себе комплексом структурних, функціональних і метаболічних порушень, а також розладами мікроциркуляції.
Назвіть зовнішні ознаки запалення.
Припухлість (tumor), почервоніння (rubor), жар (color), біль (dolor) і порушення функції (functio laesd). Ці ознаки відомі як пеншада Цельса—Голена.
З яких компонентів складається патогенез запалення?
У патогенезі запалення розрізняють:
1) альтерацію;
2) порушення мікроциркуляції'з явищами ексудації і еміграції;
3) проліферацію.
Ці компоненти іноді називають стадіями запалення (рис. 40). Однак варто пам'ятати, що зазначені процеси не є строго послідовними, оскільки вони перекриваються в часі.
14.7. У чому сутність стадії альтерації?
В основі альтерації лежать дві групи явищ:
1) ушкодження клітин і позаклітинних структур;
2) утворення медіаторів запалення.
14.8. Що таке первинна і вторинна альтерація?
Первинна альтерація — це ушкодження тканини, яке виникає внаслідок безпосередньої дії флогогенних агентів.
Вторинна альтерація — це ушкодження тканини, що виникає в результаті дії факторів, які утворилися внаслідок первинної альтерації
14.13. Що таке медіатори запалення? Назвіть основні їх класи.
Медіатори запалення — це біологічно активні сполуки, які утворюються у вогнищі запалення і визначають його патогенез.
Розрізняють медіатори клітинного (утворюються в клітинах) і плазмового (утворюються й надходять із плазми крові) походження.
До медіаторів клітинного походження відносять лізосомні фактори, продукти вільнорадикального окиснення, продукти тканинних базофілів, похідні арахідонової кислоти, цитокіни, фактори росту.
Медіаторами плазмового походження є кініни, продукти активації комплементу, продукти активації системи зсідання крові і фібринолітичної системи
Патогенез гострого запалення, стадії. Поєднання патологічних та пристосувально-компенсаторних змін в динаміці гострого запалення. Альтерація, види, причини, механізми.
У патогенезі запалення розрізняють:
1) альтерацію;
2) порушення мікроциркуляції'з явищами ексудації і еміграції;
3) проліферацію.
Ці компоненти іноді називають стадіями запалення (рис. 40). Однак варто пам'ятати, що зазначені процеси не є строго послідовними, оскільки вони перекриваються в часі.
14.7. У чому сутність стадії альтерації?
В основі альтерації лежать дві групи явищ:
1) ушкодження клітин і позаклітинних структур;
2) утворення медіаторів запалення.
14.8. Що таке первинна і вторинна альтерація?
Первинна альтерація — це ушкодження тканини, яке виникає внаслідок безпосередньої дії флогогенних агентів.
Вторинна альтерація — це ушкодження тканини, що виникає в результаті дії факторів, які утворилися внаслідок первинної альтерації.
14.9. Які фактори викликають розвиток вторинної альтерації у вогнищі запалення?
1. Медіатори запалення (лізосомні фактори, активований комплемент, лімфокіни-лімфотоксини).
2. Вільні радикали й пероксиди.
3. Гіпоксія, що виникає в результаті місцевих розладів кровообігу.
4. Місцевий ацидоз.
5. Підвищення осмотичного і онкотичного тиску у вогнищі запалення.
Назвіть причини розвитку місцевого ацидозу у вогнищі запалення.
Розрізняють первинний і вторинний ацидоз. Первинний ацидоз виникає в перші ЗО хв. унаслідок деполімеризації основної інтерстиціальної речовини і вивільнення карбоксильних і сульфатних груп.
Вторинний ацидоз розвивається пізніше й обумовлений порушеннями обміну речовин у вогнищі запалення. До його виникнення причетні накопичення молочної кислоти (активація гліколізу), вихід з ушкоджених клітин недоокиснених продуктів циклу Кребса (три- і дикарбонових кислот), вивільнення вільних жирових кислот, амінокислот і фосфорної кислоти в результаті гідролітичного розщеплення тригліце-ридів, фосфоліпідів, білків, АТФ.
14.11. Чому в осередку запалення розвиваються гіперосмія й гіперон кія?
Збільшення осмотичного тиску у вогнищі запалення (гіперосмія) зумовлене насамперед виходом іонів калію з ушкоджених клітин, а також вивільненням калію зі зв'язаного з внутрішньоклітинними білками стану. Останнє є результатом протеолізу, що відбувається в клітинах за умов їхнього ушкодження.
Збільшення онкотичного тиску (гіперонкія) обумовлене:
1) надходженням білків у тканину із крові в процесі ексудації (плазмове джерело);
2) розщепленням великих білкових молекул на дрібніші під дією лізосомних ферментів (тканинне джерело).
Зміни місцевого кровообігу при запаленні (за Ю. Конгеймом). Патогенез окремих стадій судинної реакції у вогнищі гострого запалення.
Назвіть стадії порушень місцевого кровообігу у вогнищі запалення. Хто їх уперше описав?
I. Короткочасна ішемія (тривалість від 10—20 с до кількох хвилин).
II. Артеріальна гіперемія (триває 20—30 хв, максимум до 1 год).
III. Венозна гіперемія.
IV. Стаз. Уперше зазначені зміни описав Ю. Конгейм (1867), вивчаючи кровообіг у брижі жаби під час запалення.
14.23. Який механізм лежить в основі короткочасної ішемії на початку запалення?
Короткочасну ішемію на початку запалення обумовлює рефлекторний спазм артеріол. Він пов'язаний зі збудженням судинозвужувальних адренергічних нервів і виділенням їхніми закінченнями катехоламінів. Останні, діючи на а-адренорецепто-ри, викликають скорочення гладких м'язів судинної стінки.
Ішемія, що виникає, є короткочасна, тому що швидко настає виснаження катехо-ламінових депо в нервових закінченнях і відбувається руйнування вивільнених медіаторів відповідними ферментами, зокрема, моноаміноксидазою. Крім того, вазокон-стрикція в деяких тканинах може перекриватися судинорозширювальним впливом холінергічних нервів, що реалізується за типом аксон-рефлексу.
14.24. Назвіть механізми розвитку артеріальної гіперемії у вогнищі запалення.
1. Нейрогенні механізми (нейротонічний і нейропаралітичний) (див. розд. 13). Вони мають значення в перші хвилини розвитку артеріальної гіперемії.
2. Вплив фізично-хімічних факторів: ацидозу, збільшення вмісту іонів калію в тканині, гіпоксії та ін.
3. Вплив продуктів метаболізму: молочної кислоти, АДФ, АМФ, аденозину.
4. Дія медіаторів запалення:
а) гістаміну і серотоніну;
б) кінінів (брадикініну і калідину);
в) простагландинів і простациклінів.
14.25. Які фактори викликають перехід артеріальної гіперемії у венозну в процесі розвитку запалення?
Можна виділити дві групи таких факторів.
І. Внутрішньосудинні фактори:
1) збільшення в'язкості крові;
2) мікротромбоутворення;
3) зсідання крові;
4) крайове стояння лейкоцитів;
5) агрегація еритроцитів;
6) набрякання ендотеліальних клітин. II. Позасудинні фактори:
1) здавлювання венозних судин набряковою рідиною;
2) втрата венулами еластичних властивостей внаслідок розщеплення колагену і еластину лізосомними ферментами (В. В. Воронін).
Ексудація в осередку запалення, її причини і механізми. Фази підвищення проникності судинної стінки. Види ексудатів.
Що таке ексудація? Які механізми лежать в основі виходу рідкої частини крові з судин у запалену тканину?
Ексудація - це вихід рідини й розчинених у ній компонентів плазми крові із кровоносних судин у тканину. У широкому значенні слова це поняття включає й еміграцію лейкоцитів.
В основі ексудації лежать такі механізми:
1) підвищення проникності судинної стінки;
2) збільшення гідростатичного тиску в судинах;
3) збільшення осмотичного і онкотичного тиску в тканині.
14.27. Назвіть механізми підвищення проникності судинної стінки при запаленні.
1. Активація мікровезикулярного транспорту через ендотеліальні клітини.
2. Утворення наскрізних трансклітшних каналів в ендотеліоцитах (є наслідком значного посилення мікровезикулярного транспорту).
3. Збільшення просвіту міжендотеліальних щілин (відбувається в результаті скорочення й округлення ендотеліоцитів).
4. Десквамація (злущування) ендотелію. Є проявом первинної й вторинної альтерації.
5. Деполімеризація речовин, що з'єднують ендотеліальні клітини і є компонентами базальної мембрани судинної стінки.
14.28. Що є причиною підвищення проникності судинної стінки у вогнищі запалення?
До основних причин підвищення проникності судин відносять:
а) продукти дегрануляції тканинних базофілів (гістамін і серотонін);
б) кініни (брадикінін і калідин);
в) простагландини й деякі лейкотрієни;
г) лізосомні ферменти (еластаза, колагеназа, гіалуронідаза) і неферментні катіонні білки;
ґ) фібринопептиди й продукти деградації фібрину;
д) ацидоз.
14.29. Яка динаміка підвищення проникності судин при запаленні?
Виділяють дві фази підвищення проникності судин. Пік першої (ранньої) фази припадає на 10-15 хв від початку запалення. Основною причиною її розвитку є гістамін.
Друга (пізня) фаза починається через 1 год від початку запалення й триває кілька діб. До її розвитку причетні всі інші медіатори запалення, що підвищують проникність судин.
Еміграція лейкоцитів в осередку запалення. Послідовність, причини і механізми еміграції лейкоцитів. Роль лейкоцитів у розвитку місцевих та загальних проявів запалення. Механізми знешкодження мікробів лейкоцитами.
Що таке еміграція лейкоцитів? Які лейкоцити і в якій послідовності емігрують у вогнище запалення?
Еміграція — це перехід лейкоцитів крові із кровоносних судин у тканину.
Уперше послідовність еміграції лейкоцитів описав І. Мечніков. Спочатку в запалену тканину виходять полінуклеарні фагоцити, зокрема нейтрофіли. Вони знищують мікробів, що стали причиною ушкодження тканини. Потім у вогнище виходять мононуклеарні фагоцити - моноцити. Вони фагоцитують загиблі клітини, тканинний детрит, розчищаючи тим самим "поле бою". На кінцевих етапах, особливо при імунному запаленні, у тканину надходять лімфоцити.
14.31. Що таке крайове стояння лейкоцитів? Які його механізми?
Крайове стояння лейкоцитів (маргінація) — це перехід лейкоцитів із циркулюючого пулу в пристінковий (маргінальний). Воно триває від кількох хвилин до 1 години. В основі цього явища лежать такі механізми.
1. При уповільненні течії крові (венозна гіперемія) лейкоцити як найлегші формені елементи відкидаються за законами фізики на периферію.
2. Відбувається випадання ниток фібрину на поверхні ендотелію. Гладка в нормі поверхня ендотелію стає шорсткою. "Бахрома", що утворилася, затримує лейкоцити.
3. Має місце електростатична взаємодія лейкоцитів з ендотеліальними клітинами, її пояснюють втратою лейкоцитами поверхневого негативного заряду і утворенням "кальцієвих містків" між лейкоцитами й ендотеліоцитами.
4. На поверхні лейкоцитів і ендотеліальних клітин з'являються так звані "адгезивні білки ", які специфічно взаємодіють один з одним
Що таке адгезивні білки лейкоцитів і ендотеліальних клітин? Що викликає їхню появу?
Адгезивні білки синтезуються в лейкоцитах та ендотеліальних клітинах і зберігаються у внутрішньоклітинних везикулах. При активації клітин відбувається злиття везикул з плазматичною мембраною, у результаті чого зазначені білки виявляються вмонтованими в цю мембрану. Специфічні ділянки молекул розташовуються зовні і можуть взаємодіяти з відповідними структурами адгезивних білків інших клітин.
Активаторами утворення і включення адгезивних білків у мембрану лейкоцитів є:
а) побічний продукт активації комплементу С5а;
б) лейкотрієни В4;
в) фактор активації тромбоцитів (ФАТ).
Аналогічні процеси в ендотеліальних клітинах викликають: а) ендотоксини бактерій;
б) інтерлейкін-1;
в) фактор некрозу пухлин.
14.33. Яким чином лейкоцити долають судинну стінку, емігруючи в запалену тканину?
Емігруючи в тканину, лейкоцити долають два бар'єри капілярної стінки: ендотелій і базальну мембрану. Нейтрофіли і макрофаги проходять крізь ендотелій через міжендотеліальні щілини. Вони випускають свої псевдоподії в простір між ендотелі-оцитами і "розсовують" клітини.
Подолання базальної мембрани може бути обумовлено двома механізмами. Перший з них полягає в явищі тиксотропії-при контакті нейтрофіла з базальною мембраною її колоїди переходять зі стану гелю в стан золю (відбувається розрідження мембрани). Нейтрофіл легко проходить через золь, після чого золь знову перетворюється на щільний гель. Другий механізм полягає у виділенні нейтрофілами нейтральних протеаз (еластази, колагенази), які розщеплюють волокнисті компоненти базальної мембрани.
Порушення обміну речовин в осередку запалення.
Які порушення обміну речовин закономірно виникають у вогнищі запалення?
Запалення завжди починається з посилення обміну речовин. У гострому періоді запалення переважають процеси розпаду, катаболізму. Відбувається збільшення
інтенсивності споживання кисню й активація процесів гліколізу. Під дією лізосомних гідролаз великі молекули розщеплюються на дрібні. Це все характеризують терміном "пожежа обміну". Аналогія полягає не тільки в тому, що обмін речовин у вогнищі запалення різко підвищений, але й у тому, що "горіння" іде не до кінця, а з утворенням недоокиснених продуктів.
Пізніше відбувається активація анаболічних процесів, що забезпечують явища відновлення (репарації). Збільшується синтез нуклеїнових кислот, різко зростає утворення глікозаміногліканів, глікопротеїнів, колагену й інших компонентів сполучної тканини.
Велике значення у вивченні біохімічних змін у вогнищі запалення мали роботи Менкіна і Альперна.
Медіатори запалення, їх класифікація. Механізми утворення та біологічної дії плазмових медіаторів запалення.
Медіатори запалення — це біологічно активні сполуки, які утворюються у вогнищі запалення і визначають його патогенез.
Розрізняють медіатори клітинного (утворюються в клітинах) і плазмового (утворюються й надходять із плазми крові) походження.
До медіаторів клітинного походження відносять лізосомні фактори, продукти вільнорадикального окиснення, продукти тканинних базофілів, похідні арахідонової кислоти, цитокіни, фактори росту.
Медіаторами плазмового походження є кініни, продукти активації комплементу, продукти активації системи зсідання крові і фібринолітичної системи.
14.20. Яке значення має комплемент і продукти його активації в патогенезі запалення?
Активований комплемент здатний викликати вторинну альтерацію у вогнищі імунного запалення.
Побічні продукти його активації- СЗа й С5а- викликають дегрануляцію тканинних базофілів і є сильними хемотаксинами для нейтрофілів.
Проміжні продукти, зокрема СЗЬ, мають властивості опсонінів, тобто полегшують фагоцитоз бактерій. Крім того, проміжні продукти, що виявляють протеазну активність, можуть активувати калікреїн-кінінову систему і систему зсідання крові.
14.21. Які продукти активації системи зсідання крові і фібринолітичної системи можуть впливати на патогенез запалення?
У розвитку запалення можуть мати значення:
1) фібршопептиди (відщеплюються від фібриногену при перетворенні його у фібрин) - збільшують проникність судин і активують хемотаксис лейкоцитів;
2) продукти деградації фібрину — збільшують проникність судин;
3) активні протеази (тромбін, плазмін) — активують калікреїн-кінінову систему й систему комплементу
Медіатори запалення клітинного походження; характеристика їх біологічних ефектів.
Яку роль відіграють лізосомні фактори в патогенезі запалення?
До лізосомних факторів відносять: лізосомні ферменти (кислі й нейтральні гід-ролази) і неферментні катіонні білки.
Роль лізосомних ферментів, основним джерелом яких є лейкоцити, полягає в ініціюванні таких змін.
1. Вони викликають вторинну альтерацію.
2. Беруть участь в утворенні і активації інших медіаторів запалення: стимулюють дегрануляцію тканинних базофілів, активують калікреїн-кінінову систему, систему комплементу; вивільнюють арахідонову кислоту з фосфоліпідів клітинних мембран.
3. Безпосередньо підвищують проникність капілярів завдяки дії еластази, колагена-зи і гіалуронідази на компоненти базальної мембрани судинної стінки.
4. Викликають розвиток фізико-хімічних і метаболічних змін у вогнищі запалення: активують гідролітичне розщеплення речовин, чим сприяють розвитку місцевого ацидозу й гіперонкії.
Неферментні катіонні білки лізосом викликають вторинну альтерацію, підвищують проникність судин, активують хемотаксис лейкоцитів.
14.15. Які фактори можуть викликати дегрануляцію тканинних базофілів у вогнищі запалення?
1. Безпосередня дія флогогенного агента на тканинні базофіли (механічне ушкодження, температура, продукти бактерій, хімічні речовини - лібератори гістаміну).
2. Комплекси антиген-антитіло.
3. Активні протеази, зокрема, лізосомні.
4. Побічні продукти активації комплементу - СЗа, С5а.
14.16. Які медіатори запалення вивільняються при дегрануляції тканинних базофілів ? Яку вони мають дію у вогнищі запалення?
1. Біогенні аміни — гістамін і в деяких видів тварин (зокрема щурів) - серотонін. Основні ефекти гістаміну, що мають важливе значення в патогенезі запалення: а) розширення артеріол, що веде до розвитку артеріальної гіперемії в осередку запалення;
б) підвищення проникності мікросуцин (венул) - одна з причин запального набряку;
в) подразнення нервових закінчень, що зумовлює розвиток болю;
г) спазм гладких м'язів бронхів, матки, кишок. Цим, зокрема, пояснюють порушення функції зазначених органів при їхньому запаленні.
2. Гепарин. Другий основний компонент гранул тканинних базофілів є глікозаміно-гліканом. Його вважають протизапальним медіатором, оскільки він (1) має антикоагулянту дію, (2) гальмує адгезію і агрегацію тромбоцитів, (3) зв'язує біогенні аміни, (4) пригнічує активацію комплементу та калікреїн-кінінової системи.
3. Фактори, що впливають на клітини крові. До них можна віднести поліпептиди: (1) фактор еміграції еозинофілів, (2) фактор еміграції нейтрофілів, а також сполуку фосфоліпідного походження — (3) фактор*агрегації тромбоцитів (ФАТ).
Останній відіграє особливо важливу роль у патогенезі запалення. Утворюючись одразу після стимуляції тканинних базофілів, ФАТ зумовлює такі ефекти у вогнищі запалення:
а) активує процеси агрегації тромбоцитів та вивільнення їхніх гранул. Як наслідок, з тромбоцитів в осередок запалення виходять серотонін адреналін, аденінові нуклеотиди (АТФ, АДФ, АМФ), арахідонова кислота та тромбок-сани, тромбоцитарний фактор росту та інші;
б) навіть у дуже низьких концентраціях зумовлює розширення артеріол (артеріальну гіперемію) і збільшення проникності венул. Кількісно ці ефекти ФАТ відповідно в 100 і 10000 разів сильніші за дію гістаміну;
в) значно посилює адгезію лейкоцитів до ендотелію судин (крайове стояння) і стимулює хемотаксис нейтрофілів та макрофагів у вогнищі запалення.
Крім наведених вище медіаторів запалення під час дегрануляції тканинних базофілів вивільнюються лейкотрієни (повільно реагуюча субстанція анафілаксії), гідролітичні ферменти, катіонні білки.
14.17. Як відбувається активація калікреїн-кінінової системи? Назвіть основні функціональні ефекти кінінів.
У плазмі крові є неактивний протеолітичний фермент колікреїноген. З появою в крові активних протеаз (лізосомні ферменти, фактор Хагемана, трипсин, тромбін, плазмін та ін.) відбувається відщеплення ділянки молекули калікреїногену, в результаті чого він перетворюється на активний фермент — калікреїн.
. Під дією калікреїну відбувається відщеплення від а2-глобуліну плазми крові (кі-ніногену) пептидів, які отримали назву кініни. Найважливішими кінінами є калідин і брадикінін, що складаються відповідно з 9 і 10 амінокислотних залишків (рис. 41).
У вогнищі запалення кініни викликають: 1) розширення артеріол (артеріальну гіперемію); 2) підвищення проникності судинної стінки; 3) подразнення нервових закінчень (біль).
14.18. Які медіатори запалення є похідними арахідонової кислоти? Як вони утворюються і як діють ?
Похідними арахідонової кислоти є простагландини, тромбоксани, простациклі-ни, лейкотрієни (рис. 42).
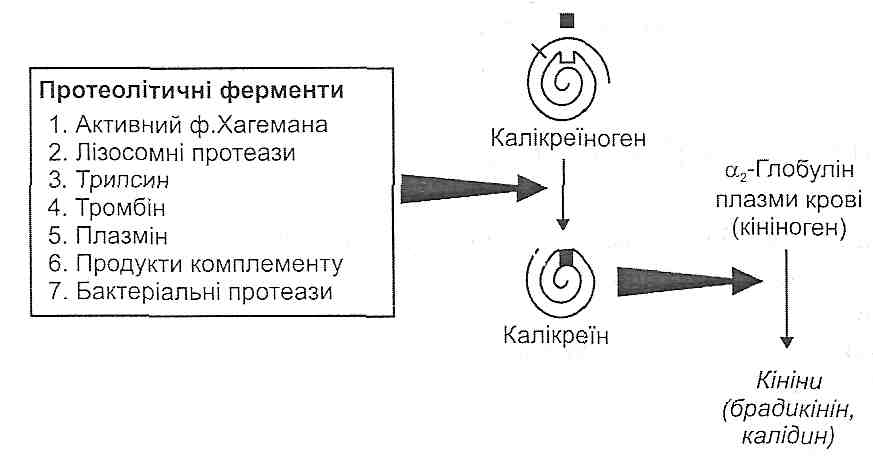
Рис. 41. Механізми активації калікреїн-кінінової системи
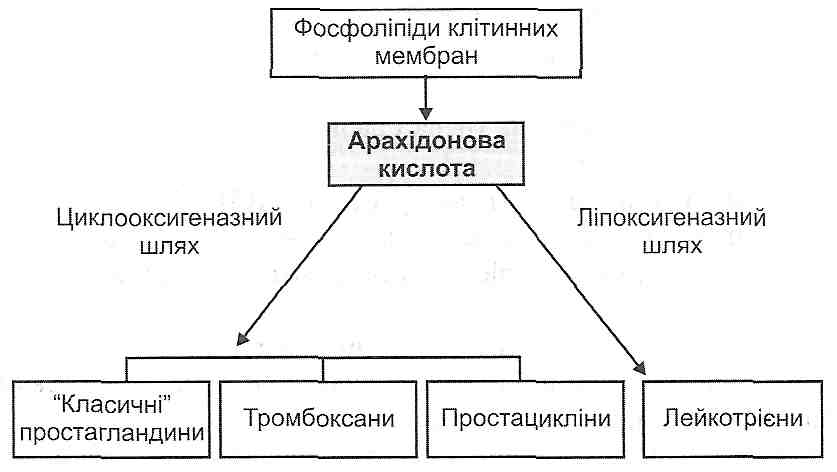
Рис. 42. Похідні арахідонової кислоти - медіатори запалення
Під дією ферменту фосфоліпази А2 (активується іонами кальцію) відбувається вивільнення арахідонової кислоти з фосфоліпідів клітинних мембран. Далі можливі два шляхи її перетворення: циклооксигеназний і ліпоксигеназний. У результаті активації першого утворюються "класичні" простагландини Е2, D2, F2 , тромбоксани й простацикліни, при активації другого - лейкотрієни С4, D4, E4.
Простагландини утворюються практично у всіх клітинах. Вони мають властивість розширювати артеріоли, звужувати венули, підвищувати проникність судинної стінки, зменшувати поріг больової чутливості нервових закінчень.
Тромбоксани утворюються в тромбоцитах. Вони викликають звуження артеріол і агрегацію тромбоцитів.
Простацикліни переважно вивільняються ендотеліальними клітинами судин і є антагоністами тромбоксанів. З їх дією пов'язано розширення артеріол і пригнічення агрегації тромбоцитів.
Місцем утворення лейкотрієнів є лейкоцити і тканинні базофіли. У вогнищі запалення вони стимулюють хемотаксис лейкоцитів і підвищують проникність кровоносних судин.
14.19. Що таке цитокіни? Яку роль вони відіграють у патогенезі запалення?
Цитокіни - це збірне поняття для позначення великої групи біологічно активних речовин білково-пептидної природи, що регулюють взаємодію між різними типами клітин.
Цитокіни синтезуються (1) активованими лімфоцитами (лшфокіни);2) моноцитами і макрофагами (монокіни), а також (3) багатьма іншими клітинами (нейтрофілами, фібробластами, ендотеліальними клітинами, тканинними базофілами, клітинами нейроглії та ін.).
На сьогодні описано понад 50 різних цитокінів. Залежно від функціональних ефектів їх поділяють на чотири групи.
I. Інтерлейкіни (ІЛ). Це сполуки, що регулюють взаємодію між різними видами лейкоцитів. Відомі нині 18 видів інтерлейкінів беруть участь у здійсненні імунних реакцій, у патогенезі алергії.
II. Інтерферони (ІНФ). Зазначена група білків здійснює природний неспецифічний противірусний захист.
III. Гемопоетичні колонієстимулятивні фактори (КСФ). Ці сполуки (гемопоети-ни) здійснюють регуляцію кровотворення в червоному кістковому мозку.
IV. Фактори, що пригнічують ріст пухлин, зокрема, фактор некрозу пухлин (ФНП)
За участю в патогенезі запалення цитокіни поділяють на (1) прозапсиїьні і (2) протизапальні. Спричинювані ними ефекти виявляють себе на місцевому рівні (в осередку запалення) і на рівні організму (системна дія).
Цитокіни є причетними до розвитку основних подій, що складають суть запального процесу, а саме:
а) вторинної альтерації. З-поміж інших цитокінів прямий стосунок до ушкодження клітин і позаклітинних компонентів має ФНП-р (лімфотоксин) — продукт активації макрофагів і Т-лімфоцитів. Високі його концентрації (1) спричинюють цитоліз (т.зв. кілінг-ефект), (2) посилюють генерацію вільних радикалів у вогнищі запалення; (3) індукують синтез колагеназ і, як наслідок, сприяють деградації колагену;
б) еміграції лейкоцитів. Ряд цитокінів (ІЛ-1, ФНП, ІНФ-у) індукують синтез адге-зивних білків в ендотеліальних клітинах, що сприяє розвиткові крайового стояння (прилипання до поверхні ендотелію) нейтрофілів, моноцитів і лімфоцитів. Крім того, деякі інтерлейкіни (ІЛ-6, ІЛ-8) значно посилюють хемотаксис лейкоцитів у вогнищі запалення;
в) проліферації. Однією з властивостей багатьох цитокінів є їхня мітогенна активність, що виявляє себе посиленням процесів проліферації в осередку запалення. Водночас, деякі цитокіни (ІЛ-1, ФНП) стимулюють синтез колагену і новоутворення кровоносних судин (ангіогенез);
г) загальних проявів запалення (див. запит. 14.37). У розвитку таких змін особливо велике значення має інтерлейкін-1 (ІЛ-1) (рис. 43).
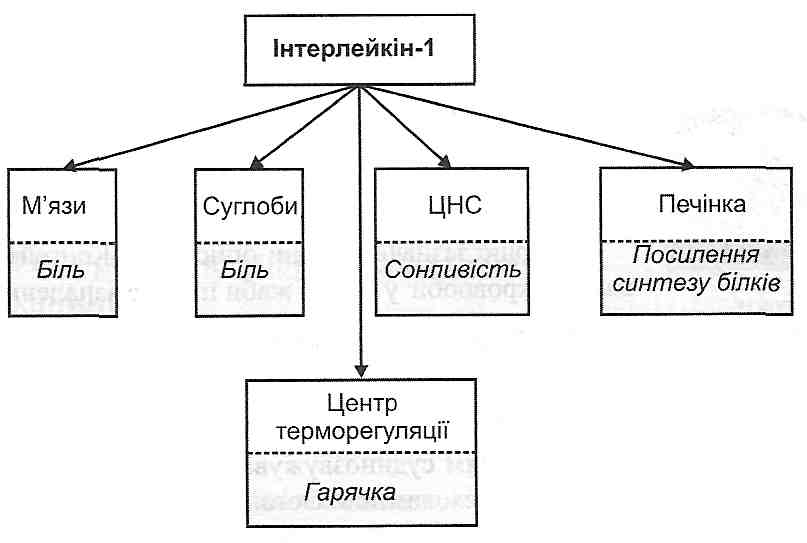
Рис. 43. Деякі системні ефекти інтерлейкіну-1
Серед відомих сьогодні цитокінів є й протизапальні медіатори, зокрема ІЛ-10, який пригнічує синтез лейкоцитами багатьох біологічно активних сполук — активних учасників запального процесу.
Проліферація клітин в осередку запалення, її механізми. Механізми мітогенної дії факторів росту і цитокінів. Регенерація та фіброплазія як способи заживлення.
У чому полягає сутність стадії проліферації в патогенезі запалення?
Стадія проліферації охоплює:
1) розмноження клітин, тобто власне проліферацію;
2) синтез позаклітинних компонентів сполучної тканини - колагену, еластину, про-теогліканів, глікопротеїнів. Ці події супроводжуються значним посиленням ана-болічних процесів.
14.35. Які фактори викликають активацію розмноження клітин у вогнищі запалення?
I. Зменшення концентрації в тканині кейлонів. Кейлоїш — це речовини білкової природи, які утворюються зрілими клітинами. Вони є інгібіторами клітинного поділу. При ушкодженні і загибелі клітин у вогнищі запалення концентрація кейлонів у тканині зменшується, а отже, знімається гальмівний вплив кейлонів на малодиференційовані (камбіальні) клітини. Вони починають ділитися. Поділ триває доти, доки концентрація кейлонів не збільшиться до рівня, що існував у неушкодженій тканині.
II. Збільшення концентрації в тканині стимуляторів проліферації— факторів росту. Фактори росту надходять у тканину з плазми крові або є продуктами клітин, що перебувають в осередку запалення. Прикладами можуть бути фактор росту епідермісу, фактор росту фібробластів, фактор росту тромбоцитарного походження, фактор росту нервів, фактор некрозу пухлин, інсуліноподібні фактори росту (соматомедини), лімфокіни (мітогенні фактори). Дія зазначених регуляторів здійснюється через активацію внутрішньоклітинних протеїнкіназ (протеїнкі-нази С і тирозинових протеїнкіназ).
Роль реактивності організму в розвитку запалення. Зв'язок між патологічною імунною відповіддю і запаленням. Вплив гормональних чинників на запалення??????????????????????????????
Гарячка: визначення поняття, принципи класифікації. Зв'язок між гарячкою і запаленням. Види пірогенів. Утворення пірогенів при інфекції, асептичному ушкодженні та імунних реакціях. Хімічна природа і походження вторинних пірогенів, механізм їх дії.
Гарячка - це типовий патологічний процес , який характерезується підвищенням темп тіла у відповідь на дію пірогенів та перебудови центру терморегуляціі.
Існує тільки у вищих теплокровних організмів та людини.
Які речовини називають пірогенними (пірогенами)? Як їх класифікують ?
Шрогени — це речовини, які є причиною розвитку гарячки. їх поділяють на:
а) інфекційні й неінфекційні;
б) природні й штучні;
в) екзогенні й ендогенні;
г) первинні й вторинні.
15.3. Наведіть приклади інфекційних і неінфекційних пірогенів.
До інфекційних пірогенів відносять:
1) ендотоксини грамнегативних бактерій (пірогенну дію має фрагмент токсину — ліпоїд А);
2) екзотоксини грампозитивних бактерій (дифтерійний, правцевий);
3) продукти діяльності патогенних грибків;
4) рикетсії;
5) віруси.
Неінфекційними пірогенами є:
1) компоненти несумісної за групами перелитої крові (трансфузійна гарячка);
2) екзогенні білки (білки молока при парентеральному його введенні);
3) продукти розпаду тканин.
15.4. Що таке природні і штучні пірогени?
Природними називають пірогени, які існують у природі або утворюються природно з непірогенних речовин.
Штучні пірогени отримують шляхом обробки нативних бактеріальних токсинів і використовують з лікувальною метою (піротерапія). Найвідомішими є пірогенал, отриманий із Pseudomonas aeruginosa, і пірексаль, виділений із Salmonella abortus equi.
15.5. Що називають екзогенними й ендогенними пірогенами?
Екзогенні пірогени надходять або їх вводять ззовні. При введенні екзогенного пірогену парентерально гарячка виникає через 45-90 хв.
Ендогенні пірогени утворюються в самому організмі. До них відносять:
1) продукти первинної і вторинної альтерації, що утворилися у вогнищі запалення;
2) продукти, які надходять у кров з осередків некрозу (наприклад, при інфаркті міокарда);
3) метаболіти стероїдних гормонів;
4) комплекси антиген-антитіло;
5) лейкоцитарні пірогени - продукти діяльності нейтрофілів і макрофагів.
15.6. Що таке первинні і вторинні пірогени?
Первинними називають пірогени, які безпосередньо не впливають на центр терморегуляції, їх пірогенна дія опосередковується утворенням і вивільненням так званих лейкоцитарних пірогенів.
Вторинними називають лейкоцитарні пірогени, які утворюються і вивільнюються лейкоцитами під впливом первинних пірогенів. Вторинні пірогени мають здатність безпосередньо впливати на центр терморегуляції й викликати розвиток гарячки.
Сьогодні відомо, що одним із вторинних лейкоцитарних пірогенів є інтерлейкін-1. Ця речовина утворюється і вивільнюється лейкоцитами (нейтрофілами й макрофагами) у процесі активації фагоцитозу. Поряд з дією на центр терморегуляції інтерлейкін-1 виявляє цілу низку інших ефектів, що визначають клінічні прояви гарячки
Які існують докази того, що дія первинних пірогенів пов'язана з утворенням вторинних?
1. Первинні пірогени мають досить високий латентний період дії (у людини 45 хв. і більше), у той час як латентний період дії вторинних пірогенів — 10 хв. і менше.
2. До дії первинних пірогенів розвивається толерантність (звикання), тобто для отримання гарячки треба збільшувати дозу пірогену з кожним наступним введенням. У той же час при повторних введеннях вторинних пірогенів толерантність не виникає.
3. Якщо фагоцитарна функція нейтрофілів і макрофагів порушена (наприклад, при лейкопенії, наповненні фагоцитів тушшю), то при введенні первинних пірогенів гарячка не виникає. ;
4. При безпосередньому введенні первинних пірогенів у терморегуляторні центри гіпоталамуса гарячка не розвивається, у той час як введення в ці центри вторинних лейкоцитарних пірогенів закономірно супроводжується підвищенням температури тіла.
15.8. Які послідовні процеси становлять сутність патогенезу гарячки?
У патогенезі гарячки можна виділити кілька етапів.
I. Індукція утворення й вивільнення вторинного лейкоцитарного пірогену (інтер-лейкіну-1) первинними пірогенами.
II. Вплив інтерлейкіну-1 на центр терморегуляції й перебудова його роботи.
III. Етап клінічних проявів гарячки (зміна температури тіла).
Які існують докази ролі простагландинів Ев патогенезі гарячки?
1. Введення простагландинів Е мікроін'єкторами в центр терморегуляції (у ділянку "установочної точки" гіпоталамуса) викликає підвищення температури тіла.
2. Фармакологічні препарати, які інгібують циклооксигеназу (фермент синтезу простагландинів), мають жарознижувальний ефект при гарячці. До таких препаратів, зокрема, належать ацетилсаліцилова кислота, індометацин.
3. Ці ж самі препарати не впливають на рівень нормальної температури тіла. Звідси висновок, що простагландини Е не беруть участі в регуляції температурного гомеостазу в нормі, а утворюються тільки при гарячці.
4. Глюкокортикоїди, що пригнічують активність фосфоліпази А2 через ліпокортино-вий механізм (див. розд. 33), зменшують утворення простагландинів Е, завдяки чому гальмують розвиток гарячки.
Гарячка: стадії розвитку, зміни терморегуляції, обміну речовин та фізіологічних функцій. Захисне значення та патологічні прояви гарячки. Принципи жарознижувальної терапії. Поняття про піротерапію.
Які послідовні процеси становлять сутність патогенезу гарячки?
У патогенезі гарячки можна виділити кілька етапів.
I. Індукція утворення й вивільнення вторинного лейкоцитарного пірогену (інтер-лейкіну-1) первинними пірогенами.
II. Вплив інтерлейкіну-1 на центр терморегуляції й перебудова його роботи.
III. Етап клінічних проявів гарячки (зміна температури тіла).
15.12. Назвіть стадії гарячки.
Клінічно виділяють три стадії гарячки (рис. 47):
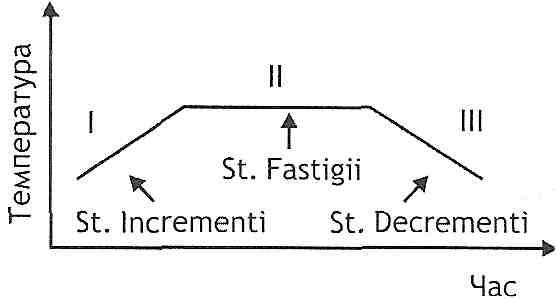
Рис. 47. Стадії гарячки
I — підвищення температури (st. incrementi);
II - стояння підвищеної температури (st. fastigii);
III - зниження температури (st. decre-menti).
. Поясніть, як відбувається підвищення температури тіла на І стадії гарячки.
На самому початку різко зменшується тепловіддача. У результаті активації сим-патоадреналової системи звужуються кровоносні судини шкіри й кінцівок, скорочуються гладкі м'язи, що піднімають волосся (у тварин піднімається шерсть, у людини виникає ознака "гусячої шкіри").
Ці зміни мають два наслідки. З одного боку, різке обмеження тепловіддачі саме по собі веде до підвищення температури ядра. З другого боку, зменшується температура шкіри, що викликає збудження холодових терморецепторів. Інформація про зниження температури "оболонки" надходить у центр терморегуляції, а звідти в кору головного мозку — у людини виникає відчуття холоду. Крім того, відбувається збудження підкіркових рухових центрів, у результаті чого підвищується тонус скелетних м'язів, розвивається тремтіння (озноб). Збільшується скоротливий термогенез.
Поряд із цим відбувається активація нескоротливого термогенезу, пов'язаного з підвищенням швидкості окисних процесів. Велике значення при цьому має підвищення інтенсивності клітинного дихання в бурій жировій тканині під дією катехо-ламінів.
Таким чином, підвищення температури тіла спочатку обумовлене зменшенням тепловіддачі, а потім збільшенням теплопродукції.
15.14. Поясніть механізми зниження температури тіла при завершенні гарячки. Які існують варіанти такого падіння?
Як тільки припиняється дія інтерлейкіну-1 на центр терморегуляції, у нейронах "установочної точки" зменшується вміст простагландинів Е, що веде до відновлення чутливості клітин до сигналів, які надходять від "термостата". Температура ядра починає сприйматися як підвищена, у результаті чого активується центр тепловіддачі й пригнічується центр теплопродукції. Найбільше значення при цьому мають дві фізіологічні реакції: розширення кровоносних судин шкіри й кінцівок та збільшення потоутворення і потовиділення. Ці реакції ведуть до збільшення тепловіддачі і, як наслідок, до зменшення температури тіла.
Буває два варіанти падіння температури:
1) критичне падіння — різке зменшення температури протягом кількох годин;
2) літичне падіння - поступове зменшення температури протягом кількох діб.
Критичне падіння може бути небезпечним, особливо у літніх і у хворих на серцево-судинні недуги людей у зв'язку з можливим падінням артеріального тиску і розвитком колапсу.
15.15. Які типи температурних кривих можуть бути характерні для гарячки? Які фактори впливають на динаміку зміни температури тіла при гарячці?
До основних типів температурних кривих відносять:
1) febris intermittent- температура нормалізується один або кілька разів на добу
(гнійна інфекція, абсцеси, туберкульоз);
2) febris remittens — коливання температури становлять більше 1 °С на добу, однак вона не повертається до норми (більшість вірусних і багато бактеріальних інфекцій);
3) febris continua - добові коливання температури становлять менше 1 °С (черевний і висипний тиф);
4) febris recurrens — приступи підвищення температури чергуються з періодами її нормалізації, що тривають кілька діб (поворотний тиф, малярія).
Динаміка змін температури при гарячці визначається, з одного боку, властивостями й особливостями життєвого циклу збудника, з другого - добовими (циркадни-ми) біологічними ритмами організму
У чому полягає захисно-пристосувальне значення гарячки?
При гарячці в організмі створюються несприятливі умови для розвитку збудників інфекційних хвороб і підвищується активність механізмів неспецифічної й специфічної резистентності організму. Зокрема: а) пригнічується розмноження багатьох вірусів, посилюється утворення інтерферонів;
б) збільшується фагоцитарна активність макрофагів і нейтрофілів;
в) підвищується інтенсивність синтезу антитіл;
г) зростає чутливість багатьох інфекційних збудників до дії лікарських речовин.
15.18. Які власне патологічні зміни можуть виникати при гарячці?
При температурі тіла, що перевищує 39 °С, можуть розвиватися:
а) порушення загального стану - нездужання, головний біль, відчуття жару та ін.;
б) розлади обміну речовин;
в) підвищення навантаження на серце (тахікардія, збільшення серцевого виштовху) або зменшення артеріального тиску при критичному падінні температури;
г) розлади центральної нервової системи, які можуть виявлятися маренням, галюцинаціями, а в дітей від 5 міс. до 5 років розвитком фебрильних судом. Можуть провокуватися напади епілепсії;
ґ) якщо температура перевищує 40 °С, послаблюється фагоцитоз, порушується життєдіяльність і функціональна активність лімфоцитів, збільшується чутливість організму до дії деяких екзотоксинів;
д) у вагітних можливе порушення розвитку плода, тератогенні ефекти.
15.19. Чим виявляє себе вплив гарячки на обмін речовин?
Насамперед, збільшується основний обмін (на 10-12 % при збільшенні температури на 1°С). Це викликає збільшення споживання кисню і поживних речовин. Оскільки апетит у хворого відсутній, іде використання ендогенних джерел енергії, людина втрачає у вазі.
Зростає потреба організму у воді, оскільки збільшуються нечутливі втрати води через шкіру й дихальні шляхи. У зв'язку з цим у маленьких дітей і важкохворих може розвиватися зневоднення. З метою його попередження особам з гарячкою рекомендують багато пити.
При помірній гарячці (38-39 °С) частота дихання й альвеолярна вентиляція зростають у більшій мірі, ніж утворення вуглекислого газу. У зв'язку із цим може розвиватися гіпокапнія й, як наслідок, газовий алкалоз. З метою його попередження рекомендують пити підкислену рідину.
При високій гарячці (понад 39 °С) відбувається мобілізація вільних жирових кислот і посилюється утворення в печінці кетонових тіл. Кетонемія може вести до розвитку негазового ацидозу.
При гарячці розвивається негативний азотистий баланс, обумовлений переважанням катаболізму білків над їх синтезом.
Що є основним патогенетичним принципом жарознижувальної терапії?
Основним патогенетичним принципом жарознижувальної терапії є зменшення "установочної точки" центру терморегуляції, що досягається пригніченням утворення простагландинів Е з допомогою інгібіторів циклооксигенази (ацетилсаліцилова кислота, індометацин, парацетамол) та інгібіторів фосфоліпази А2.
Основні відмінності між гарячкою, екзогенним перегріванням та іншими видами гіпертермії.
При гарячці має місце не порушення, а перебудова терморегуляції. Організм сам підтримує високу температуру, оскільки "установочна точка" терморегуляторного центру налаштована на більш високий рівень. Якщо тварину з гарячкою охолоджувати, то її температура не зменшується, а продовжує зберігатися високою.
При гіпертермії терморегуляція порушена. Температура тіла підвищується всупереч прагненням організму підтримувати температурний гомеостаз. "Установочна
точка" терморегуляторного центру не міняється. Якщо тварину з гіпертермією охолоджувати, то в результаті різкого збільшення тепловіддачі температура тіла починає зменшуватися (рис. 48).
Рис. 48. "Установочна точка " і температура прирізних видах порушень терморегуляції
Пухлина: визначення поняття, принципи класифікації пухлин. Загальні закономірності пухлинного росту. Молекулярно-генетичні основи безмежного росту і потенційного безсмертя пухлинних клітин.
Пухлина (тумор, бластома)- типовий патологічний процес, який характерезується неконтрольваним безмежним розмноженням клітин внаслідок порушення генів, які регулюють клітинний поділ, та розростання тканини, не повязаним із загальною структурою органу та його функціі.
Класифікація пухлин
злоякісні ( малігнус)
доброякісні ( бенінгус)
злоякісні з епітеліальноі тканини. -рак, канцер
з сполучноі тканини - саркома
Залежно від числа вогнищ виникнення пухлини:
уніцентричний ріст;
мультицентричний ріст.
В залежності від характеру взаємодії зростаючої пухлини з елементами навколишньої тканини:
експансивний ріст;
інфільтруючий (інвазивний) ріст;
апозиційний ріст.
Залежно від ставлення до просвіту порожнистого органа:
екзофітний ріст;
ендофітний ріст.
Загальною характеристикою злоякісних пухлин є їх виражені тканинні аплазія, атипізм (тобто втрата клітинами здатності до диференціювання із порушенням структури тканини, з якої розвивається пухлина), агресивне зростання з ураженням, як самого органу, так і інших прилеглих органів, схильність до метастазування, тобто до поширення клітин пухлини із током лімфи або крові по всьому організму з утворенням нових осередків пухлинного росту в багатьох органах, віддалених від первинного осередка. За темпами зростання більшість злоякісних пухлин перевершують доброякісні і, як правило, можуть досягати значних розмірів у короткі терміни. Розрізняють також вид місцедеструктуриючих злоякісних пухлин, що ростуть з утворенням інфільтрату в товщі тканини, призводячи до її руйнування, але, як правило, не метастазують. До таких форм відносять, наприклад, базаліому шкіри.
Типові властивості доброякісних та злоякісних пухлин. Види анаплазії. Шляхи й механізми метастазування.
1. Доброякісні пухлини складаються з добре диференційованих клітин. Ці пухлини зберігають типову структуру тканини, з якої утворилися. У той же час злоякісні пухлини характеризуються втратою диференціювання клітин, спрощенням і ати-повістю своєї будови.
2. Доброякісні пухлини часто ростуть повільно, їх ріст може зупинитися, а іноді спостерігається й зворотний розвиток (регресія). Для злоякісних пухлин, як правило, характерний швидкий ріст, що спонтанно не зупиняється. Мимовільна регресія таких пухлин невідома.
3. Доброякісні пухлини мають капсулу й ростуть експансивно, тобто не проростають у навколишні здорові тканини, а розсовують їх. Ріст злоякісних пухлин інвазивний (інфільтративний). Вони не мають капсули і проростають у навколишні тканини.
4. Доброякісні пухлини не метастазують, у той час як злоякісні зазвичай дають метастази.
5. Доброякісні пухлини добре лікуються хірургічними методами, летальних наслідків, як правило, не буває. Злоякісні пухлини при відсутності лікування призводять до смерті
Назвіть основні особливості росту злоякісних пухлин in vivo.
1. Моноклональнісшь походження. Пухлина росте "сама із себе", тобто походить із однієї первинно трансформованої клітини. Інші, розташовані поруч, клітини в процес пухлинного переродження не втягуються:.
2. Автономність, росту — незалежність росту пухлини від регуляторних впливів організму (нервової системи, гормонів, факторів росту, інгібіторів проліферації). Для деяких пухлин така автономність ле абсолютна, а відносна.
3. Анаплазія- набуття пухлинними клітинами властивостей, характерних для ембріонального етапу розвитку організму. Інакше кажучи, пухлинні клітини своїми структурними, функціональними, біохімічними, імунологічними властивостями нагадують ембріональні клітини.
4. Поява в пухлинних клітинах великої кількості хромосомних аберацій. У клітинах злоякісної пухлини закономірно виявляють такі види хромосомних порушень, як транслокація, делеція, інверсія, дуплікація (докладно див. розд. 6).
5. Пухлинна прогресія (див. запит. 16.19 і 16.20).
6. Інвазивність.
7. Метастазування.
16.41. Що таке метастазування? Як воно здійснюється?
Метастазування — це здатність злоякісної пухлини давати вторинні пухлинні вузли (метастази) у віддалених від первинної пухлини частинах організму.
Процес метастазування складається з трьох етапів. І. Вихід пухлинних клітин із тканини в лімфатичні або кровоносні судини - інтра-
вазація. Цьому передує прикріплення пухлинних клітин до компонентів базаль-
ної мембрани судин. В основі такого прикріплення лежить утворення "ламініно-вих містків" між поверхнею клітин пухлини і колагеном IV типу базальної мембрани. Глікопротеїд ламінін є компонентом інтерстиціальної речовини базальної мембрани. Деякі найбільш злоякісні клітини самі здатні продукувати цю речовину. Ламінін, з одного боку, взаємодіє з так званими ламініновими рецепторами пухлинних клітин, а з другого - з хімічними групами колагену IV типу.
II. Транспорт пухлинних клітин лімфою й кров'ю - дисемінація. Із клітин пухлини, що проникли в судини, утворюються пухлинні емболи. їх утворення пов'язане з агрегацією пухлинних клітин. У цьому процесі обов'язково беруть участь тромбоцити.
III. Вихід пухлинних клітин з судин у тканини й утворення вторинних вогнищ (метастазів)— екстравазація. Для локалізації метастазів мають значення місцезнаходження первинної пухлини, шлях метастазування (лімфогенний чи гематогенний), рецепторна взаємодія пухлинних клітин з ендотелієм судин. Останнім, зокрема, пояснюється факт специфічної локалізації метастазів деяких пухлин (раку простати в кістки, бронхокарциноми в головний мозок і надниркові залози, не-йробластоми в печінку і т. д.).
Етіологія пухлин. Загальна характеристика канцерогенів. Фактори ризику (генетичні/хромосомні дефекти, аномалії конституції) і умови виникнення та розвитку пухлин.?????????????
Назвіть основні причини виникнення злоякісних пухлин.
Існує три групи етіологічних факторів: хімічні (канцерогенні речовини), фізичні (іонізуюча радіація, ультрафіолетове випромінювання, висока температура, механічний вплив), біологічні (онкогенні віруси).
16.8. Хто і як уперше довів роль хімічних факторів у виникненні злоякісних пухлин?
У 1775 р. англійський лікар П. Ttomm уперше припустив, що хімічні сполуки можуть бути причиною раку. До такого висновку він дійшов на підставі своїх спостережень численних випадків раку шкіри мошонки у молодих людей, які багато років до виникнення хвороби, будучи дітьми, працювали сажотрусами. Було вперше встановлено зв'язок між виникненням раку сажотрусів і дією на шкіру сажі та смоли.
Однак тільки в 1915 p. японські вчені Ямагіва та Ішикава підтвердили цей висновок в експерименті. Вони вперше відтворили злоякісну пухлину (рак шкіри) шляхом тривалого, протягом 6-ти місяців утирання кам'яновугільної смоли в шкіру вуха кролів.
На початку 30-х років з кам'яновугільної смоли був виділений 3,4-бензпірен — речовина, з дією якої й був пов'язаний канцерогенний ефект продуктів неповного згоряння вугілля.
Фізичні канцерогени. Хімічні канцерогени: принципи класифікації, характеристика основних груп. Механізми хімічного канцерогенезу. Коканцерогенез та синканцерогенез.
Які фізичні фактори можуть мати значення у виникненні злоякісних пухлин?
Серед факторів фізичної природи до етіології пухлин можуть мати стосунок:
1) іонізуюча радіація;
2) ультрафіолетове випромінювання;
3) механічний вплив (тривалий тиск на тканини);
4) висока температура.
Роль іонізуючоїрадіації доводять в експериментах і численними клінічними спостереженнями. Радіаційний канцерогенез було виявлено вже через 7 років після відкриття рентгенівського випромінювання. Першою його жертвою став перший виробник рентгенівських трубок Фрікен, що перевіряв якість своєї продукції на власних руках. У нього виник рак шкіри.
При тривалому перебуванні білих щурів на сонці у них часто розвивається рак шкіри, що пов'язують із впливом ультрафіолетового випромінювання.
Розвиток пухлини в ділянці імплантації тваринам пластинок, виготовлених з хі-Вачно інертного матеріалу ("пластмасовий " канцерогенез), виникнення раку в місці тривалого тиску на шкіру кантрі (спеціальної сумки, у якій жителі Індії носять вугілля) - це приклади можливого значення механічних факторів у канцерогенезі.
Нарешті, часте виникнення пухлин порожнини рота у пастухів гірських районів, які п'ють дуже гарячий чай, наводить на думку про можливу роль високої температури в розвитку пухлин.
16.22. Назвіть основні закономірності канцерогенної дії іонізуючого випромінювання
Хоча для розвитку пухлин мають значення доза й вид випромінювання, немає лінійної залежності між частотою виникнення пухлинного росту і поглиненою дозою. Немає також і мінімальної порогової дози, так що будь-яке випромінювання в будь-якій дозі є потенційно небезпечним. За інших рівних умов тривалий і постійний вплив низьких доз, з погляду канцерогенезу, значно небезпечніший, ніж короткочас-ний вплив великих доз.
16.23. Що таке "пластмасовий" канцерогенез? У чому його особливості?
В експерименті було показано, що після імплантації пластмасових пластинок під шкіру в цій ділянці часто розвивається злоякісна пухлина. Таке явище отримало назву "пластмасового" канцерогенезу. Було встановлено такі його закономірності. 1. Для виникнення пухлини матеріал, з якого зроблено пластинки, не має особливого значення. Такий висновок зроблено на підставі результатів дослідження багатьох речовин, серед яких целофан, поліетилен, тефлон, скло, золото, платина та ін. | 2. Якщо з речовини зробити пластинку й імплантувати її - пухлина виникає, якщо пластинку подрібнити і ввести в тканину отриманий порошок - пухлина не розвивається.
3. Вирішальне значення має розмір пластинки. Пухлина виникає при імплантації пластинок розмірами не менше 0,5 х 0,5 см.
4. Пластинка має бути суцільною, тобто не мати отворів (перфорацій).
Відповідно до однієї з гіпотез, навколо пластинки утворюється сполучнотканинна капсула, серед колагенових волокон якої перебувають одиничні клітини. Вони є відірваними від інших клітин і сигналів, що регулюють інтенсивність проліфератив-них процесів. У зв'язку з цим створюються умови, які сприяють переродженню нормальних клітин у пухлинні.
16.9. Як класифікують хімічні канцерогени?
Хімічні канцерогени — це речовини, здатні викликати розвиток злоякісних пухлин. ї. За походженням розрізняють канцерогени природні і штучні. її. За хімічною будовою канцерогени можуть бути представлені:
а) поліциклічними ароматичними вуглеводнями (ПАВ);
б) ароматичними амінами;
в) нітрозосполуками;
г) мікотоксинами;
ґ) гетероциклічними вуглеводнями;
д) аміноазосполуками;
є) простими сполуками (миш'як, азбест та ін.).
III. Відносно організму хімічні канцерогени можуть бути екзогенними й ендогенними.
IV. За механізмом канцерогенного впливу розрізняють канцерогени прямої дії й канцерогени непрямої дії.
16.10. Наведіть приклади канцерогенів природного і штучного походження.
До канцерогенів природного походження відносять продукти життєдіяльності деяких грибів (мікотоксини) і продукти вулканічної діяльності.
Джерелами канцерогенів штучного походження є:
1) викиди промислових підприємств;
2) вихлопні гази автомобілів;
3) тютюновий дим;
4) продукти неправильної кулінарної обробки їжі (використання пересмажених жирів, порушення технології копчення та ін.).
16.11. Охарактеризуйте канцерогенну дію поліциклічних ароматичних вуглеводнів (ПАВ).
ЛАВ - це сполуки, які мають у структурі своїх молекул кілька сполучених бензольних кілець. Найпоширенішими і найбільш вивченими є такі їхні представники, як 3,4-бензпірен, диме-
тилбензантрацен (рис. 50). їх можна виявити в продуктах вулканічної діяльності, викидах промислових підприємств, вихлопних газах автомобілів, тютюновому димі, харчових продуктах при неправильному їх копченні та ін.
Головна особливість канцерогенного впливу ПАВ полягає в їх місцевій дії. Це означає, що вони викликають розвиток пухлин у тих органах і тканинах, з якими контактують. Так, при втиранні в шкіру виникає рак шкіри, при підшкірному введенні - саркома, при вдихуванні - рак легень, при надходженні з їжею - рак стравоходу, шлунка і кишок, при виділенні з молоком - рак молочних залоз.
16.12. У чому полягає особливість канцерогенної дії ароматичних амінів?
Ароматичні аміни — це сполуки, що містять у своїй структурі бензольні кільця й аміногрупи. Канцерогенні властивості мають анілін та його похідні, зокрема, 2-на-фтиламін, диметиламіноазобензол (ДАБ) (рис. 51). Ці сполуки широко використовуються в промисловості як основа барвників, вихідні речовини для синтезу лікарських препаратів і виробництва вибухових речовин.
Характерною рисою канцерогенної дії ароматичних амінів є їх органотропність. Незалежно від шляху надходження або введення речовини пухлини виникають у певних органах. Так, при введенні 2-нафтиламіну розвивається рак сечового міхура, а при введенні ДАБ - рак печінки.
16.13. Чим характеризується канцерогенний вплив нітрозосполук?
До нітрозосполук належать нітрозаміни і нітрозаміди (рис. 52). У дослідах на тваринах показано, що 90 % всіх вивчених нітрозосполук мають канцерогенну дію. Багато речовин цього класу викликають утворення пухлин після одноразового вве-
дення в порівняно невеликих дозах. Не виявлено жодного виду тварин (вивчалося 40 видів, від риб до приматів), які були б стійкі до канцерогенної дії нітрозосполук. Дуже чутливим до їх впливу є й організм людини.
Нітрозаміни можуть утворюватися в шлунку людини з неканцерогенних попере-дників (нітритів, амінокислот, амідопірину) при наявності хлороводневої (соляної) кислоти.
Для канцерогенної дії нітрозосполук характерна органотропність. Є речовини, які вибірково викликають розвиток пухлин головного мозку, нирок, печінки, шлунка та ін.
16.14. Наведіть приклади канцерогенно)'дії продуктів життєдіяльності грибів.
Серед продуктів життєдіяльності грибів (цвілі), що мають канцерогенну дію, найбільш вивчено афлашоксин В. Цей мікотоксин є гетероциклічною сполукою і продукується грибом Aspergillus flavum (див. рис. 52). Канцерогенна дія афлатоксину виявляє себе в дуже малих дозах - 0,02 мг/кг маси. Для порівняння, ціанід калію спричиняє токсичну дію в дозах, що перевищують зазначену в кілька десятків разів.
Із впливом мікотоксинів пов'язують розвиток первинного раку печінки у представників племені Банту в Африці. Клімат території, на якій проживає це плем'я, дуже вологий, тому продукти харчування швидко покриваються цвіллю. За традицією, що існує в тих місцях, такі продукти придатні до використання. Аналізи показали, що у споживаній їжі міститься афлатоксин В.
16.15. Який зміст вкладають у поняття "ендогенні канцерогени"?
Ендогенними називають канцерогени, які утворюються в організмі з його нормальних компонентів.
Остаточно не доведено, однак є підстави думати, що в організмі при порушенні обміну речовин можуть утворюватися канцерогенні поліциклічні ароматичні вуглеводні, їхніми джерелами можуть бути холестерол, жовчні кислоти, стероїдні гормони. Зокрема, показано, що обробка in vitro жовчних кислот призводить до появи ме-тилхолантрену — представника ПАВ, що має сильну канцерогенну дію.
Крім того, ендогенні канцерогени можуть з'являтися при порушенні обміну деяких амінокислот, зокрема, триптофану.
16.16. Які досліди доводять можливу участь гормонів у виникненні злоякісного пухлинного росту?
При порушенні регуляції секреції тронних гормонів аденогіпофіза (при порушенні механізмів зворотного зв'язку) їхня кількість у крові може істотно зростати. Впливаючи на органи-мішені, вони можуть стимулювати проліферацію клітин і розвиток пухлини.
Подібну можливість демонструють у такому досліді. У тварини видаляють один яєчник, а другий пересаджують у селезінку. Кров від селезінки надходить у печінку, де естрогени, що утворилися в яєчнику, руйнуються. До гіпофіза надходить кров, у якій мало естрогенів. Це викликає стимуляцію утворення гонадотропних гормонів. Останні, діючи на яєчник, посилюють секрецію статевих гормонів, однак їх, як
і раніше, мало в крові, що надходить у гіпофіз, оскільки відбувається руйнування естрогенів у печінці. Постійна стимулятивна дія гонадотропних гормонів на яєчник викликає проліферацію його клітин і у великій кількості випадків - розвиток злоякісних пухлин.
16.17. Що таке хімічні канцерогени прямої і непрямої дії? Дайте їх порівняльну характеристику.
Канцерогени прямої дії— це високоактивні хімічні сполуки, зокрема лактони, хлоретиламіни, епоксиди. Вони здатні безпосередньо взаємодіяти зі структурами клітин і викликати розвиток пухлини. Ці сполуки не вимагають жодних перетворень в організмі для прояву своєї канцерогенної дії.
Маючи високу реакційну здатність, прямі канцерогени не можуть накопичуватися в навколишньому середовищі (при взаємодії з компонентами зовнішнього середовища вони руйнуються). Тому, з погляду гігієни, вони не являють великої небезпеки для людини як фактори канцерогенезу.
Канцерогени непрямої дії— це інертні за своїми хімічними властивостями сполуки. До них, зокрема, належать поліциклічні ароматичні вуглеводні, ароматичні аміни, нітрозосполуки, афлатоксини. Маючи низьку реакційну здатність, зазначені канцерогени можуть накопичуватися в навколишньому середовищі, а тому становлять велику небезпеку для людини.
Канцерогенними ці сполуки стають в організмі тільки після ряду хімічних ферментативних перетворень, у результаті яких утворюються їхні активні форми - власне канцерогени (рис. 53). Подібним чином з ПАВ утворюються епоксиди, з ароматичних амінів- гідроксиламіни, з нітрозамінів - алкильний радикал. Ці форми канцерогенів впливають на генетичний апарат клітини й викликають її перетворення на пухлинну.
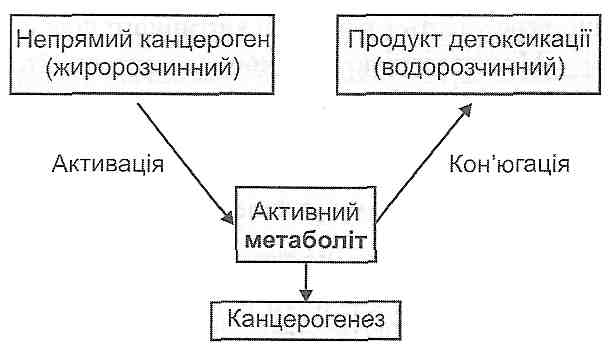
Рис. 53. Перетворення канцерогенів непрямої дії в печінці
16.18. Які властивості хімічних речовин обумовлюють їхню канцерогенну дію ?
У 60-ті роки подружжя Міллерів показало, що канцерогенна дія хімічних сполук пов'язана з наявністю в їхній структурі позитивно зарядженої (електрофільної) групи. У молекулах канцерогенів прямої дії така група з'являється відразу ж при розчиненні речовини у воді. У випадку канцерогенів непрямої дії електрофільна група утворюється в процесі ферментативних перетворень речовини в організмі.
Так, продуктами перетворення ПАВ, що містять позитивно заряджену групу, є епоксиди. Подружжя Пульманів показало, що канцерогенні властивості мають епоксиди, у молекулі яких атом кисню приєднаний до так званої К-зони (від нім. слова
Krebs - рак). Пізніше було встановлено, що найбільш канцерогенними є епоксиди з атомом кисню в так званій бей-ділящі молекули ПАВ (від англ. bay - затока) (рис. 54).
Таким чином, знаючи хімічну будову молекули ПАВ, можна передбачити канцерогенні властивості речовини.
16.19. Які стадії проходить хімічний канцерогенез? У чому їхня сутність ?
У хімічному канцерогенезі виділяють три стадії: ініціацію, промоцію і пухлинну прогресію.
Сутність ініціації полягає в тому, що під впливом хімічного канцерогену відбувається
трансформація клітини, тобто перетворення її з нормальної на пухлинну.
Під час стадії промоціїтрансформована клітина отримує стимул до розмноження, пухлина починає рости. Речовини, що стимулюють розмноження трансформованих клітин, отримали назву промоторів. Самі вони можуть бути й неканцерогенни-ми. Дуже сильними промоторами є форболові ефіри, які активують протеїнкіназу С, що бере участь у регуляції клітинного поділу.
Між ініціацією і промоцією може пройти багато часу (іноді роки). Більшість вивчених хімічних канцерогенів є повними, тобто виявляють властивості й ініціаторів, і промоторів.
Пухлинна прогресія — це якісні зміни властивостей пухлини в процесі її розвитку. Як правило, згодом пухлина набуває усе більш і більш злоякісних властивостей.
16.20. Чим пояснюється явище пухлинної прогресії?
Пухлинним клітинам притаманна висока мінливість, тому їхня популяція неоднорідна. Іде постійна боротьба клітин за виживання в несприятливих умовах існування (дефіцит субстратів, кисню і т. д.). Це є основою природного їх добору - виживають тільки ті, що найбільш пристосовані до існування в таких умовах. А найбільш пристосованими є найпростіше влаштовані пухлинні клітини, які втратили свої спеціалізовані функції й зберегли тільки властивість безмежного поділу. У такий спосіб іде подальше збільшення злоякісності пухлини (малігнізація) — головний напрям пухлинної прогресії.
Біологічні канцерогенні фактори. Класифікація онкогенних вірусів. Вірус-асоційовані пухлини у тварин і людей. Механізми вірусного канцерогенезу.
Ким і в яких експериментах було доведено роль вірусів у виникненні пухлин?
Експериментальними доказами вірусного походження пухлин вважають їх виникнення після введення тваринам безклітинних фільтратів пухлинної тканини. Такі фільтрати готують із суспензії пухлинних клітин, пропускаючи її через порцелянові фільтри, що затримують бактерії і клітини тканини (рис. 55).
Уперше в 1908 р. В. Елерман і О. Банг у такий спосіб відтворили лейкоз у курей, уводячи безклітинні фільтрати лейкозних клітин.
У 1910 р. П. Раус викликав саркому у курей введенням безклітинного фільтрату саркоматозної тканини. За ці дослідження його в 1966 р. було удостоєно Нобелівської премії.
У 1932 p. Р. Шоуп повідомив про виділення вірусу з доброякісної пухлини кролів - фіброми, а трохи пізніше - з папіломи.
У 1934 р. Б. Люке виявив у ядрах ракових клітин нирок леопардової жаби тільця-включення, що нагадують такі при вірусних інфекціях. Потім він відтворив пухлину введенням тваринам висушеного екстракту пухлинних клітин.
У 1936 р.Дж. Біттнер відкрив "фактор молока" (фактор Біттнера), що викликав рак молочних залоз у мишей. Подальші дослідження показали, що цей фактор є ві- . русом, який передається від матері дитинчатам з молоком.
У 1951 p. JT. Гросс уперше відтворив лейкоз у мишей введенням безклітинних фільтратів пухлинних клітин новонародженим тваринам. Ці дослідження довели можливість вірусної етіології злоякісних пухлин і у представників ссавців.
Рис. 55. Перевивання пухлини клітинним і безклітинним матеріалом: а — клітинне перевивання; б - перевивання фільтратом, що не містить клітин
16.25. Які ДНК-вмісні віруси є онкогенними для тварин і людини?
До онкогенних ДНКчвмісних вірусів відносять:
а) nanoea-eipycu. Вони викликають розвиток у тварин трьох видів пухлин: папіломи, поліоми і пухлини, що виникає під дією вакуолізуючого вірусу мавп SV-40;
б) аденовіруси. Онкогенними для тварин є аденовіруси 12,18 і 31-го типів;
в) герпес-віруси, зокрема, вірус Епшгпейна — Барр.
Що стосується людини, то в одних випадках доведено, а в інших є підстави думати, що причиною деяких пухлин є:
а) вірус Епштейна - Барр (викликає лімфому Беркіта, назофарингеальний рак);
б) вірус гепатиту В (може бути причиною раку печінки);
в) вірус папіломи людини (викликає доброякісні пухлини шкіри, жіночих геніталій, гортані).
16.26. Які РНК-вмісні віруси є онкогенними для тварин і людини?
Всі онкогенні РНК-вмісні віруси належать до сімейства pempoeipycie. Загальною їхньою властивістю є наявність у вірусному геномі гена, що кодує структуру ферменту ревертази, відомого ще як зворотна транскриптаза, або РНК-залежна ДНК-полімераза. Цей фермент забезпечує синтез двоспіральної ДНК на матриці односпіральноїРНК. Унаслідок цього утворюється ДНК-копія ретровірусу, що отримала назву ДНК-провірусу.
Залежно від онкогенності ретровіруси поділяють на дві групи.
I. Гостротрансформуючі ретровіруси. Дуже онкогенні, викликають розвиток пухлин після короткого латентного періоду. Ці віруси мають у своєму геномі онкоген, тому в основі трансформації клітин у пухлинні лежить епігеномнш механізм. До зазначеної групи, зокрема, відносять віруси гострих лейкозів у птахів, мишей і саркоми Рауса у курей.
II. Повільнотрансформуючі ретровіруси. Викликають розвиток пухлин після тривалого латентного періоду. Ці віруси не мають у своєму складі онкогена, тому основний механізм їх трансформуючої дії — мутаційний. До вірусів цієї групи відносять віруси лімфолейкозів.
Онкогенним ретровірусом людини є вірус Т-клітинноїлімфоми-лейкемії. Він передається від людини до людини при тривалих інтимних контактах, переливанні крові. Слід зазначити, що цей лімфотропний вірус дуже схожий на віруси імунодефіциту людини (ВІЛ), що викликають СНІД.
16.27. Назвіть етапи вірусного онкогенезу.
I. Рецепція вірусу. Відбувається взаємодія вірусної частки з певними структурами плазматичної мембрани клітини (рецепторами). Відсутністю відповідних рецепторів пояснюється видовий імунітет до вірусної інфекції.
II. Роздягання й проникнення вірусу в клітину (інтернолізація).
III. Об'єднання (інтеграція) вірусного геному з геномом клітини. Це центральний і обов'язковий етап вірусного онкогенезу. У випадку ДНК-вмісних онковірусів відбувається вбудовування вірусної ДНК у ДНК клітини; у випадку РНК-вмісних вірусів - вбудовується ДНК-провірус, який утворюється під впливом ферменту ревертази.
IV. Постійне перебування (персистенція) вірусу в геномі клітини. При цьому вірус розмножується разом з клітиною. Такий перебіг вірусної інфекції називається абортивним. Абортивний перебіг є неодмінною умовою перетворення клітини в пухлинну під впливом вірусу.
V. Трансформація клітини.
VI. Промоція.
VII. Пухлинна прогресія (див. запит. 16.19 і 16.20).
16.28. Назвіть фактори, від яких залежить трансформуюча дія вірусів на клітину.
I. Фактори, що їх визначають властивості вірусу. До них, зокрема, відносять так звану структурну дефектність вірусу. Структурно дефектними називаються віруси, які в процесі розмноження й формування вірусних часток втратили частину свого геному. Вони здатні проникати в клітину й об'єднуватися з її геномом, однак не здатні до репродукції. У цьому випадку єдино можливим варіантом вірусної інфекції є абортивний її перебіг. Це збільшує ймовірність трансформації клітини під впливом структурно дефектних вірусів.
Крім того, для механізмів онкогенної дії вірусів має значення наявність або відсутність у їхньому геномі вірусного онкогена.
II. Фактори, що їх визначають властивості клітин. До них відносять наявність віщіовіщтх. рецепторів на поверхні клітин, період клітинного циклу, під час якого вірус потрапляє в клітину. Інтеграція геному вірусу з геномом клітини можлива тільки в синтетичну фазу (фазу S) або в період, що безпосередньо передує їй (кінець фази G,). Цим, зокрема, пояснюється та обставина, що високодиференційова-ні клітини, які втратили здатність до поділу, є стійкими до дії онкогенних вірусів. Крім того, клітина стає чутливою до онкогенних вірусів, якщо в ній нема умов для синтезу всіх білків, необхідних для формування вірусних часток. За таких умов неможлива репродукція вірусу й вірусна інфекція має абортивний перебіг, при якому збільшується ймовірність трансформації клітини. Таке явище отримало назву функціональної дефектності вірусу.
16.29. Що таке вірусні онкогєни?
Вірусні онкогєни - це гени вірусу, з функціонуванням яких пов'язане перетворення нормальних клітин у пухлинні. Білки-продукти вірусних онкогенів порушують регуляцію процесів клітинного поділу і в такий спосіб викликають трансформацію клітини (епігеномний механізм канцерогенезу).
16.30. Що таке протоонкогени?
Про то онкогєни - це власні гени клітин, які несуть інформацію про структуру білків, що беруть участь у регуляції клітинного поділу. Протоонкогени є клітинними аналогами вірусних онкогенів.
Вважають, що вірусні онкогєни являють собою протоонкогени, які потрапили в геном вірусів у результаті тривалої еволюції останніх. Це пояснюється тим, що віруси, проходячи через клітини, можуть "красти", тобто прихоплювати із собою клітинні гени.
Протоонкогени є у всіх клітинах. Однак в одних клітинах вони все життя репресовані, тобто "мовчать", в інших вони "працюють" тільки в період ембріогенезу, а в інших - функціонують відповідно до регуляторних сигналів, що надходять.
16.31. Як класифікують вірусні онкогени і протоонкогени?
Залежно від продуктів, інформацію про які несуть онкогени вірусів і протоонкогени клітин, їх поділяють на такі групи (рис. 56):
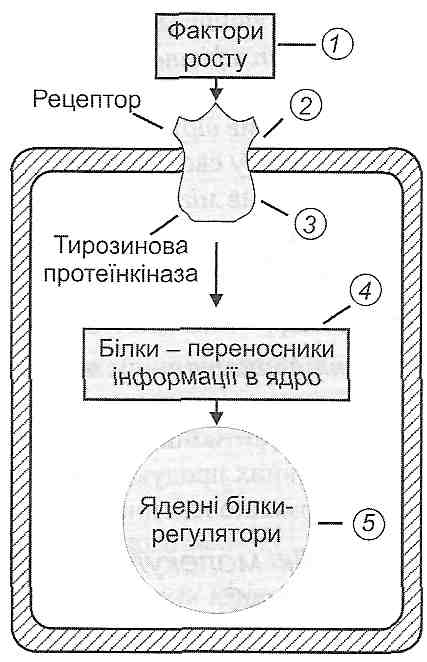
Рис. 56. Продукти протоонкогенів і вірусних онкогенів
1. Онкогени, що кодують фактори росту або їхні аналоги. Наприклад, sis-онкоген вірусу саркоми мавп і аналогічний протоонкоген клітин кодують структуру фактора росту тромбоцитар-ного походження.
2. Онкогени, що кодують клітинні рецептори до факторіє росту.
Наприклад, онкоген вірусу лейкозу курей erb-B несе інформацію про видозмінений рецептор до епідермального фактору росту.
3. Онкогени, що кодують тирозинспецифічні про-теїнкінази. Прикладом може бути scr-онкоген вірусу саркоми Рауса та відповідний протоонкоген клітин.
4. Онкогени, що кодують структуру білків, які переносять інформацію від рецепторів плазматичної мембрани клітини до ядра. Прикладом є ras-онкоген вірусу саркоми щурів, який несе інформацію про цитоплазматичний білок, що зв'язує ГТФ.
5. Онкогени, що кодують структуру ядерних білків-регуляторів.
До них, зокрема, належить mic-онкоген вірусу мієлоцитозу курей. Продукти вірусних онкогенів є аналогами продуктів протоонкогенів, тобто власних білків клітин, що беруть участь у регуляції клітинного поділу. Однак є деякі, хоча й невеликі, відмінності, Вони стосуються певних структурних характеристик вірусних онкогенів і протоонкогенів. Саме ці відмінності і роблять продукти вірусних онкогенів власне онкогенними для клітини.
16.32. Що таке клітинні онкогени? Назвіть основні механізми перетворення протоонкогенів у клітинні онкогени.
Клітинні онкогени (трансформуючі гени) - це протоонкогени, які набули здатності трансформувати клітину, тобто перетворювати її в пухлинну. Перенесення таких генів в інші, здорові клітини викликає трансформацію останніх.
Нині доведено існування таких механізмів перетворення протоонкогенів у клітинні онкогени.
І. Дерепресія протоонкогена. Може відбуватися або в результаті порушення структури, а отже, і функції відповідного антионкогена, або внаслідок мутацій у генах-репресорах, що блокують діяльність (експресію) протоонкогена.
її. Підвищення експресії протоонкогена. Спостерігається тоді, коли білок-про-дукт протоонкогена утворюється і в нормі, однак його дуже мало. Під впливом певних генетичних факторів утворення такого продукту іноді істотно зростає. Зазначене явище може бути обумовлене такими конкретними механізмами:
а) ампліфікація генів (збільшення кількості їхніх копій);
б) хромосомні мутації — транслокація;
в) вплив вірусних промоторів і підсилювачів (так діють ретровіруси, що не мають у своєму складі онкогена);
г) вплив мігруючих генів самої клітини (транспозонів).
Ш. Якісні зміни протоонкогена, що викликають утворення видозміненого продукту. Ці зміни, як правило, обумовлені точковими мутаціями протоонкогена.
16.33. Що таке антионкогени?
AnmuoHKOzenu — це клітинні гени, продукти яких викликають репресію прото-онкогенів.
Втрата антионкогенів (делеція) або мутації в них, що призводять до утворення неактивних продуктів, можуть мати своїм наслідком дерепресію протоонкогенів і трансформацію клітини, тобто утворення злоякісної пухлини.
16.34. Які молекулярні механізми можуть лежати в основі вірусного онкогенезу?
I. Продукти вірусного онкогена порушують регуляцію клітинного поділу й у такий спосіб викликають трансформацію клітини. Цей механізм отримав назву епігеном-ного. Він реалізує себе у випадку дії вірусів, що містять у своєму геномі онкогени. Із цим механізмом, зокрема, пов'язаний вплив на клітини гостротрансформуючих ретровірусів.
II. Під дією промоторів і підсилювачів вірусів, що потрапили в геном клітини, відбувається активація власних протоонкогенів, і вони перетворюються на клітинні онкогени, що трансформують клітину. Це так звання мутаційний механізм вірусного канцерогенезу. Він виявляє себе при дії вірусів, позбавлених онкогенів, зокрема, повільнотрансформуючих ретровірусів (рис. 57).
16.35. З якими молекулярними механізмами може бути пов'язаний канцерогенез, що його викликають хімічні і фізичні фактори?
При дії хімічних і фізичних канцерогенних факторів можливими є такі молекулярні зміни, що призводять до перетворення клітини в пухлинну.
1. Точкові мутації протоонкогенів.
2. Мутації антионкогенів, що регулюють експресію протоонкогенів.
3. Хромосомні аберації.
4. Утворення дефектних вірусів, унаслідок чого вони отримують онкогенні властивості.
5. Вплив на системи природної і специфічної протипухлинної резистентності, зокрема, розвиток стану імунодепресії.
Патогенез пухлинного росту. Роль порушень молекулярних (генетичних) механізмів регуляції клітинного поділу в процесі пухлинної трансформації. Способи перетворення протоонкогенів на онкогени. Властивості онкобілків.
Назвіть фактори, від яких залежить трансформуюча дія вірусів на клітину.
I. Фактори, що їх визначають властивості вірусу. До них, зокрема, відносять так звану структурну дефектність вірусу. Структурно дефектними називаються віруси, які в процесі розмноження й формування вірусних часток втратили частину свого геному. Вони здатні проникати в клітину й об'єднуватися з її геномом, однак не здатні до репродукції. У цьому випадку єдино можливим варіантом вірусної інфекції є абортивний її перебіг. Це збільшує ймовірність трансформації клітини під впливом структурно дефектних вірусів.
Крім того, для механізмів онкогенної дії вірусів має значення наявність або відсутність у їхньому геномі вірусного онкогена.
II. Фактори, що їх визначають властивості клітин. До них відносять наявність віщіовіщтх. рецепторів на поверхні клітин, період клітинного циклу, під час якого вірус потрапляє в клітину. Інтеграція геному вірусу з геномом клітини можлива тільки в синтетичну фазу (фазу S) або в період, що безпосередньо передує їй (кінець фази G,). Цим, зокрема, пояснюється та обставина, що високодиференційова-ні клітини, які втратили здатність до поділу, є стійкими до дії онкогенних вірусів. Крім того, клітина стає чутливою до онкогенних вірусів, якщо в ній нема умов для синтезу всіх білків, необхідних для формування вірусних часток. За таких умов неможлива репродукція вірусу й вірусна інфекція має абортивний перебіг, при якому збільшується ймовірність трансформації клітини. Таке явище отримало назву функціональної дефектності вірусу.
Що таке клітинні онкогени? Назвіть основні механізми перетворення протоонкогенів у клітинні онкогени.
Клітинні онкогени (трансформуючі гени) - це протоонкогени, які набули здатності трансформувати клітину, тобто перетворювати її в пухлинну. Перенесення таких генів в інші, здорові клітини викликає трансформацію останніх.
Нині доведено існування таких механізмів перетворення протоонкогенів у клітинні онкогени.
І. Дерепресія протоонкогена. Може відбуватися або в результаті порушення структури, а отже, і функції відповідного антионкогена, або внаслідок мутацій у генах-репресорах, що блокують діяльність (експресію) протоонкогена.
її. Підвищення експресії протоонкогена. Спостерігається тоді, коли білок-про-дукт протоонкогена утворюється і в нормі, однак його дуже мало. Під впливом певних генетичних факторів утворення такого продукту іноді істотно зростає. Зазначене явище може бути обумовлене такими конкретними механізмами:
а) ампліфікація генів (збільшення кількості їхніх копій);
б) хромосомні мутації — транслокація;
в) вплив вірусних промоторів і підсилювачів (так діють ретровіруси, що не мають у своєму складі онкогена);
г) вплив мігруючих генів самої клітини (транспозонів).
Ш. Якісні зміни протоонкогена, що викликають утворення видозміненого продукту. Ці зміни, як правило, обумовлені точковими мутаціями протоонкогена.
Що таке вірусні онкогєни?
Вірусні онкогєни - це гени вірусу, з функціонуванням яких пов'язане перетворення нормальних клітин у пухлинні. Білки-продукти вірусних онкогенів порушують регуляцію процесів клітинного поділу і в такий спосіб викликають трансформацію клітини (епігеномний механізм канцерогенезу).
Патогенез пухлин:
Включає 3 стадіІ розвитку:
1) Трансформація( ініціація) - перетворення нормальноі клітини у пухлину
2) Промоція( активізація)- розмноження пухлинноі клітини і утворення пухлинного вузла
3) Пухлинна прогресія- набуття пухлиною більшоі злоякісності.
Механізми онкогенезу:
1) мутаційний - повязані з генними мутаціями ( змінюється структура геному), спричинюять фізичні та хімічні канцерогени
2) епігеномний - відбувається зміна експресіі генів, що веде до стійкого порушення норм регуляціі геному, спричиняють онковіруси
Протоонкогени - це власні гени клітини, які несуть інформацію про структуру білків які беруть участь в регуляціі клітинного поділу.
Вірусні онкогени - це гени вірусу, які індукують перетворення нормальноі клітини в пухлину.
Клітинні онкогени - це протоонкогени, які набули здатності трансформувати нормальну клітину в пухлинну.
Антионкогени - це клітинні гени, продукти яких репресуюють протоонкогени.
Механізми перетворення протонкогенів в онкогени
1) Дерепресія протоонкогену
2) Підвищення експресіі протоонкогенів
3) Якісні зміни протооногену внаслідок мутаціі та утворення видозміненого онкобілка
Онкобілки - білки, які впливають на власну клітину і активують клітинний поділ
Можуть бути: 1) мембранними - фактори росту, рецептори до факторівросту, активатори аденілатциклази та гуанілатциклази; 2) цитоплазматичні білки - передають сигнал з мембран до ядра; 3) ядерні білки - викликають імморталізацію
4)?Білки які діють на інші клітини - фактори росту фібробластів, ангіогенін, еритропоетин.
Особливості вірусного канцерогенезу
Вірусний онкоген, потрапляючи а геном клітини заставляє синтезуватись онкобілки ( епігеномний механізм канцерогенезу)
Фізичні та хімічні канцерогени призводять до зниження активності антионкогенів, або відбвувається дерепресія протоонкогенів > активація клітинних онкобілків> синтез онкобілків (мутаційний механізм канерогенезу)
Пухлинна прогресія: визначення поняття, причини і механізми, типові ознаки. Механізми інвазійного росту і метастазування. Набуття резистентності до хіміопрепаратів. Вплив пухлини на організм. Патогенез ракової кахексії.
Особливості етапу пухлинноі прогресіі
Пухлинна прогресія - це стійкі якісні зміни властивостей пухлини в бік малігнезаціів процесі іі росту.
1) анаплазія
2) інвазивний ріст
3) утворення метастазів
4) біохімічний атипізм
5) хіміорезистентність
Пухлинна прогресія — це якісні зміни властивостей пухлини в процесі її розвитку. Як правило, згодом пухлина набуває усе більш і більш злоякісних властивостей.
16.20. Чим пояснюється явище пухлинної прогресії?
Пухлинним клітинам притаманна висока мінливість, тому їхня популяція неоднорідна. Іде постійна боротьба клітин за виживання в несприятливих умовах існування (дефіцит субстратів, кисню і т. д.). Це є основою природного їх добору - виживають тільки ті, що найбільш пристосовані до існування в таких умовах. А найбільш пристосованими є найпростіше влаштовані пухлинні клітини, які втратили свої спеціалізовані функції й зберегли тільки властивість безмежного поділу. У такий спосіб іде подальше збільшення злоякісності пухлини (малігнізація) — головний напрям пухлинної прогресії
Що таке інвазивність пухлини? Як злоякісні клітини проростають у навколишню тканину?
Інвазивність - це здатність злоякісної пухлини проростати у навколишні тканини. Основу інвазії становлять три процеси.
1. Прикріплення пухлинних клітин до колагенових волокон інтерстиціальної тканини. Це відбувається завдяки наявності на поверхні пухлинних клітин рецепторів до фібронектину, що сприяє утворенню "фібронектинових містків" між клітинами пухлини і колагеном І і III типів.
2. Деградація інтерстиціальної сполучної тканини. Після прикріплення до колагену пухлинні клітини активуються й починають виділяти ферменти: колагенази І і III типів, еластазу, катепсини, плазмін. Усі вони спричиняються до гідролітичного розщеплення волокнистих структур сполучної тканини і компонентів основної інтерстиціальної речовини. Цим створюється шлях для міграції клітин, для просування їх у здорову тканину.
3. Власне міграція пухлинних клітин. Тиск, що створюється в процесі поділу, направляє пухлинні клітини в інтерстиціальну тканину, що зазнала змін. Крім того, частина клітин пухлини має власну рухливість і має властивість хемотаксису. їхній хемотаксис стимулюється продуктами деградації сполучної тканини.
16.41. Що таке метастазування? Як воно здійснюється?
Метастазування — це здатність злоякісної пухлини давати вторинні пухлинні вузли (метастази) у віддалених від первинної пухлини частинах організму.
Процес метастазування складається з трьох етапів. І. Вихід пухлинних клітин із тканини в лімфатичні або кровоносні судини - інтра-
вазація. Цьому передує прикріплення пухлинних клітин до компонентів базаль-
ної мембрани судин. В основі такого прикріплення лежить утворення "ламініно-вих містків" між поверхнею клітин пухлини і колагеном IV типу базальної мембрани. Глікопротеїд ламінін є компонентом інтерстиціальної речовини базальної мембрани. Деякі найбільш злоякісні клітини самі здатні продукувати цю речовину. Ламінін, з одного боку, взаємодіє з так званими ламініновими рецепторами пухлинних клітин, а з другого - з хімічними групами колагену IV типу.
II. Транспорт пухлинних клітин лімфою й кров'ю - дисемінація. Із клітин пухлини, що проникли в судини, утворюються пухлинні емболи. їх утворення пов'язане з агрегацією пухлинних клітин. У цьому процесі обов'язково беруть участь тромбоцити.
III. Вихід пухлинних клітин з судин у тканини й утворення вторинних вогнищ (метастазів)— екстравазація. Для локалізації метастазів мають значення місцезнаходження первинної пухлини, шлях метастазування (лімфогенний чи гематогенний), рецепторна взаємодія пухлинних клітин з ендотелієм судин. Останнім, зокрема, пояснюється факт специфічної локалізації метастазів деяких пухлин (раку простати в кістки, бронхокарциноми в головний мозок і надниркові залози, не-йробластоми в печінку і т. д.).
16.42. Як впливає пухлина на організм у цілому? Чому розвивається ракова кахексія?
Розвиток злоякісної пухлини може виявляти себе трьома групами загальних порушень в організмі.
ї. Ракова кахексія - загальне виснаження. Характеризується різким зменшенням маси тіла, слабкістю, втратою апетиту, анемією. Виникнення ракової кахексії пояснюють такими явищами:
а) пухлина захоплює із крові великі кількості глюкози ( "пастка глюкози"), у результаті чого розвивається гіпоглікемія і відбувається "енергетичне обкрадання" організму;
б) пухлина захоплює із крові великі кількості амінокислот ("пастка азоту"). Відбувається "пластичне обкрадання" організму;
в) з пухлинних клітин, що гинуть, у кров надходять токсичні продукти — ток-согормони. Вони обумовлюють явища загальної інтоксикації;
г) з пухлинних клітин виходить багато недоокиснених продуктів - розвивається негазовий ацидоз;
ґ) у результаті виходу ферментів із загиблих пухлинних клітин у кров розвивається ферментемія;
д) при локалізації пухлини у травному каналі порушуються функції травної системи.
II. Загальні прояви, пов'язані з місцевими змінами тканин. До цієї групи відносять утворення виразок, вторинні інфекції, кровотечі, больовий синдром.
III. Паранеонластичні синдроми. Вони часто супроводжують розвиток пухлин, однак їх патогенез і зв'язок зі злоякісним пухлинним ростом залишаються нез'ясованими.
До цієї групи відносять:
а) ендокринопатії;
б) гіперкальціємію;
в) нервово-м'язовий синдром (міастенію, порушення центральної й периферичної нервової системи);
г) дерматологічні порушення; ґ) ураження кісток і суглобів;
д) судинні й гематологічні порушення (тромбози, анемію, лейкемоїдну реакцію).
Великий внесок у вивчення складних механізмів впливу пухлини на організм зробив Р. Є. Кавецький- учень
0. О. Богомольця
Механізми природного протипухлинного захисту, імунні та неімунні.
16.44. Які механізми протипухлинного захисту існують в організмі?
1. Механізми природної неспецифічної резистентності організму до пухлин. Не мають імунологічної специфічності і не вимагають попередньої імунізації. Вони здійснюються такими клітинами:
а) NK-wiimuHOMU (природними кілерами). Це великі гранулярні лімфоцити, що є різновидом О-лімфоцитів. Вони розпізнають пухлинні клітини і знищують їх;
б) LAK-wiimuHaMU (лімфокін-активованими кілерами). Вони, як і NK-клітини, здійснюють цитоліз пухлинних клітин;
в) макрофагами. Знищення клітин пухлини макрофагами здійснюється за допомогою фагоцитозу й механізмів позаклітинної цитотоксичності. Механізми природного неспецифічного протипухлинного захисту ефективні, якщо кількість пухлинних клітин в організмі не перевищує 103.
II. Реакції набутого (специфічного) протипухлинного імунітету. Обумовлені специфічними пухлинними антигенами і складаються як з клітинних, пов'язаних з функцією Т-лімфоцитів, так і гуморальних, пов'язаних з утворенням антитіл, імунних реакцій.
Зазначені реакції ефективні, якщо кількість клітин у пухлині становить від 103 до 10б. Якщо ж їхня кількість перевищує 106, то розвивається стан імунологічної депресії й описані вище механізми протипухлинного захисту пригнічуються
Експериментальне вивчення етіології і патогенезу пухлин: методи індукції, трансплантації, експлантації. Патофізіологічні основи профілактики і лікування пухлин.
Методами експериментального моделювання пухлин є індукція, трансплантація і експлантація.
Метод індукції передбачає відтворення злоякісних пухлин шляхом введення в організм канцерогенних факторів. Найчастіше з цією метою використовують хімічні канцерогенні сполуки й безклітинні фільтрати пухлинної тканини, що містять онкогенні віруси. Крім того, з метою індукції пухлин іноді використовують фізичні впливи (рентгенівське випромінювання, радіонукліди, ультрафіолетове опромінення).
Метод трансплантації - це пересадження пухлини від однієї тварини іншій. Уперше здійснена М. Новинським у 1876 р. Для успішної трансплантації пухлини важливими є такі умови:
а) пересадження має здійснюватися в межах одного виду тварин;
б) пересаджувати треба живі життєздатні пухлинні клітини;
в) трансплантацію слід робити в стерильних умовах, щоб уникнути запального процесу в тканині.
Метод експлантації— це вирощування пухлини в культурі тканини поза організмом. Цей метод дає можливість вивчати вплив різних факторів на пухлинний ріст, здійснювати пошук засобів терапії злоякісних пухлин.
Голодування: визначення поняття, класифікація. Зовнішні та внутрішні причини голодування. Характеристика порушень основного обміну і обміну речовин у різні періоди повного голодування (з водою).
Голодування - це типовий патологічний процес, що виникає внаслідок повної відсутності їжі або недостатнього надходження в організм поживних речовин, а також в умовах різкого порушення складу їжі та її засвоєння.
18.2. Як класифікують голодування?
За походженням виділяють фізіологічне, патологічне і лікувальне голодування. Фізіологічне голодування характерне для деяких видів тварин під час зимової (бабаки, ховрашки) або літньої сплячки (плазуни).
Залежно від змісту виділяють такі види голодування.
1. Повне голодування:
а) із уживанням води;
б) без уживання води (абсолютне).
2. Неповне голодування (недоїдання).
3. Часткове голодування (якісне).
18.3. На які періоди поділяють патогенез повного голодування із уживанням води?
I. Період неекономного витрачання енергії.
II. Період максимального пристосування.
III. Термінальний період.
На які періоди поділяють патогенез повного голодування із уживанням води?
I. Період неекономного витрачання енергії.
II. Період максимального пристосування.
III. Термінальний період.
18.4. Чим характеризується період неекономного витрачання енергії?
Його тривалість— 2-4 доби. Характерне сильне відчуття голоду, обумовлене збудженням харчового центру. При повному голодуванні воно триває до 5-ти діб, а потім зникає. Відбувається швидке падіння маси тіла (схуднення). Основним джерелом енергії у цей період є вуглеводи, про що свідчить дихальний коефіцієнт, який дорівнює 1,0. Виникає гіпоглікемія, яка збільшує секрецію глюкокортикоїдів корою надниркових залоз. Наслідком цього є посилення катаболізму білків у периферичних тканинах, зокрема м'язовій, і активація глюконеогенезу в печінці.
Основний обмін спочатку трохи збільшується, а потім поступово зменшується і стає на 10—20 % менше вихідного. Розвивається негативний азотистий баланс.
18.5. Що характерно для другого періоду голодування - періоду максимального пристосування?
Середня його тривалість — 40-50 діб. Темпи зменшення маси тіла уповільнюються і становлять 0,5-1 % на добу. Відчуття голоду зникає. Основним джерелом енергії є жири, про що свідчить дихальний коефіцієнт, який дорівнює 0,7.
Гіпоглікемія збільшує надходження в кров ліполітичних гормонів (адреналіну, глюкокортикоїдів, глюкагону). Унаслідок цього відбувається мобілізація жиру з депо — розвивається гіперліпацидемія. Вона, у свою чергу, є причиною посиленого утворення кетонових тіл у печінці. Кетонемія, що виникає, може призводити до не-газового ацидозу.
Основний обмін у цей період на 10-20 % нижче вихідного рівня. Азотистий баланс негативний.
18.6. Як втрачають масу різні органи і тканини в другому періоді голодування?
Втрата маси в другому періоді голодування становить: жирова тканина — 97 %,' селезінка — 60 %, печінка — 54 %, сім'яники — 40 %, м'язи - 31 %, кров - 26 %, нирки - 26 %, нервова система - 4 %, серце - 3,6 %.
18.7. Дайте характеристику третього періоду голодування.
Цей період називають термінальним, тому що він передує смерті. Його тривалість 2—3 доби. Відбувається інтенсивний розпад тканин, розвивається інтоксикація. Основним джерелом енергії є білки, про що свідчить дихальний коефіцієнт, який дорівнює 0,8. Збільшується виділення із сечею азоту, калію, фосфатів (ознаки деструкції клітин і тканинних білків).
Смерть настає при зменшенні маси тіла до 50 % від вихідної.
Патофізіологія неповного та часткового (якісного) голодування. Види, причини та механізми найбільш важливих проявів. Поняття про лікувальне голодування.
18.8. Які фактори визначають максимально можливу тривалість повного голодування із уживанням води?
І. Ендогенні фактори:
а) вид тварини-
б) вік;
в) стать;
г) кількість і якість жирових та білкових резервів організму; ґ) загальний функціональний стан організму;
д) м'язова робота.
Усі зазначені чинники впливають на тривалість голодування, змінюючи величину основного обміну. Що вищий рівень основного обміну, то менша тривалість голодування, і навпаки. її. Екзогенні фактори. Це чинники зовнішнього середовища, які збільшують енер-говитрати організму. Такими є:
а) низька температура навколишнього середовища;
б) висока вологість повітря;
в) велика швидкість руху повітря.
18.11. Назвіть особливості неповного голодування.
Неповне голодування (енергетична недостатність) розвивається, коли енергетична цінність їжі не задовольняє енергетичні потреби організму.
Неповне голодування від повного відрізняється такими особливостями:
1) тривалістю (неповне голодування може тривати місяці, роки);
2) більш вираженими деструктивними змінами в тканинах;
3) значно більшим зменшення основного обміну (на 30—40 %);
4) розвитком виражених набряків унаслідок зменшення вмісту білків у плазмі крові;
5) великого падіння маси тіла не відбувається через затримку рідини в організмі;
6) відновити життєдіяльність систем організму після неповного голодування набагато складніше.
Білково-калорійна недостатність, форми. Патогенез основних клінічних проявів.
18.12. Що таке білково-енергетична недостатність? Наведіть приклади.
Білково-енергетична недостатність — це стан, що виникає як результат поєднання неповного і якісного білкового голодування. Прикладами є:
а) аліментарна дистрофія. Описана в Ленінграді під час облоги в роки другої світової війни. У її патогенезі, крім білкової й енергетичної недостатності, мають значення й додаткові чинники: холод, фізичне стомлення, нервово-психічна напруга;
б) аліментарний маразм. Розвивається в дітей віком до одного року. На перше місце виступає енергетична недостатність;
в) квашіоркор. Розвивається в дітей віком 3-6 років. Головним у патогенезі є білкова недостатність. Енергетичний дефіцит компенсується надлишковим споживанням вуглеводів.
18.13. Якими клінічними синдромами виявляє себе білково-енергетична недостатність?
І. Недостатнє надходження в організм білків призводить до порушення білоксин-тетичної функції печінки. Це є причиною гіпопротеїнемії, що, у свою чергу, обумовлює розвиток онкотичних набряків.
II. Енергетична недостатність є причиною зменшення основного обміну. Це виявляється зниженням температури тіла (гіпотермією).
III. Атрофічні синдроми. їх розвиток пов'язаний з порушеннями пластичного й енергетичного забезпечення клітин.
Гіпоксія: визначення поняття, класифікація, етіологія, патогенез. Патологічні зміни та пристосувально-компенсаторні реакції при гіпоксії.
Гіпоксія (кисневе голодування) — це типовий патологічний процес, що виникає внаслідок недостатнього постачання тканин киснем або в результаті порушення його використання клітинами.'
19.2. Як класифікують кисневе голодування?
I. Етіологічна класифікація:
а) гіпоксична (екзогенна);
б) дихальна (респіраторна);
в) серцево-судинна (циркуляторна);
г) кров'яна (гемічна); ґ) тканинна (гістотоксична) гіпоксія.
II. За темпами розвитку і тривалістю виділяють:
а) блискавичну;
б) гостру;
в) підгостру;
г) хронічну гіпоксію Ні. Залежно від поширеності процесу гіпоксія може бути загальною і місцевою.
Назвіть причини зменшення доставки кисню кров'ю.
![]()
де С. — вміст кисню в артеріальній крові; Q — об'ємна швидкість течії крові. Причинами порушення доставки кисню кров'ю можуть бути:
а) зменшення вмісту кисню в артеріальній крові;
б) зменшення об'ємної швидкості течії крові в тканині (порушення кровопостачання);
в) зменшення віддачі кисню гемоглобіном (зменшення дисоціації оксигемоглобіну).
19.6. Чим може бути обумовлене зменшення вмісту кисню в артеріальній крові?
![]()
де - Ск - вміст кисню в артеріальній крові; [НЬ] - концентрація гемоглобіну в крові; S — насичення гемоглобіну киснем; 1,34 - число Хюфнера.
Причиною зменшення вмісту кисню в артеріальній крові можуть бути:
а) зменшення концентрації гемоглобіну, здатного зв'язувати кисень (зменшення кисневої ємності крові). Це може бути обумовлено або анемією (зменшується загальний вміст гемоглобіну), або інактивацією гемоглобіну;
б) зменшення насичення гемоглобіну киснем. Закономірно виникає при зменшенні напруги кисню в артеріальній крові нижче 60 мм рт. ст.
19.7. Які зміни можуть зменшувати об'ємну швидкість течії крові в тканинах і призводити до гіпоксії?
Оскільки
![]()
де Q — об'ємна швидкість течії крові; Ра — артеріальний тиск на початку, а Рвсн — венозний тиск наприкінці перфузованої ділянки; (Р т — Р ) перфузійний тиск; R — гемодинамічний опір, то зменшення кровопостачання тканин може бути обумовлене такими групами причин.
І. Зменшення перфузійного тиску в судинах органа або тканини:
а) зменшення артеріального тиску;
б) збільшення венозного тиску.
IJ. Збільшення гемодинамічного опору судин даної ділянки:
а) звуження судин;
б) підвищення в'язкості крові.
19.8. Які фактори викликають зміщення кривої дисоціації оксигемоглобіну?
Крива дисоціації оксигемоглобіну відображує залежність між напругою кисню в артеріальній крові і насиченням гемоглобіну киснем (рис. 61).
Рис. 61. Крива дисоціації оксигемоглобіну
Зміщення цієї кривої вліво відбувається при:
а) зниженні температури;
б) алкалозі;
в) гіпокапнії;
г) зменшенні в еритроцитах вмісту 2,3-дифос- -фогліцерату;
ґ) отруєннях оксидом вуглецю (II);
д) появі спадково обумовлених патологічних форм гемоглобіну, які не віддають кисень тканинам.
При зміщенні кривої вліво гемоглобін легше приєднує кисень у капілярах легень, але гірше віддає його тканинам.
Причиною зміщення кривої дисоціації оксигемоглобіну вправо можуть бути:
а) підвищення температури;
б) ацидоз;
в) гіперкапнія;
г) збільшення вмісту в еритроцитах 2,3-дифосфогліцерату.
Вплив ацидозу і гіперкапнії на дисоціацію оксигемоглобіну відомий як ефект Бора.
При зміщенні кривої вправо гемоглобін гірше приєднує кисень у капілярах легень, але краще віддає його тканинам. З цим, зокрема, пов'язане захисно-компенсаторне значення ефекту Бора при кисневому голодуванні.
19.9. Які фактори зумовлюють порушення дифузії кисню в тканинах? Відповідно до закону Фіка
![]()
де m — кількість газу, що дифундує (дифузійний потік); k — коефіцієнт дифузії; S - загальна площа поверхні, через яку здійснюється дифузія; 1 - відстань дифузії; (Pj —Р2) — різниця між напругою О, у капілярах і клітинах.
Звідси випливає, що причинами порушення дифузії кисню в тканинах можуть бути:
1) зменшення коефіцієнта дифузії кисню (наприклад, при відкладеннях у тканині ліпідів, гіаліну, амілоїду, солей кальцію);
2) зменшення загальної площі поверхні кровоносних капілярів при зменшенні їхньої кількості;
3) збільшення дифузійної відстані (наприклад, при набряку);
4) зменшення напруги кисню в капілярах;
5) збільшення напруги кисню в клітинах.
19.10. Що таке гіпоксія навантаження?
Гіпоксія навантаження — це кисневе голодування, що виникає при збільшенні функціонального навантаження. Воно пов'язане з посиленим використанням кисню клітинами. При цьому доставка кисню тканинам може навіть зростати.
Якщо все-таки доставка кисню не покриває його посиленого використання клітинами, напруга кисню в тканинах падає й розвивається гіпоксія.
19.11. Що таке гіпоксична гіпоксія? Коли вона виникає?
Тіпоксичною (екзогенною) називають гіпоксію, причиною якої є зменшення парціального тиску кисню у вдихуваному повітрі.
Гіпоксична гіпоксія може наставати при: а) зниженні атмосферного тиску (при підніманні на висоту в гори);
6) зменшенні вмісту кисню у вдихуваному повітрі (робота в шахтах, несправність систем кисневого забезпечення в літальних апаратах, на підводних човнах, у скафандрах).
19.12. Назвіть патогенетичні фактори розвитку гірської хвороби.
Гірська хвороба виникає при підніманні неадаптованого організму в гори. Вона являє приклад підгострої і хронічної гіпоксії.
Провідне значення в патогенезі гірської хвороби має зменшення парціального тиску кисню у вдихуваному повітрі (гіпоксична гіпоксія).
Крім того, при підніманні в гори патогенну дію чинять й інші фактори, зокрема, зменшення власне атмосферного тиску (синдром декомпресії), сонячна радіація, зниження температури зовнішнього середовища, сухість вдихуваного повітря, збільшення фізичного навантаження.
19.13. Які зони виділяють при підніманні в гори з урахуванням ознак гіпоксії, що розвивається?
I. Нейтральна зона (висота від 0 до 2000 м над рівнем моря). Функції організму не страждають.
II. Зона повної компенсації (висота від 2000 до 4000 м над рівнем моря). Відзначається збільшення частоти пульсу, дихання, підвищення артеріального тиску. У той же час зменшується фізична й розумова працездатність, розвивається ейфорія, порушується тонка координація рухів, послаблюється увага.
III. Зона неповної компенсації (висота від 4000 до 6000 м над рівнем моря). Розвиваються важкі, але оборотні зміни. Тахікардія змінюється брадикардією, падає артеріальний тиск, дихання стає частим і поверхневим, іноді розвивається дихання Чейна-Стокса, характерні сонливість, млявість, нудота.
IV. Критична зона (понад 7000 м над рівнем моря). Розвиваються необоротні зміни. Артеріальний тиск падає до 0, пульс стає ниткоподібним, з'являється термінальне дихання, людина непритомніє, розвиваються судоми й настає смерть.
19.14. Що таке висотна хвороба?
Висотна хвороба - це гостра або блискавична форма гіпоксичної гіпоксії, що виникає під час висотних польотів у літальних апаратах з кабінами відкритого типу або при порушенні герметичності кабін закритого типу.
19.15. Які зміни показників газового стану крові характерні для гіпоксичної гіпоксії?
Зменшення р02 і рС02 артеріальної крові, розвиток газового алкалозу.
19.16. Що таке дихальна гіпоксія?
Дихальна гіпоксія - це кисневе голодування, причиною якого є недостатність зовнішнього дихання. Причини її розвитку див. розд. 29.
19.17. Які зміни показників газового стану крові характерні для дихальної гіпоксії?
Зменшення р02 і збільшення рС02 артеріальної крові, розвиток газового ацидозу.:
19.18. Що таке циркуляторна гіпоксія?
Циркуляторна гіпоксія — це кисневе голодування, причиною якого є розлади загальної гемодинаміки або порушення місцевого кровообігу.
В основі порушень системного кровообігу можуть лежати недостатність серця й недостатність судин (шок, колапс).
До місцевої гіпоксії призводять ішемія, тромбоз, емболія, венозна гіперемія.
Залежно від механізмів розвитку деякі автори виділяють дві форми циркулятор-ної гіпоксії: ішемічну і застійну.
19.19. Які зміни показників газового стану крові характерні для циркуляторної гіпоксії?
Збільшення артеріовенозної різниці за киснем за рахунок повнішого вилучення його з артеріальної крові.
19.20. Що таке кров'яна (гемічна) гіпоксія? Назвіть її види.
Кров 'яна (гемічна) гіпоксія — це кисневе голодування, що виникає внаслідок зменшення кисневої ємності крові.
Виділяють дві форми кров'яної гіпоксії:
а) анемічну — виникає як наслідок анемії (див. розд. 26);
б) гіпоксію, пов'язану з інактивацією гемоглобіну.
19.21. Назвіть форми інактивованого гемоглобіну.
1. Карбоксигемоглобін - продукт взаємодії гемоглобіну з оксидом вуглецю (II) (чадним газом, CO).
2. Метгемоглобін — гемоглобін, у якому залізо перебуває в окисненому, тривалентному стані.
3. Сульфгемоглобін — сполука гемоглобіну із сірководнем.
19.22. Які механізми обумовлюють розвиток порушень в організмі при отруєнні оксидом вуглецю (II)?
У патогенезі порушень, спричинюваних оксидом вуглецю (II), мають значення такі фактори: |< -*•
а) інактивація гемоглобіну, що зменшує кисневу ємність крові, — розвивається кров 'яна гіпоксія;
б) зміщення кривої дисоціації оксигемоглобіну вліво - навіть той гемоглобін, що не зазнав інактивації, погано віддає кисень тканинам;
в) зв'язування оксиду вуглецю (II) із залізом інших білків, що містять у собі гем, зокрема цитохромів — розвивається тканинна гіпоксія.
19.23. Які фактори можуть бути причиною утворення метгемоглобіну, а отже й розвитку кров'яної гіпоксії?
Причини утворення метгемоглобіну:
1) екзогенніречовши-окислювачі (метгемоглобіноутворювачі). До них відносять:
а) нітросполуки (оксид азоту (II), нітрити, нітрати);
б) аміносполуки (анілін, фенілгідразин);
в) окислювачі (хлорати, перманганати, хінони);
г) окисно-відновні барвники (метиленовий синій у високих концентраціях); ґ) лікарські препарати (нітрогліцерин, амілнітрит, сульфаніламіди, барбітурати);
2) недостатність антиоксидантних систем еритроцитів, що відновлюють метгемоглобін. Це спостерігається при порушеннях пентозного циклу і глютатіон-редуктази. Описано генетично обумовлений дефект ферменту - НАДФ-залежної метгемоглобінредуктази.
19.24. Які зміни показників газового стану крові характерні для кров 'яної гіпоксії?
Зменшення кисневої ємності крові
Етіологія і патогенез тканинної гіпоксії. Принципи терапії гіпоксії. Можливі негативні наслідки кисневої терапії.
19.25. Що таке тканинна гіпоксія?
Тканинна гіпоксія — це кисневе голодування, що виникає в результаті порушення утилізації кисню клітинами.
В її основі лежать два типи порушень:
а) пригнічення біологічного окиснення;
б) роз'єднання окиснення й фосфорування (див. розд. 17).
19.26. Які зміни показників газового стану крові характерні для тканинної гіпоксії?
Зменшення артеріовенозної різниці за киснем і збільшення рО, венозної крові.
19.27. Дайте порівняльну характеристику основних показників газового стану крові при різних видах гіпоксії.
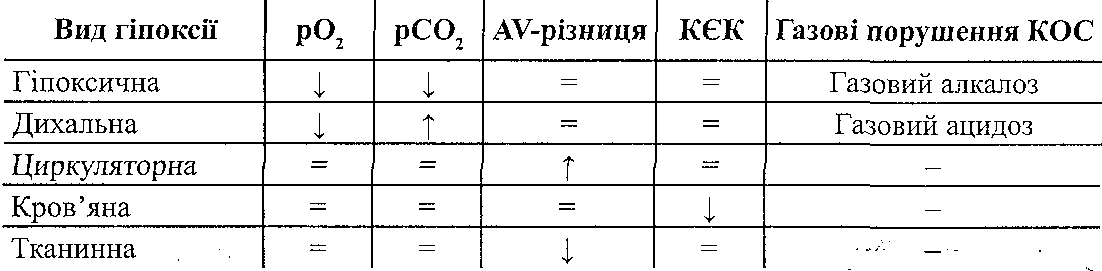
р02 — напруга кисню в артеріальній крові; рС02 — напруга вуглекислого газу в артеріальній крові; AV-різниця — артеріовенозна різниця за киснем; КЄК — киснева ємність крові; КОС — кислотно-основний стан.
19.28. На які групи можна поділити всі захисно-компенсаторні реакції, що виникають при гіпоксії?
I. Реакції, спрямовані на збільшення доставки кисню кров 'ю.
II. Місцеві (тканинні) реакції, спрямовані на поліпшення забезпечення клітин киснем.
III. Реакції в системах утилізації кисню.
19.29. Назвіть захисно-компенсаторні реакції організму, спрямовані на збільшення доставки кисню тканинам.
1. Реакції зовнішнього дихання. Спрямовані на збільшення р02 артеріальної крові, тому можуть бути ефективними тільки при гіпоксичній і дихальній гіпоксії. Вони виявляють себе:
а) збільшенням глибини дихання;
б) збільшенням частоти дихання;
в) мобілізацією резервних альвеол.
Комплекс зазначених змін отримав назву гіпервенпгиляції.
2. Реакції системи кровообігу. Спрямовані на збільшення кровопостачання тканин і є ефективними при всіх видах гіпоксії, крім тканинної. До цих реакцій відносять:
а) збільшення хвилинного об'єму крові за рахунок збільшення сили й частоти серцевих скорочень;
б) підвищення артеріального тиску;
в) перерозподіл течії крові - зменшення кровообігу в шкірі, скелетних м'язах, органах черевної порожнини і збільшення — у серці й головному мозку.
3. Реакції системи крові. Спрямовані на збільшення кисневої ємності крові і виявляють себе збільшенням кількості еритроцитів та концентрації гемоглобіну в периферичній крові. Це досягається за рахунок:
а) виходу додаткової кількості еритроцитів з депо;
б) активації еритропоезу (при гіпоксії посилюється утворення ниркових ери-тропоетинів).
Крім того, захисне значення має зміщення кривої дисоціації оксигемоглобіну вправо — ефект Бора (див. запит. 19.8).
19.31. Назвіть місцеві (тканинні) реакції, спрямовані на поліпшення забезпечення клітин киснем в умовах гіпоксії.
1. Посилення місцевого кровообігу — артеріальна гіперемія. Розвивається як наслідок безпосереднього впливу зменшення р02 на гладкі м'язи судин і дії на судини вазо-активних метаболітів (аденозину, молочної кислоти, іонів калію й водню та ін.)
2. Збільшення кількості капілярів, що функціонують. У результаті зменшується відстань дифузії кисню і збільшується загальна площа дифузійної поверхні.
3. Підвищення вмісту в клітинах міоглобіну, який у м'язах є внутрішньоклітинним депо кисню.
19.32. Назвіть захисно-компенсаторні реакції в системах утилізації кисню при гіпоксії.
1. Зниження функціональної активності клітин, унаслідок чого зменшується їхня потреба в кисні.
2. Збільшення кількості дихальних ферментів і мітохондрій у клітинах.
3. Збільшення спорідненості цитохромоксидази з киснем.
4. Підвищення ступеня спряженості окиснення й фосфорування до максимально можливої величини, що дорівнює 3.
5. Активація гліколізу
Порушення енергетичного обміну: етіологія, патогенез, наслідки. Поняття про енергетичні потреби організму, позитивний та негативний енергетичний баланс. Зміни основного обміну при патології.
17.1. Що таке позаклітинні і внутрішньоклітинні порушення енергетичного обміну?
Залежно від причин розвитку розрізняють поза- і внутрішньоклітинні розлади енергетичного обміну.
Позаклітинними називають розлади енергетичного обміну, пов'язані з первинними порушеннями забезпечення клітин поживними речовинами і киснем. Це голодування і більшість видів гіпоксії.
При внутрішньоклітинних розладах енергетичного обміну доставка поживних речовин і кисню не страждає, а порушується їх використання клітинами.
17.2. Назвіть основні причини порушення постачання клітин поживними речовинами.
1. Повна відсутність їжі або дефіцит поживних речовин у ній.
2. Порушення процесів травлення і всмоктування.
3. Порушення мобілізації поживних речовин з депо (наприклад, ураження печінки, розлади нервово-гуморальної регуляції жирового обміну).
4. Порушення транспорту поживних речовин кров'ю (загальні й місцеві розлади кровообігу).
5. Порушення дифузії поживних речовин у тканинах.
6. Втрата поживних речовин або їх використання не за призначенням (протеїнурія, глюкозурія, опікова хвороба, злоякісні пухлини).
17.3. Які порушення хімічного складу крові свідчать про порушення постачання клітин поживними речовинами?
1. Гіпоглікемія - зменшення концентрації глюкози в крові.
2. Гіполіпацидемія й гіполіпопротеїнемія — зменшення вмісту в крові вільних жирових кислот і ліпопротеїнів.
3. Гіпопротеїнемія й гіпоаміноацидемія - зменшення вмісту білків і вільних амінокислот у крові.
17.4. Назвіть причини внутрішньоклітинних порушень енергетичного обміну.
1. Порушення транспорту поживних речовин через клітинну мембрану (наприклад, глюкози при цукровому діабеті).
2. Порушення центральних внутрішньоклітинних катаболічних шляхів.
3. Розлади процесів біологічного окиснення в мітохондріях.
4. Роз'єднання процесів окиснення й фосфорування.
5. Порушення транспорту АТФ із мітохондрій до місць використання.
6. Порушення використання енергії АТФ.
17.5. Порушення яких центральних внутрішньоклітинних катаболічних шляхів можуть призводити до розладів енергозабезпечення клітин?
1. Пригнічення гліколізу.
2. Порушення циклу Кребса.
3. Розлади пентозного циклу.
4. Пригнічення окиснення жирових кислот.
5. Пригнічення процесів дезамінування і окиснення амінокислот.
17.6. Назвіть основні причини порушень центральних катаболічних шляхів у клітинах.
I. Зменшення вмісту вітамінів і вітаміноподібних речовин у клітинах (вітамінна недостатність).
II. Набуте зменшення активності ферментів:
а) зменшення активності окремих молекул ферментів (дія метаболічних отрут);
б) зменшення кількості молекул ферментів (розлади білоксинтетичної функції клітин).
III. Спадково обумовлені ензимопатії. Описано численні генетичні дефекти ферментів гліколізу, пентозного циклу, катаболічних перетворень амінокислот. Нині не відомі які-небудь ензимопатії, безпосередньо пов'язані з циклом Кребса й (3-окис-ненням жирових кислот.
IV. Дефіцит АТФ. АТФ використовується клітиною для активації субстратів, які надходять у центральні катаболічні шляхи (наприклад, фосфорування глюкози й фрук-тозо-6-фосфату, активація жирових кислот). Дефіцит АТФ створює "зачароване коло" — він порушує катаболічні перетворення поживних речовин, що веде до порушення ресинтезу АТФ. Це, у свою чергу, збільшує дефіцит макроергічних сполук.
17.7. Які причини можуть викликати розвиток вітамінної недостатності у клітинах?
Розвиток вітамінної недостатності в клітинах викликають:
а) гіпо- і авітамінози;
б) порушення транспорту вітамінів у клітину;
в) порушення перетворення вітамінів у коферменти;
г) порушення утворення холоферментів — комплексів коферментів з апоферментами.
17.8. Назвіть основні причини порушення біологічного окиснення в мітохондріях клітин.
1. Дефіцит коферментів: НАД, ФМН, убіхінону.
2. Дефіцит мікроелементів: Fe (входить до складу залізо-сірчаних центрів НАДН-дегі-дрогеназного комплексу і цитохромів) і Си (входить до складу цитохромоксидази).
3. Блокада транспорту електронів по дихальному ланцюгу (дія отрут— ротенону, антиміцину А, аміталу, ціанідів, оксиду вуглецю (II), сірководню; рис. 58).
Рис. 58. Блокада транспорту електронів по дихальному ланцюгу
4. Порушення акцепторного контролю дихання (АДФ не контролює швидкість транспорту електронів по дихальному ланцюгу).
17.9. Що таке роз 'єднання окиснення й фосфорування? Які його механізми?
Роз'єднання окиснення й фосфорування — це стан, при якому енергія, що вивільняється в процесі транспорту електронів по дихальному ланцюгу, не здатна акумулюватися в макроергічних зв'язках АТФ і тому виділяється у вигляді теплоти.
Для цього стану характерне зменшення ресинтезу АТФ і збільшення споживання кисню клітинами.
В основі роз'єднання окиснення й фосфорування можуть лежати такі механізми:
а) зменшення градієнта концентрацій іонів водню між матриксом мітохондрій і цитоплазмою;
б) зменшення трансмембранного електричного потенціалу на внутрішній мітохон-дріальній мембрані;
в) порушення АТФ-синтетазного ферментного комплексу;
г) використання енергії градієнта концентрацій іонів водню не на синтез АТФ, а на інші цілі (транспорт іонів кальцію із цитоплазми в мітохондрії, транспорт фосфату, АДФ, АТФ та ін.).
17.10. При порушенні яких біохімічних реакцій зменшується ресинтез АТФ у клітинах?
Утворення АТФ у клітинах зменшується за умов пригнічення: а) гліколітичного (субстратного) фосфорування:
![]()
б) окисного фосфорування в мітохондріях:
![]()
в) креатинкіназної реакції:
![]()
г) аденілаткіназної реакції:
![]()
ґ) нуклеозиддифосфокіназної реакції:
![]()
17.11. Які наслідки для клітини має дефіцит АТФ?
Дефіцит АТФ у клітині призводить до:
1. Порушення механічної роботи - скорочення контрактильних структур клітин. Це виявляє себе розладами елементарних клітинних функцій: скорочення, міграції, екзо- і ендоцитозу, клітинного поділу, руху війок, джгутиків (рис. 59).
2. Порушення осмотичної роботи — процесів активного транспорту іонів. При дефіциті АТФ страждають механізми як первинного, так і вторинного активного транспорту: натрій-калієвий і кальцієвий насоси, натрій-кальцієвий і натрій-водневий обмінні механізми. Це викликає порушення внутрішньоклітинного гомеостазу й ушкодження клітин.
3. Порушення хімічної роботи — біосинтезу речовин. Наслідком цього є порушення самовідновлення й самовідтворення клітин, механізмів їх довгострокової адаптації до дії факторів навколишнього середовища. В остаточному підсумку відбувається повільна загибель клітин.
4. Порушення реакцій клітинного метаболізму.
Рис. 59. Шляхи використання АТФ у клітинах
17.12. Які порушення метаболізму в клітинах можуть бути пов'язані з первинним дефіцитом АТФ?
В умовах дефіциту АТФ порушуються не тільки реакції біосинтезу речовин (анаболізм), але й реакції їх розщеплення (катаболізм).
Це пов'язано з тим, що при дефіциті АТФ безпосередньо порушуються:
а) активація субстратів (фосфорування) і залучення їх у катаболічні шляхи (гліколіз, Р-окиснення);
б) активація багатьох ферментів, здійснювана протешкіназами;
в) утворення циклічного АМФ.
17.13. Наведіть приклади "зачарованих кіл" у розвитку енергодефіцитного стану клітин.
Зменшення вмісту АТФ у клітині призводить до пригнічення функції Са-насосів, внаслідок чого збільшується концентрація іонів кальцію в цитоплазмі. Це викликає роз'єднання окиснення й фосфорування - у результаті дефіцит АТФ зростає (див. розд. 11).
Недостатність АТФ у клітині є причиною порушень її білоксинтетичних функцій. Страждає синтез усіх білків, у тому числі й білків-ферментів. Це веде до порушення катаболічних перетворень поживних речовин у клітині й процесів біологічного окиснення - дефіцит АТФ збільшується (рис. 60).
Рис. 60. Одне з "зачарованих кіл" у розвитку енергодефіцитного стану клітини
17.14. Що таке основний обмін?
Основний обмін — це мінімальні енергетичні витрати організму, визначені ранком у стані повного спокою, натще (через 12-14 год. після останнього прийому їжі), в умовах температурного комфорту (t = 18-20 °С, вологість повітря - 60-80 %, швидкість його руху - 0,1-0,2 м/с).
17.15. Які зовнішні і внутрішні фактори впливають на величину основного обміну?
Зовнішні фактори:
а) кліматичні умови;
б) час доби;
в) висота над рівнем моря;
г) характер харчування; ґ) характер виробничої діяльності.
Внутрішні фактори:
а) ріст і маса тіла;
б) площа поверхні тіла;
в) вік;
г) стать.
17.16. Наведіть приклади збільшення і зменшення основного обміну в умовах патології.
Збільшення основного обміну характерне для гіперфункції щитоподібної залози, аденогіпофіза, статевих залоз.
Зменшення основного обміну реєструють при гіпофункції щитоподібної залози, аденогіпофіза, статевих залоз; при хронічній недостатності кори надниркових залоз, при голодуванні.
Характеристика порушень вуглеводного обміну; критерії еуглікемії, гіпоглікемії, гіперглікемії, порушень толерантності до глюкози. Роль змін нейрон-гуморальної регуляції вуглеводного обміну в патогенезі гіпо- і гіперглікемічних станів.
20.1. Назвіть основні причини порушень вуглеводного обміну.
1. Порушення перетравлювання вуглеводів та їх усмоктування в кишках (див. розд. ЗО).
2. Порушення вуглеводної функції печінки (див. розд. 31).
3. Порушення катаболізму глюкози в периферичних клітинах (див. розд. 17).
4. Порушення нейрогормональної регуляції вуглеводного обміну.
20.2. Які гормони беруть участь у регуляції вуглеводного обміну?
Гормони, що беруть участь у регуляції вуглеводного обміну, поділяють на дві групи: інсулін і контрінсулярні гормони.
Контрінсулярними називають гормони, які за своїми біологічними ефектами є антагоністами інсуліну. До них належать адреналін, глюкагон, глюкокортикоїди, соматотропний гормон.
20.3. На які органи і тканини діє інсулін?
Залежно від чутливості до інсуліну всі структури організму поділяють на три групи.
I. Абсолютно залежні від інсуліну. Такими є печінка, м'язи (скелетні, міокард), жирова тканина.
II. Абсолютно нечутливі. Це головний мозок, мозкова речовина надниркових залоз, еритроцити, сім'яники.
III. Відносно чутливі (всі інші органи й тканини).
20.4. Назвіть основні біологічні ефекти інсуліну.
1. Гіпоглікемична дія. Інсулін зменшує вміст глюкози в крові за рахунок:
а) пригнічення процесів, що забезпечують вихід глюкози з печінки в кров (глі-когенолізу і глюконеогенезу);
б) посиленого використання глюкози інсулінозалежними тканинами (м'язовою, жировою).
2. Анаболічна дія. Інсулін стимулює ліпогенез у жировій тканині, глікогенез у печінці і біосинтез білків у м'язах.
3. Мітогенна дія. У великих дозах інсулін стимулює проліферацію клітин in vitro та in vivo.
Залежно від швидкості виникнення ефекти інсуліну поділяють на:
а) дуже швидкі (виникають протягом секунд) - зміна мембранного транспорту глюкози, іонів;
б) швидкі (тривають хвилини) — алостерична активація анаболічних ферментів і пригнічення ферментів катаболізму;
0:#:«
в) повільні (тривають від кількох хвилин до кількох годин) - індукція синтезу анаболічних ферментів і репресія синтезу ферментів катаболізму;
г) дуже повільні (від кількох годин до кількох діб) — мітогенна дія.
20,5. Які зміни вуглеводного обміну викликає адреналін?
Під впливом адреналіну збільшується вміст глюкози в крові. В основі цього ефекту лежать такі механізми:
а) активація глікогенолізу в печінці. Вона пов'язана з активацією аденілатциклазної системи гепатоцитів і утворенням, в кінцевому підсумку, активної форми фосфорилази;
б) активація глікогенолізу в м'язах з наступною активацією глюконеогенезу в печінці. При цьому молочна кислота, що вивільняється з* м'язової тканини в кров, іде на утворення глюкози в гепатоцитах (рис. 62);
в) пригнічення поглинання глюкози інсулінозалежними тканинами з одночасною активацією ліпрлізу в жировій тканині;
г) пригнічення секреції інсуліну β-клітинами і стимуляція секреції глюкагону а-клі-тинами острівців підшлункової залози.
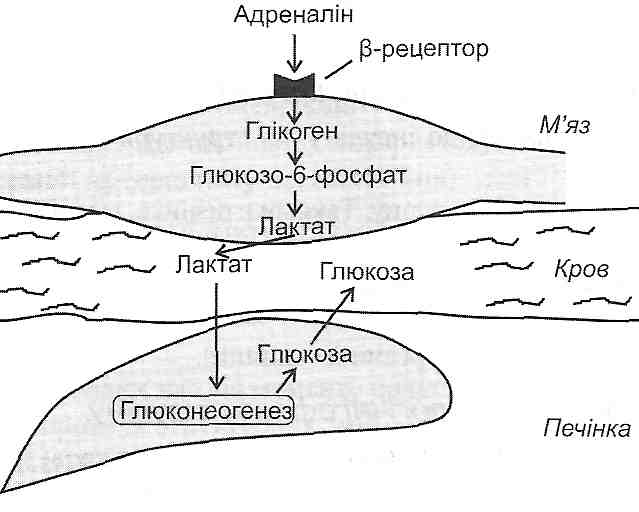
Рис. 62. Один з механізмів гіперглікемічної дії адреналіну
20.6. Які механізми лежать в основі гіперглікемічної дії глюкагону?
1. Активація глікогенолізу в печінці.
2. Активація глюконеогенезу в гепатоцитах.
Обидва механізми є цАМФ-опосередкованими.
20.7. Яким чином глюкокортикоїди підвищують рівень глюкози в крові?
Глюкокортикоїди активують процеси глюконеогенезу в печінці, збільшуючи:
а) синтез відповідних ферментів (вплив на транскрипцію);
б) вміст у крові субстратів глюконеогенезу — амінокислот — за рахунок посилення протеолізу в м'язах.
Крім того, глюкокортикоїди зменшують поглинання глюкози інсулінозалежними тканинами.
20.8. Чому при дії великих доз соматотропного гормону підвищується вміст глюкози в крові?
Тривалий вплив великих доз соматотропного гормону супроводжується розвитком інсулінорезистентності м'язів і жирової тканини — вони стають нечутливими до дії інсуліну. Результат цього — гіперглікемія.
20.9. Яка тривалість гіперглікемічного ефекту контрінсулярних гормонів?
Гіперглікемічна дія адреналіну триває до 10 хв, глюкагону—3 0-60 хв, глюкокортико-ї'дів — від кількох годин до кількох діб, соматотропного гормону—тижні, місяці, роки.
20.10. Як змінюється вміст глюкози в крові при порушенні балансу між інсуліном і контрінсулярними гормонами?
При збільшенні вмісту інсуліну розвивається гіпоглікемія, а при зменшенні його концентрації - гіперглікемія.
При збільшенні вмісту контрінсулярних гормонів розвивається гіперглікемія, а . при зменшенні їх концентрації - гіпоглікемія.
20.11. Як здійснюється нервова регуляція вуглеводного обміну?
Існує ряд доказів того, що нервова система бере участь у регуляції вмісту глюкози в крові.
Так, Клод Бернар уперше показав, що укол у дно IV шлуночка спричиняється до гіперглікемії ("іскровийукол"). Збільшення концентрації глюкози крові може виникати внаслідок подразнення сірого горба гіпоталамуса, сочевицеподібного ядра і смугастого тіла базальних ядер великого мозку. Кеннон спостерігав, що психічне перенапруження, емоції можуть підвищувати рівень глюкози в крові. Гіперглікемія виникає також при больових відчуттях, під час нападів епілепсії і т. д.
Нині доведено, що вплив нервової системи на рівень глюкози крові опосередковується гормонами. Можливі такі варіанти:
1) ЦНС → симпатична нервова система → мозкова речовина надниркових залоз → адреналін → гіперглікемія (укол К. Бернара);
2) ЦНС →• парасимпатична нервова система → острівці підшлункової залози → інсулін і глюкагон;
3) ЦНС → симпатична нервова система → мозкова речовина надниркових залоз → адреналін → Β-клітини острівців підшлункової залози → пригнічення секреції інсуліну;
4) ЦНС щ гіпоталамус → аденогіпофіз кі АКТГ → глюкокортикоїди → гіперглікемія
Причини і механізми розвитку гіпоглікемічних станів. Патогенез гіпоглікемічної коми.
20.12. Дайте визначення поняття "гіпоглікемія".
Гіпоглікемія - це зменшення концентрації глюкози в плазмі крові до рівня, який обумовлює появу клінічних симптомів, що зникають після нормалізації вмісту цієї речовини.
Ознаки гіпоглікемії виникають, як правило, при зменшенні вмісту глюкози нижче 4 ммоль/л.
20.13. Які механізми можуть лежати в основі розвитку гіпоглікемії?
1. Зменшення надходження глюкози в кров. Це буває при голодуванні, порушеннях травлення (недостатність амілолітичних ферментів, розлади всмоктування), при спадкових і набутих порушеннях глікогенолізу і глюконеогенезу в печінці.
2. Посилене використання глюкози на енергетичні потреби організму (наприклад, важка фізична робота).
3. Втрата глюкози (глюкозурія) або використання її не за призначенням (злоякісні пухлини).
20.14. Якими клінічними ознаками виявляє себе гіпоглікемія? Що таке гіпоглікемічна кома?
Клінічні ознаки гіпоглікемії пов'язані з двома групами порушень в організмі.
I. Порушення постачання глюкозою тканин головного мозку. Залежно від ступеня гіпоглікемії розвиваються такі симптоми, як головний біль, неможливість зосередитися, стомлюваність, неадекватна поведінка, галюцинації, суцоми, гіпоглікемічна кома.
II. Активація симпатоадреналової системи. Цим обумовлені серцебиття, посилене потовиділення, тремтіння, почуття голоду.
Гіпоглікемічна кома є найтяжчим проявом гіпоглікемії, і якщо вчасно не надати допомогу (введення глюкози), призводить до смерті. Вона характеризується втратою свідомості, зникненням рефлексів, порушеннями життєво важливих функцій (див. розд. 12).
Визначення поняття, класифікація цукрового діабету (ВООЗ). Загальна характеристика основних типів цукрового діабету (тип інсулінової недостатності, її походження, особливості перебігу, типові прояви, ускладнення і принципи лікування).
20.17. Дайте визначення поняття "цукровий діабет".
Цукровий діабет - це хвороба, яка у нелікованому стані виявляє себе хронічним збільшенням вмісту глюкози в крові - гіперглікемією (визначення ВООЗ, 1987).
20.18. Які існують експериментальні моделі цукрового діабету?
1. Панкреатичний цукровий діабет- видалення в собак 9/10 підшлункової залози (Мерінг і Мінковський, 1889).
2. Алоксановий цукровий діабет: однократне введення тваринам алоксану — речовини, що вибірково ушкоджує Β-клітини острівців підшлункової залози.
3. Стрептозотоциновий цукровий діабет: введення тваринам антибіотика - стреп-тозотоцину, що вибірково ушкоджує )3-клітини острівців.
4. Дитизоновий цукровий діабет: введення тваринам дитизону — речовини, що зв'язує цинк і в такий спосіб порушує депонування й секрецію інсуліну.
5. Імунний цукровий діабет - введення тваринам антитіл проти інсуліну.
6. Метагіпофізарний цукровий діабет — тривале введення тваринам гормонів адено-гіпофіза - соматотропного гормону, АКТГ.
7. Метастероїдиий цукровий діабет — тривале введення тваринам глюкокортикоїдів.
8. Генетичні моделі цукрового діабету — виведення чистих ліній мишей та інших тварин зі спадково обумовленою формою хвороби.
20.19. Наведіть патогенетичну класифікацію цукрового діабету.
1. Спонтанний цукровий діабет (первинний) - являє собою самостійну нозологічну одиницю. На його частку припадає близько 90 % усіх випадків цукрового діабету. Виділяють два різновиди спонтанного діабету: тип І, або інсулінозалеоісний (юнацький), і тип II, або інсулінонезолежний (діабет дорослих).
2. Вторинний цукровий діабет — є тільки ознакою інших захворювань. Він, зокрема, розвивається при ураженнях підшлункової залози, ендокринних хворобах, що супроводжуються збільшенням секреції контрінсулярних гормонів, при складних спадково обумовлених синдромах (наприклад, атаксія-телеангіектазія).
20.20. Дайте порівняльну характеристику цукрового діабету і і II типів.
Цукровий діабет І типу — інсулінозалежшй. Він характеризується абсолютною інсуліновою недостатністю, що виникає в результаті загибелі β-клітин панкреатичних острівців. Розвивається в осіб молодого віку, зазвичай до 30 років. Тому його ще називають ювенільним, або юнацьким. Супроводжується кетозом — накопиченням кетонових тіл. Необхідне лікування інсуліном.
Цукровий діабет II типу — інсулінонезолежний. Він характеризується відносною недостатністю інсуліну або інсулінорезистентністю. Розвивається у дорослих, найчастіше після 40 років. Не супроводжується кетозом. У його лікуванні інсулін не застосовують.
Етіологія цукрового діабету 1-го типу (значення спадкових факторів та факторів середовища в розвитку абсолютної інсулінової недостатності). Патогенез цукрового діабету 1 типу: порушення білкового, вуглеводного, жирового, водно-електролітного обмінів і кислотно-основного стану. Клінічні прояви.
20.21. Які причини розвитку цукрового діабету І типу?
Цукровий діабет І типу — захворювання з генетичною схильністю, у виникненні якого важливу роль відіграють фактори зовнішнього середовища (рис. 63).
Спадкова схильність до діабету І типу обумовлена антигенасоційованим характером цього захворювання. Так, доведений зв'язок між виникненням цукрового діабету І типу і наявністю певних HLA-генів (В8, Bwl5, Dw3, Dw4) у головному комплексі гістосумісності (МНС). Це наводить на думку про значення аутоімунних механізмів у розвитку цієї хвороби.
Серед факторів зовнішнього середовища, здатних ушкоджувати β-клітини панкреатичних острівців, велике значення мають р-цитотропні віруси (віруси Коксакі,
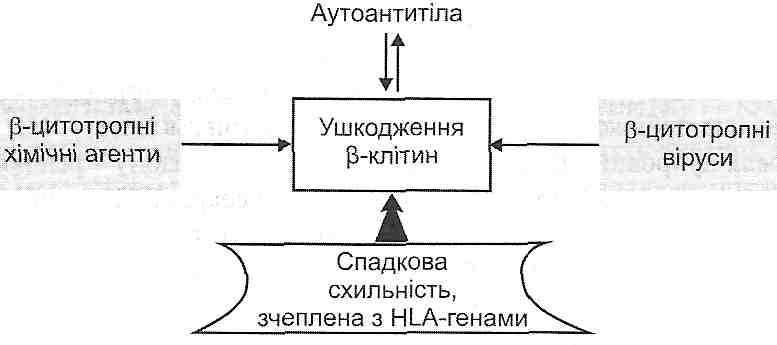
Рис. 63. Причини цукрового діабету І типу
епідемічного паротиту, кору) і fi-цитотропні хімічні агенти (наприклад, алоксан, стрептозотоцин).
20.22. Які механізми лежать в основі розвитку абсолютної інсулінової недостатності при цукровому діабеті і типу?
Генетична схильність, пов'язана з генами МНС, виявляє себе, з одного боку, утворенням аутоантитіл, здатних вибірково ушкоджувати Β-клітини острівців підшлункової залози, з другого - зменшенням резистентності клітин до дії екзогенних ушкоджувальних факторів (Р-цитотропних вірусів і хімічних агентів).
Усе це разом призводить до ушкодження інсулярного апарату й загибелі β-клітин (рис. 64). Утворення інсуліну припиняється.
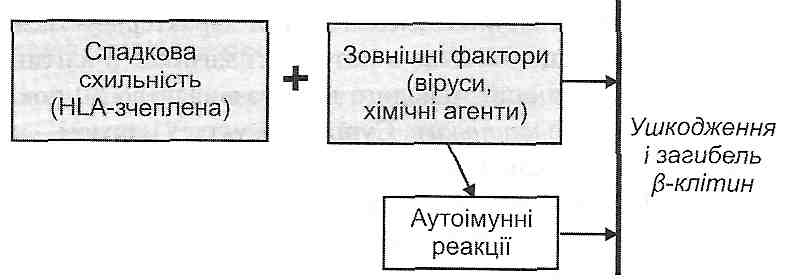
Рис. 64. Механізми абсолютної інсулінової недостатності при цукровому діабеті І типу
20.26. Які види обміну речовин порушуються при цукровому діабеті?
Цукровий діабет — це захворювання, при якому порушуються всі види обміну речовин: вуглеводний, жировий, білковий, водно-електролітний обмін, кислотно-основний стан.
20.27. Поясніть механізми розвитку гіперглікемії при цукровому діабеті.
Абсолютна або відносна недостатність інсуліну при цукровому діабеті викликає розвиток гіперглікемії, в основі якої лежать такі механізми.
I. Збільшення надходження глюкози в кров із печінки. Це пояснюється тим, що знімається гальмівний вплив інсуліну на ферменти глікогенолізу й глюконеогенезу, унаслідок чого збільшується інтенсивність цих процесів у печінці.
II. Зменшення використання глюкози інсулінозалежшми тканинами. Це пов'язане з тим, що при дефіциті інсуліну:
а) зменшується проникність клітинних мембран для глюкози у м'язовій (при обох типах цукрового діабету) і жировій (тільки при діабеті І типу) тканині;
б) зменшується утворення глікогену в печінці і м'язах;
в) падає активність пентозного циклу в печінці й жировій тканині;
г) зменшується активність гліколізу в усіх інсулінозалежних тканинах; ґ) відбувається пригнічення ферментів циклу Кребса в печінці і м'язах;
д) порушується перетворення глюкози в жири у печінці й жировій клітковині.
20.28. Які клінічні ознаки цукрового діабету обумовлені гіперглікемією ?
Можна виділити три групи таких ознак.
I. Гіперглікемія, глюкозурія, поліурія, полідипсія (спрага). Глюкозурія виникає тоді, коли концентрація глюкози в крові перевищує так званий "нирковий поріг", тобто 10 ммоль/л.
Унаслідок появи глюкози у вторинній сечі в ній збільшується осмотичний тиск.
Це викликає осмотичний діурез і поліурію.
Як результат поліурії розвивається зневоднення й спрага.
II. Висока гіперглікемія (понад ЗО ммоль/л) викликає збільшення осмотичного тиску крові, унаслідок чого розвивається дегідратація тканин, особливо мозку. Це є причиною так званої гіперосмолярної коми.
III. При гіперглікемії істотно зростає швидкість неферментпативного глікозіїлювання білків (хімічної взаємодії білків з глюкозою крові). Це спричиняється до структурних та функціональних порушень багатьох білків — як наслідок, виникають різні зміни в організмі, серед яких деформація й гемоліз еритроцитів, порушення зсідання крові, підвищення проникності судинної стінки, помутніння кришталика та ін.
20.29. Які порушення свідчать про розлади жирового обміну при цукровому діа б є ті?
1. Гіперліпацидемія — збільшення вмісту в крові вільних жирових кислот. Пов'язана з активацією ліполізу і пригніченням ліпогенезу в жировій тканині внаслідок порушення балансу між інсуліном і контрінсулярними гормонами (рис. 67).
2. Кетоз (гіперкетонемія й кетонурія). Збільшення вмісту кетонових тіл у крові й поява їх у сечі пов'язані з гіперліпацидемією (див. розд. 21).
3. Гіперліпопротеїнемія. Характеризується збільшенням вмісту в крові ліпопротеї-дів дуже низької густини (ЛПДНГ) (див. розд. 21).
4. Жирова інфільтрація печінки. Як і гіперліпопротеїнемія, вона є наслідком надмірного надходження в печінку вільних жирових кислот. Останні виводяться з печінки, перетворюючись у тригліцериди, з наступним утягуванням у формування ЛПДНГ, — розвивається гіперліпопротеїнемія. Якщо можливості гепатоцитів утворювати міцели ЛПДНГ вичерпуються, надлишок тригліцеридів відкладається в печінкових клітинах.
5. Схуднення. При нелікованому цукровому діабеті порушується здатність жирової тканини перетворювати вільні жирові кислоти плазми крові в тригліцериди. Це пов'язано з гальмуванням ліпогенезу при відсутності інсуліну й пригніченням реакцій гліколізу, які необхідні для цього процесу (рис. 68).
Посилення ліполізу під дією контрінсулярних гормонів також сприяє зменшенню маси жирової клітковини. 6. Атеросклероз (див. запит. 20.33).
20.30. Чим виявляють себе порушення білкового обміну при цукровому діабеті?
1. Аміноацидемією — збільшенням вмісту амінокислот у плазмі крові (рис. 69).
В основі цього лежить зменшення транспорту амінокислот у м'язові клітини (при відсутності інсуліну зменшується проникність клітинних мембран для амінокислот) і посилення протеолізу в м'язах, унаслідок чого вивільнені амінокислоти надходять у кров.
Надлишок вільних амінокислот поглинається печінкою, де посилюються процеси їх перетворення в глюкозу (глюконеогенез). Це, в кінцевому підсумку, призводить до подальшого збільшення рівня гіперглікемії.
2. Порушеннями біосинтезу білків. Це прямо пов'язано з випадінням анаболічної дії
Клінічно пригнічення білоксинтетичних процесів виявляє себе порушеннями фізичного й розумового розвитку дітей, уповільненням загоєння ран, порушеннями утворення антитіл, унаслідок чого збільшується чутливість до інфекцій, часто розвивається фурункульоз.
20.31. Які порушення водно-електролітного обміну характерні
для цукрового діабету? Який їх
патогенез?
1. Зневоднення (дегідратація). Є наслідком поліурії. Посилює дегідратацію блювота, яка часто супроводжує ацидоз, що розвивається у хворих на цукровий діабет (рис. 70).
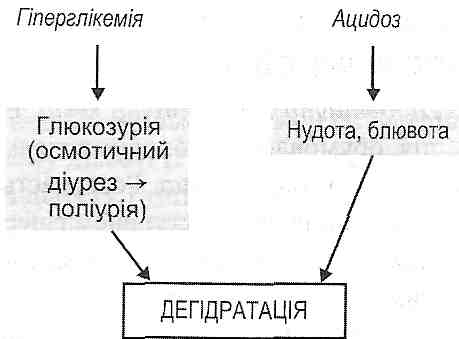
Рис. 70. Механізми дегідратації при цукровому діабеті
2. Гіперкаліємія. Є наслідком активації внутрішньоклітинного протеолізу. Відбувається вивільнення зв'язаного з білками калію, і його іони виходять з клітин у тканинну рідину і кров.
3. Гіпонатріємія. Якщо процеси ацидогене-зу в дистальних звивистих канальцях ниркових нефронів не забезпечують повного відтитровування гідрокарбонатного буфера, то якась частина іонів натрію втрачається із сечею разом з аніонами органічних кислот (ацетооцтової, р-оксимасля-ної).
20.32. Які порушення кислотно-основного стану розвиваються при цукровому діабеті?
Для цукрового діабету характерний негазовий ацидоз. Залежно від механізмів
його розвитку виділяють:
а) кетонемічний метаболічний ацидоз - пов'язаний з накопиченням кетонових тіл;
б) лактацидемічний метаболічний ацидоз — пов'язаний з накопиченням молочної кислоти. Причиною утворення останньої є зневоднення, що призводить до гіпо-волемії, згущення крові (гемоконцентрації) і, як наслідок, — до гіпоксії (рис. 71).
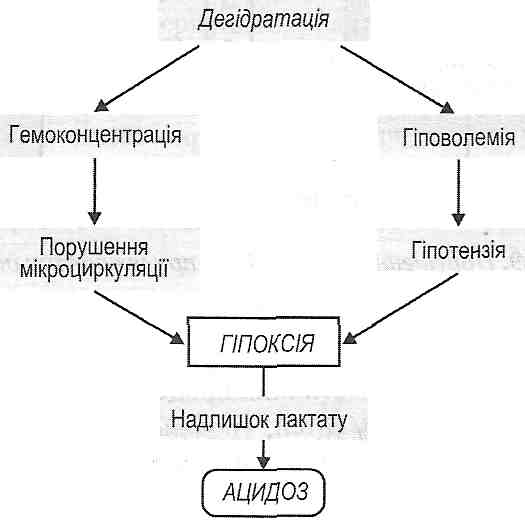
Рис. 71. Механізми лактацидеміїпри цукровому діабеті
20.33. Які варіанти коматозних станів можуть розвиватися при цукровому діабеті?
1. Діабетична кетонемічна кома. В основі її розвитку лежать ацидоз та інтоксикація, обумовлені кетоновими тілами.
2. Гіперосмолярна кома. Розвивається внаслідок дегідратації головного мозку, обумовленої високим ступенем гіперглікемії (див. запит. 20.26).
3. Лактацидемічна кома. Обумовлена накопиченням молочної кислоти й пов'язаним із цим ацидозом.
4. Гіпоглікемічна кома. Може розвиватися в результаті передозування інсуліну при лікуванні цукрового діабету.
Етіологія, патогенез цукрового діабету 2-го типу. Роль спадкових факторів. Причини відносної інсулінової недостатності. Порушення обміну речовин і фізіологічних функцій. Клінічні прояви.
20.23. Які причини розвитку цукрового діабету // типу?
1. Спадкова схильність. Вона, однак, на відміну від діабету І типу, не пов'язана з генами головного комплексу гістосумісності (МНС).
2. Ожиріння. Його відзначають у 80 % хворих. Тому виділяють дві форми цукрового діабету II типу:
а) з ожирінням;
б) без ожиріння.
На відміну від діабету І типу фактори зовнішнього середовища не мають особливого значення в етіології діабету II типу.
20.24. Опишіть патогенез цукрового діабету II типу з ожирінням.
Умовно виділяють два етапи патогенезу. І. Гіперінсулінемічний етап (рис. 65). Споживання великої кількості їжі особами з ожирінням викликає збільшення секреції інсуліну (гіперінсулінемія). Ця реакція спрямована на активацію процесів депонування поживних речовин у жировій тканині.
Рис. 65. Гіперінсулінемічний етап патогенезу цукрового діабету II типу з ожирінням
М'язи ж не мають потреби в дії інсуліну. Тому вони оберігають себе від надлишку цього гормону зменшенням кількості рецепторів на поверхні м'язових клітин. Розвивається явище інсулінорезистентності м'язової тканини — її чутливість до дії інсуліну падає. II. Гіпоїнсулінемічний етап (рис. 66). Підвищене навантаження на інсулярний апарат може призводити до функціонального виснаження β-клітин. Цьому сприяють генетично обумовлені їхні дефекти і надлишок в організмі контрінсулярних гормонів. Як наслідок, секреція інсуліну падає й розвивається його відносна недостатність. При цьому дія інсуліну на жирову тканину зберігається (на жирових клітинах багато рецепторів до інсуліну), а на м'язову тканину - зменшується внаслідок інсулінорезистентності.
Клінічно це виявляє себе розвитком гіперглікемії (немає дії інсуліну на м'язову тканину) і відсутністю кетозу (зберігається дія інсуліну на жирову тканину).
20.25. Назвіть можливі причини позапанкреатичної недостатності інсуліну.
Позапанкреатичну недостатність інсуліну можуть викликати такі причини:
а) порушення перетворення проінсуліну в інсулін;
б) утворення аномального інсуліну;
в) висока активність печінкових інсуліназ;
г) зв'язування інсуліну сироватковими білками;
ґ) утворення антитіл проти інсуліну;
д) аномалії інсулінових рецепторів на поверхні периферичних клітин.
Рис. 66. Гіпоінсулінемічний етап патогенезу цукрового діабету II типу з
20.26. Які види обміну речовин порушуються при цукровому діабеті?
Цукровий діабет — це захворювання, при якому порушуються всі види обміну речовин: вуглеводний, жировий, білковий, водно-електролітний обмін, кислотно-основний стан.
20.27. Поясніть механізми розвитку гіперглікемії при цукровому діабеті.
Абсолютна або відносна недостатність інсуліну при цукровому діабеті викликає розвиток гіперглікемії, в основі якої лежать такі механізми.
I. Збільшення надходження глюкози в кров із печінки. Це пояснюється тим, що знімається гальмівний вплив інсуліну на ферменти глікогенолізу й глюконеогенезу, унаслідок чого збільшується інтенсивність цих процесів у печінці.
II. Зменшення використання глюкози інсулінозалежшми тканинами. Це пов'язане з тим, що при дефіциті інсуліну:
а) зменшується проникність клітинних мембран для глюкози у м'язовій (при обох типах цукрового діабету) і жировій (тільки при діабеті І типу) тканині;
б) зменшується утворення глікогену в печінці і м'язах;
в) падає активність пентозного циклу в печінці й жировій тканині;
г) зменшується активність гліколізу в усіх інсулінозалежних тканинах; ґ) відбувається пригнічення ферментів циклу Кребса в печінці і м'язах;
д) порушується перетворення глюкози в жири у печінці й жировій клітковині.
20.28. Які клінічні ознаки цукрового діабету обумовлені гіперглікемією ?
Можна виділити три групи таких ознак.
I. Гіперглікемія, глюкозурія, поліурія, полідипсія (спрага). Глюкозурія виникає тоді, коли концентрація глюкози в крові перевищує так званий "нирковий поріг", тобто 10 ммоль/л.
Унаслідок появи глюкози у вторинній сечі в ній збільшується осмотичний тиск.
Це викликає осмотичний діурез і поліурію.
Як результат поліурії розвивається зневоднення й спрага.
II. Висока гіперглікемія (понад ЗО ммоль/л) викликає збільшення осмотичного тиску крові, унаслідок чого розвивається дегідратація тканин, особливо мозку. Це є причиною так званої гіперосмолярної коми.
III. При гіперглікемії істотно зростає швидкість неферментпативного глікозіїлювання білків (хімічної взаємодії білків з глюкозою крові). Це спричиняється до структурних та функціональних порушень багатьох білків — як наслідок, виникають різні зміни в організмі, серед яких деформація й гемоліз еритроцитів, порушення зсідання крові, підвищення проникності судинної стінки, помутніння кришталика та ін.
20.29. Які порушення свідчать про розлади жирового обміну при цукровому діа б є ті?
1. Гіперліпацидемія — збільшення вмісту в крові вільних жирових кислот. Пов'язана з активацією ліполізу і пригніченням ліпогенезу в жировій тканині внаслідок порушення балансу між інсуліном і контрінсулярними гормонами (рис. 67).
Рис. 67. Перетворення жирових кислот (ЖК) у печінці при цукровому діабеті: ТГ — тригліцериди; ЛПДНГ - ліпопротеїди дуже низької густини
2. Кетоз (гіперкетонемія й кетонурія). Збільшення вмісту кетонових тіл у крові й поява їх у сечі пов'язані з гіперліпацидемією (див. розд. 21).
3. Гіперліпопротеїнемія. Характеризується збільшенням вмісту в крові ліпопротеї-дів дуже низької густини (ЛПДНГ) (див. розд. 21).
4. Жирова інфільтрація печінки. Як і гіперліпопротеїнемія, вона є наслідком надмірного надходження в печінку вільних жирових кислот. Останні виводяться з печінки, перетворюючись у тригліцериди, з наступним утягуванням у формування ЛПДНГ, — розвивається гіперліпопротеїнемія. Якщо можливості гепатоцитів утворювати міцели ЛПДНГ вичерпуються, надлишок тригліцеридів відкладається в печінкових клітинах.
5. Схуднення. При нелікованому цукровому діабеті порушується здатність жирової тканини перетворювати вільні жирові кислоти плазми крові в тригліцериди. Це пов'язано з гальмуванням ліпогенезу при відсутності інсуліну й пригніченням реакцій гліколізу, які необхідні для цього процесу (рис. 68).
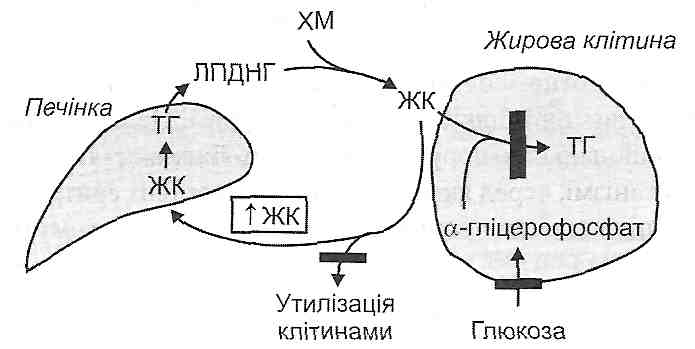
Рис. 68. Порушення депонування жирових кислот (ЖК) у жировій тканині при цукровому діабеті: ТГ— тригліцериди; ХМ — хіломікрони; ЛПДНГ'—ліпопротеїди дуже низької
густини
Посилення ліполізу під дією контрінсулярних гормонів також сприяє зменшенню маси жирової клітковини. 6. Атеросклероз (див. запит. 20.33).
20.30. Чим виявляють себе порушення білкового обміну при цукровому діабеті?
1. Аміноацидемією — збільшенням вмісту амінокислот у плазмі крові (рис. 69).
В основі цього лежить зменшення транспорту амінокислот у м'язові клітини (при відсутності інсуліну зменшується проникність клітинних мембран для амінокислот) і посилення протеолізу в м'язах, унаслідок чого вивільнені амінокислоти надходять у кров.
Надлишок вільних амінокислот поглинається печінкою, де посилюються процеси їх перетворення в глюкозу (глюконеогенез). Це, в кінцевому підсумку, призводить до подальшого збільшення рівня гіперглікемії.
2. Порушеннями біосинтезу білків. Це прямо пов'язано з випадінням анаболічної дії
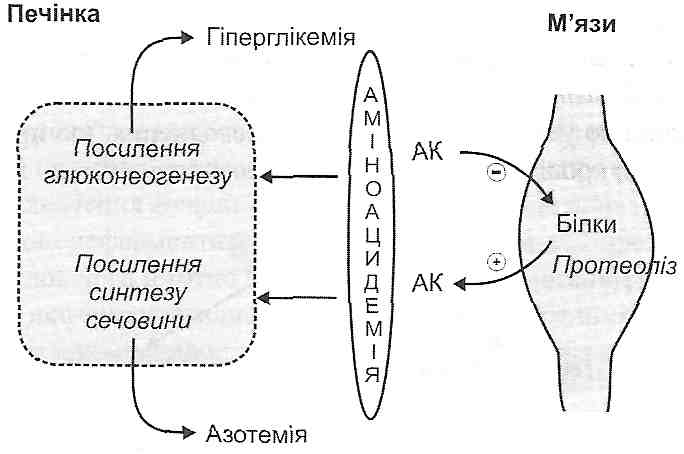
Рис. 69. Порушення обміну білків при цукровому діабеті: АК—амінокислоти
інсуліну.
Клінічно пригнічення білоксинтетичних процесів виявляє себе порушеннями фізичного й розумового розвитку дітей, уповільненням загоєння ран, порушеннями утворення антитіл, унаслідок чого збільшується чутливість до інфекцій, часто розвивається фурункульоз.
20.31. Які порушення водно-електролітного обміну характерні
для цукрового діабету? Який їх
патогенез?
1. Зневоднення (дегідратація). Є наслідком поліурії. Посилює дегідратацію блювота, яка часто супроводжує ацидоз, що розвивається у хворих на цукровий діабет (рис. 70).
Рис. 70. Механізми дегідратації при цукровому діабеті
2. Гіперкаліємія. Є наслідком активації внутрішньоклітинного протеолізу. Відбувається вивільнення зв'язаного з білками калію, і його іони виходять з клітин у тканинну рідину і кров.
3. Гіпонатріємія. Якщо процеси ацидогене-зу в дистальних звивистих канальцях ниркових нефронів не забезпечують повного відтитровування гідрокарбонатного буфера, то якась частина іонів натрію втрачається із сечею разом з аніонами органічних кислот (ацетооцтової, р-оксимасля-ної).
20.32. Які порушення кислотно-основного стану розвиваються при цукровому діабеті?
Для цукрового діабету характерний негазовий ацидоз. Залежно від механізмів
його розвитку виділяють:
а) кетонемічний метаболічний ацидоз - пов'язаний з накопиченням кетонових тіл;
б) лактацидемічний метаболічний ацидоз — пов'язаний з накопиченням молочної кислоти. Причиною утворення останньої є зневоднення, що призводить до гіпо-волемії, згущення крові (гемоконцентрації) і, як наслідок, — до гіпоксії (рис. 71).
Рис. 71. Механізми лактацидеміїпри цукровому діабеті
20.33. Які варіанти коматозних станів можуть розвиватися при цукровому діабеті?
1. Діабетична кетонемічна кома. В основі її розвитку лежать ацидоз та інтоксикація, обумовлені кетоновими тілами.
2. Гіперосмолярна кома. Розвивається внаслідок дегідратації головного мозку, обумовленої високим ступенем гіперглікемії (див. запит. 20.26).
3. Лактацидемічна кома. Обумовлена накопиченням молочної кислоти й пов'язаним із цим ацидозом.
4. Гіпоглікемічна кома. Може розвиватися в результаті передозування інсуліну при лікуванні цукрового діабету.
Ускладнення цукрового діабету. Причини та механізми різних видів ком при цукровому діабеті. Віддалені ускладнення діабету.
Які ускладнення характерні для цукрового діабету?
... . . . 'Лі
Макроангюпатії, мікроангюпатії, нейропатії.
20.35. Які механізми можуть лежати в основі розвитку макроангіопатій при цукровому діабеті?
Макроангіопшпії характеризуються прискореним розвитком атеросклерозу в артеріях хворих на цукровий діабет. Найчастіше вражаються вінцеві артерії серця, артерії головного мозку й нижніх кінцівок. Це може призводити до розвитку таких
ускладнень, як інфаркт міокарда, інсульт, гангрена пальців ніг і всієї стопи. Існують дві концепції, що пояснюють патогенез макроангіопатій.
I. Концепція порушеного гомеостазу (власне діабетична). Головне значення в розвитку атеросклерозу при цукровому діабеті надається загальним порушенням обміну речовин в організмі, а саме: гіперглікемії, гіперліпопротеїнемії й ацидозу. Патогенетичне значення гіперглікемії полягає в тому, що вона:
1) є причиною неферментативного глікозилювання ліпопротеїдів плазми крові, унаслідок чого істотно збільшується їхня атерогенність;
2) викликає неферментативне глікозилювання мембранних білків ендотеліаль-них клітин і, як наслідок, призводить до підвищення проникності судинної стінки;
3) активує сорбітоловий шлях перетворення глюкози в гладких м'язових клітинах судин. Останнє відбувається, якщо концентрація глюкози в крові перевищує 20 ммоль/л.
Результатом активації сорбітолоеого шляху є утворення в клітинах фруктози. , Оскільки плазматична мембрана непроникна для цієї речовини, вона накопичується в цитоплазмі, підвищуючи осмотичний тиск внутрішньоклітинної рідини, викликаючи набряк і ушкодження клітин.
Гіперліпопротеїнемія при цукровому діабеті характеризується збільшенням вмісту в крові ліпопротеїдів дуже низької густини (ЛПДНГ) і появою "модифікованих" ліпопротеїдів (ЛП): глікозильованих і ацетоацетильованих ЛІХ Про значення цих порушень у розвитку атеросклерозу див. розд. 28.
З виникненням ацидозу пов'язане підвищення проникності судинної стінки й ушкодження її гладком'язових і ендотеліальних клітин - фактори, що сприяють атеросклерозу.
II. Інсулінова концепція. її прихильники вважають, що провідною ланкою в патогенезі діабетичних макроангіопатій є гіперінсулінемія. Збільшення вмісту інсуліну в крові може бути ендогенним, як при цукровому діабеті II типу, і екзогенним, як результат передозувань інсуліну при лікуванні діабету І типу.
Інсулін у великих кількостях, маючи мітогенну дію, викликає проліферацію гладком'язових клітин артеріальної стінки, що призводить до формування фіброзних атеросклеротичних бляшок.
20.36. Як пояснюють розвиток мікроангіопатій при цукровому діабеті? Чим вони можуть виявляти себе?
Мікроангіопатїї— це ураження судин мікроциркуляторного русла (артеріол, капілярів), що виникають як ускладнення цукрового діабету. Сутність цих уражень полягає в значному збільшенні товщини базальної мембрани мікросудин, що утруднює обмін речовин між кров'ю й тканинами.
Серед механізмів розвитку мікроангіопатій велике значення мають збільшення синтезу глікопротеїнів базальної мембрани і неферментативне глікозилювання її компонентів.
Мікроангіопатії найчастіше виявляють себе ураженням судин нирок (діабетич-
на нефропатія) і сітківки очей (діабетичнаретинопатія). Як наслідок, можуть розвиватися хронічна ниркова недостатність, відшарування сітківки.
20.37. Який патогенез нейропатій при цукровому діабеті?
Heuponamu — це специфічні ураження нервових провідників у хворих на цукровий діабет. Вони виявляють себе розладами чутливості, вегетативних і рухових функцій, нервової трофіки.
В основі патогенезу діабетичних нейропатій лежать процеси демієлінізації нервів і порушення аксоплазматичного транспорту.
Суть демієлінізації полягає в руйнуванні мієлінової оболонки нервових волокон і порушенні утворення мієліну. Ці розлади пов'язують із:
а) активацією сорбітолового шляху перетворення глюкози у шваннівських клітинах, що спричиняє їхнє ушкодження й загибель;
б) пригніченням міоінозитолового шляху, внаслідок чого порушується утворення мі-оінозитолу - речовини, необхідної для побудови мієліну.
Експериментальне моделювання цукрового діабету. Принципи профілактики і терапії його основних типів. Профілактика ускладнень цукрового діабету.
20.38. Назвіть основні патогенетичні принципи лікування цукрового діабету.
1. Уведення інсуліну при інсулінозалежному цукровому діабеті І типу. Перспективними в цьому плані є трансплантація (3-клітин острівців підшлункової залози й застосування автоматизованих систем дозування і введення інсуліну.
2. Уведення фармакологічних препаратів, що усувають гіперглікемію, - гіпоглікемічних засобів (бігуаніди, похідні сульфонілсечовини).
3. Дієтотерапія, що забороняє вживати харчові продукти з високим вмістом цукру, регулює енергетичну цінність їжі та режим її споживання.
4. Фізичні навантаження (тренування). Вони зменшують рівень гіперглікемії й збільшують чутливість м'язової тканини до інсуліну.
Порушення ліпідного обміну: причини, механізми, прояви. Залежність розвитку дисліпопротеїнемій від факторів середовища, спадковості, супутніх захворювань. Принципи класифікації дисліпопротеїнемій. Етіологія і патогенез первинних (спадкових) і вторинних гіперліпопротеїнемій.
Назвіть основні причини порушень жирового обміну в організмі.
Причинами розладів жирового обміну можуть бути порушення:
1) перетравлювання і всмоктування ліпідів у тонкій кишці;
2) транспорту ліпідів кров'ю;
3) депонування ліпідів у жировій тканині;
4) жирової функції печінки (див. розд. 31);
5) проміжного обміну ліпідів у периферичних тканинах;
6) нервової й гормональної регуляції жирового обміну.
27.2. Що може бути причиною порушень перетравлювання і всмоктування ліпідів у кишках?
1. Порушення емульгування жирів:
а) недостатнє надходження жовчі в кишки (механічна жовтяниця);
б) передчасне руйнування жовчних кислот бактеріальною флорою при порушенні моторної функції кишок.
2. Порушення гідролітичного розщеплення жирів:
а) недостатнє надходження панкреатичної ліпази у дванадцятипалу кишку;
б) порушення активації цього ферменту за умов недостатньої секреції жовчі.
3. Порушення утворення ліпідних міцел у порожнині тонкої кишки:
а) недостатнє надходження жовчі в тонку кишку;
б) зв'язування жовчних кислот деякими лікарськими препаратами (холестира-мін, неоміцин та ін.);
в) утворення кальцієвих солей жирових кислот при надмірному надходженні кальцію з їжею й водою.
4. Порушення всмоктування міцел:
а) швидка евакуація вмісту тонкої кишки (проноси);
б) ушкодження епітелію слизової оболонки тонкої кишки (ентерити, радіаційні ураження).
5. Порушення ресинтезу тригліцеридів і формування хіломікронів в епітеліальних клітинах кишок:
а) зменшення кількості або пригнічення активності відповідних ферментів;
б) дефіцит АТФ.
21.3. Які зміни складу крові можуть бути проявом порушень транспорту ліпідів в організмі?
Гіперліпопротеїнемія і гіполіпопротеїнемія — відповідно збільшення й зменшення вмісту ліпопротеїдів у плазмі крові; дисліпопротеїнемія — порушення співвідно-
шення між окремими класами ліпопротеїдів плазми крові; гіперліпацидемія — збільшення вмісту вільних жирових кислот у крові.
21.4. Які існують класи ліпопротеїдів плазми крові?
У плазмі крові містяться хіломікрони (ХМ), ліпопротеїди дуже низької густини (ЛПДНГ), ліпопротеїди проміжної густини (ЛППГ), ліпопротеїди низької густини (ЛПНГ), ліпопротеїди високої густини (ЛПВГ).
21.5. Дайте порівняльну характеристику різних класів ліпопротеїдів плазми крові.
Показник |
хм |
ЛПДНГ |
ЛПНГ |
ЛПВГ |
Діаметр міцел, нм |
Близько 500 |
Близько 50 |
15-20 |
6-9 |
Основний ліпідний компонент |
Тригліцериди |
Тригліцериди |
Холестерол |
Фосфоліпіди |
Місце утворення |
Епітелій тонкої кишки |
Печінка |
Кровоносні капіляри печінки |
Печінка, тонка кишка |
Функції |
Транспорт екзогенних тригліцеридів у/Ьмнтіітло .ж |
Транспорт ендогенних тригліцеридів Of'V |
Транспорт холестеролу до периферичних тканин |
Транспорт холестеролу від периферичних тканин до печінки |
21.6. Як класифікують гіперліпопротеїнемії?
За походженням гіперліпопротеїнемії бувають первинними (спадковими) і вторинними (набутими).
Класифікація ВООЗ передбачає поділ гіперліпопротеїнемій на типи залежно від того, вміст ліпопротеїдів якого класу збільшений у крові (див. запит. 21.9).
Залежно від механізмів розвитку гіперліпопротеїнемія може бути продукційною і ретенційною.
21.7. Які генетичні дефекти можуть бути причиною розвитку первинних (спадкових) гіперліпопротеїнемій?
1. Генетично обумовлені зміни структури апопротеїнів — білкової частини ліпопро-теїдних міцел, унаслідок чого ліпопротеїди плазми крові не можуть взаємодіяти з відповідними рецепторами або зазнавати ферментативних перетворень.
2. Спадкові дефекти ферментів, що беруть участь в обміні ліпопротеїдів, зокрема, дефіцит ліпопротеїнліпази, печінкової ліпази, лецитинхолестеролацилтрансфера-зи (ЛХАТ).
3. Аномалії клітинних рецепторів до ліпопротеїдів.
21.8. Назвіть можливі причини розвитку вторинних (набутих) . гіперліпопротеїнємій.
1. Ендокринні хвороби (цукровий діабет, гіпотиреоз).
2. Метаболічні розлади (ожиріння, подагра).
3. Хвороби нирок (нефротичний синдром).
4. Хвороби печінки (обтураційна жовтяниця).
5. Інтоксикації (алкоголізм).
21.9. Наведіть класифікацію гіперліпопротеїнємій, запропоновану експертами ВООЗ.

Умовні позначення див. запит. 21.4.
21.10. Наведіть приклади розвитку різних типів гіперліпопротеїнємій за класифікацією ВООЗ.
Тип гшерліпопротеїнемії |
Спадкові порушення |
Набуті порушення |
Типі |
Дефіцит ліпопротеїнліпази |
Системний червоний вовчак |
Тип Па |
Сімейна гіперхолестеролемія (дефіцит рецепторів до ЛПНГ) |
Гіпотиреоз |
Тип ПЬ |
Комбінована сімейна гіперхолестеролемія |
Нефротичний синдром |
Тип III |
Сімейна гіперліпопротеїнемія ПІ типу |
Ожиріння |
Тип IV |
Комбінована сімейна гіперліпідемія |
Цукровий діабет |
ТипУ |
Сімейна гшертриглщеридемія |
Алкогольна інтоксикація |
21.11. Що таке продукційна і ретенційна гіперліпопротеїнемія?
Продукційна гіперліпопротеїнемія розвивається внаслідок збільшення утворення ліпопротеїдів, а ретенційна — у результаті порушення їх утилізації.
21.12. У чому полягає патогенетичне значення гіперліпопротеїнемій ?
Гіперліпопротеїнемії сприяють розвитку атеросклерозу (див. розд. 28).
21.13. Наведіть приклади гіполіпопротеїнемій. Дайте коротку характеристику.
Зменшення вмісту в крові ліпопротеїдів (гіполіпопротеїнемія) спостерігається значно рідше, ніж гіперліпопротеїнемія. Причиною гіполіпопротеїнемій найчастіше є спадково обумовлені порушення. Серед них:
1) абеталіпопротеїнемїя. В організмі нема апопротеїну В, унаслідок чого не утворюються хіломікрони і ЛПНГ. Клінічно виявляє себе стеатореєю (поява жиру в калі) і авітамінозами жиророзчинних вітамінів. Успадковується аутосомно-рецесивно;
2) гіпобеталіпопротеїнемія. Зменшено вміст ЛПНГ. Вважають, що це доброякісний стан, який сприяє довголіттю, оскільки перешкоджає розвитку атеросклерозу й ішемічної хвороби серця. Успадковується аутосомно-домінантно;
3) танжерська хвороба (від назви острова Танжер на східному узбережжі США). Характеризується повною відсутністю ЛПВГ або наявністю їх аномальних форм. Порушується транспорт холестеролу від тканин у печінку, він накопичується в периферичних клітинах. Клінічно це виявляє себе гепатомегалією, спленомегалією, збільшенням лімфатичних вузлів.
21.14. Що таке "модифіковані" ліпопротеїди? Наведіть приклади. Яке їх патогенетичне значення?
"Модифікованими" називають якісно змінені ліпопротеїди (ЛП). До них, зокрема, відносять глікозильовані ЛП (зв'язані з глікозильними групами, утворюються при гіперглікемії), ацетоацетильовані ЛП (зв'язані з ацетооцтовою кислотою, утворюються при цукровому діабеті); ЛП, зв'язані з продуктами пероксидного окиснення ліпідів; ліпопротеїн X (з'являється при обтураційній жовтяниці); комплекси ліпопро-теїд-антитіло.
"Модифіковані" ЛП є атерогенними. їх накопичення сприяє розвитку атеросклерозу.
Ожиріння: визначення поняття, класифікації; етіологія і патогенез окремих форм. Експериментальне моделювання ожиріння. Медичні проблеми, пов'язані з ожирінням.
Що таке первинне і вторинне ожиріння?
Первинним називають ожиріння, що являє собою самостійний патологічний процес.
Вторинне ожиріння є ознакою тих чи тих захворювань. Найпоширенішими варіантами вторинного ожиріння є церебральне і гормональне ожиріння.
21.16. Назвіть основні причини первинного ожиріння.
1. Надмірне споживання їжі, що перевищує енергетичні витрати організму.
2. Гіподинамія — обмеження фізичної активності людини.
3. Генетична схильність. Може виявляти себе особливостями харчової поведінки людини або особливостями регуляції жирового обміну.
21.17. У яких випадках виникає церебральне ожиріння?
Церебральне ожиріння виникає при ураженнях гіпоталамуса, де зосереджені центри, що регулюють харчову поведінку. Такі ураження виникають при травмах, пухлинах, енцефаліті. Провідним механізмом ожиріння в цьому випадку є поліфагія (підвищення апетиту).
21.18. Які гормональні порушення можуть бути причиною вторинного ожиріння?
Гормональне ожиріння розвивається як одна з ознак ендокринних хвороб. Воно супроводжує розвиток:
а) гіпотиреозу;
б) аденоми острівців підшлункової залози (гіперінсул інізм);
в) синдрому Іценка—Кушинга;
г) гіпофункції статевих залоз.
21.19. Чим відрізняється гіперпластичний тип ожиріння від гіпертрофічного?
Гіперпластичне ожиріння пов'язане з гіперплазією жирових клітин, тобто зі збільшенням їхньої кількості. Для нього характерними є початок у ранньому дитячому віці і велика надлишкова вага.
В основі гіпертрофічного ожиріння лежить збільшення маси окремих жирових клітин, при цьому їхня кількість не міняється. Ожиріння цього типу має більш пізній початок і не настільки виражене, як у попередньому випадку.
21.20. Які існують експериментальні моделі ожиріння?
I. Експериментальні моделі первинного ожиріння. Отримано чисті лінії мишей і щурів з генетично обумовленим ожирінням, що є ознакою, яка передається від потомства до потомства.
II. Експериментальні моделі вторинного церебрального ожиріння:
а) руйнування вентромедіальних ядер гіпоталамуса, що утворюють "центр насичення";
б) електростимуляція латеральних ядер гіпоталамуса, що становлять "центр апетиту".
III. Експериментальні моделі вторинного гормонального ожиріння:
а) вимикання функції деяких ендокринних залоз (видалення щитоподібної залози, кастрація);
б) введення в організм великої кількості деяких гормонів (інсуліну, глюкокор-тикоїдів).
IV. Експериментальні моделі місцевого ожиріння — перетинання симпатичних нервів. При цьому в тканині з порушеною іннервацією збільшується маса жирової клітковини, оскільки припиняється ліполітична дія катехоламінів.
21.21. Які механізми лежать в основі збільшення маси жирової тканини при ожирінні?
I. Посилення ліпогенезу. Цей механізм є провідним при:
а) посиленому надходженні жирових кислот у жирові клітини з хіломікронів і ліпопротеїдів дуже низької густини (переїдання, церебральне ожиріння, гіперінсулінізм);
б) посиленому утворенні жирових кислот в адипоцитах із глюкози (надмірне споживання вуглеводів, гіперінсулінізм, гіперфункція кори надниркових залоз);
в) збільшенні активності ферментів ліпогенезу (гіперінсулінізм).
II. Пригнічення ліполізу. В основі цього механізму зменшення активності гормончут-ливої ліпази адипоцитів. Це буває при:
а) гіподинамії;
б) гіпотиреозі;
в) порушенні симпатичної іннервації.
21.22. У чому полягає патогенетичне значення ожиріння?
При ожирінні значно зростає ризик виникнення багатьох соматичних хвороб. Серед них — атеросклероз, ішемічна хвороба серця, інфаркт міокарда, цукровий діабет II типу.
Позитивний і негативний баланс азоту. Види гіперазотемії. Зміни білкового складу крові. Спадкові порушення обміну амінокислот.
У дорослої здорової людини кількість азотистих речовин, що виводяться з організму, дорівнює кількості, що він її отримує з їжею. Такий стан називається азопшстою рівновагою.
В організмі, що росте, при вагітності, при введенні або надмірному утворенні ! анаболічних гормонів, при посиленому годуванні після виснажливих хвороб азоту виводиться менше, ніж його надходить. У цьому випадку йдеться про позитивний азотистий баланс.
І навпаки, якщо азоту виводиться більше, ніж надходить, то розвивається негативний азотистий баланс. Це може бути при голодуванні, втраті білків через нирки (протеїнурія), шкіру (опіки), кишки (проноси); при тиреотоксикозі, інфекційній гарячці
22.2. Назвіть основні причини аліментарної білкової недостатності.
Аліментарнії білкова недостатність розвивається внаслідок порушень надходження в організм білків, їх перетравлювання і всмоктування.
Основними її причинами є голодування, незбалансоване за амінокислотним і складом харчування, запальні й дистрофічні зміни різних відділів кишок, що супроводжуються порушеннями їх секреторної та моторної функцій.
22.3. Назвіть основні причини порушення біосинтезу білків у клітинах.
1. Порушення структури генів, що кодують інформацію про будову білків (мутації).
2. Отрути й специфічні інгібітори мультиферментних комплексів, що забезпечують ] процеси транскрипції, трансляції й посттрансляційної модифікації білків.
3. Дефіцит незамінних амінокислот.
4. Дефіцит АТФ.
5. Порушення утворення транспортних і рибосомної РНК, білків рибосом.
22.4. Порушення яких етапів можуть спричиняти розлади біосинтезу білків у клітинах?
I. Порушення транскрипції- утворення інформаційної РНК на матриці ДНК.
II. Порушення трансляції— синтезу поліпептидних ланцюгів з амінокислот.
III. Порушення посттрансляційної модифікації білків — формування їхньої третин- ; ної й четвертинної структур.
22.5. Які зміни білкового складу крові можуть виникати в умовах патології?
I. Гіпопротеїнемія - зменшення вмісту білків у плазмі крові. Виникає головним чином за рахунок зниження кількості альбумінів і може бути набутою і спадковою. До гіпопротеїнемії спричиняються голодування, аліментарна білкова недостатність, захворювання печінки, вихід білків із кровоносного русла (крововтрата, плазмовтрата, ексудація, транссудація) і втрата білків із сечею (протеїнурія). Гіпопротеїнемія веде до зменшення онкотичного тиску плазми крові, у результаті чого рідина виходить з кровоносних судин в інтерстиціальну тканину - розвиваються набряки.
II. Гіперпротеїнемія — збільшення вмісту білків у плазмі крові. Буває відносною (згущення крові) і абсолютною. Абсолютна гіперпротеїнемія найчастіше обумовлена збільшенням синтезу білків плазми крові і головним чином у-глобулінів (антитіл).
Клінічні прояви гіперпротеїнемії пов'язані зі збільшенням в'язкості крові, зміною її реологічних властивостей і, як наслідок, порушеннями мікроциркуляції.
III. Диспротеїнемія - зміна співвідношення між окремими білковими фракціями крові. Може бути спадковою і набутою. Часто пов'язана зі зміною спектру а- і у-глобулінів. Характерна для гострих запальних процесів ("білки гострої фази запалення"), дифузних захворювань сполучної тканини, аутоімунних захворювань. Іноді в крові з'являються якісно змінені білки, зокрема, парапротеїни і кріогло-буліни. Парапротеїни — це імуноглобуліни, що є продуктами одиничних клонів лімфоцитів. їх поява обумовлена проліферацією окремих антитілосинтезуючих клітин, що буває при патологічних процесах пухлинної природи (мієломна хвороба, макроглобулінемія Вальденстрема).
Кріоглобуліни — це різновид парапротеїнів. Вони являють собою патологічні білки із властивостями імуноглобулінів, що преципітують при охолодженні.
22.6. Що таке продукційна іретенційна гіперазотемія?
Пперазотемія — це збільшення залишкового (небілкового) азоту в крові. У нормі залишковий азот на 50 % складається з азоту сечовини, близько 25 % його припадає на частку амінокислот, інша частина — на інші азотисті продукти.
Продукційна (печінкова) гіперазотемія виникає внаслідок порушення утворення сечовини, детоксикації азотистих продуктів у печінці.
Ретенційна (ниркова) гіперазотемія є наслідком порушення видільної функції нирок.
22.7. Які фактори можуть викликати порушення утворення сечовини в печінці?
Порушення синтезу сечовини може спостерігатися на завершальних етапах розвитку недостатності печінки як одна з ознак порушення її детоксикаційної функції.
Можливі спадково обумовлені порушення сечовиноутворення. їх причиною може бути недостатній синтез цілого ряду ферментів: аргінінсукцинатліази (аргінін-
сукцинатурія), карбамоїлфосфатсинтетази і орнітинкарбамоїлтрансферази (амоніє-мія) і аргінінсукцинатсинтетази (цитрулінурія).
Наслідком порушення синтезу сечовини є накопичення аміаку в крові й цілий ряд пов'язаних з цим тяжких клінічних проявів (докладно див. розд. 31).
22.8. У чому сутність і чим виявляють себе спадково обумовлені порушення обміну фенілаланіну?
Спадково обумовленим порушенням обміну фенілаланіну є фенілкетонурія -за-хворювання з аутосомно-рецесивним типом спадкування. Його причиною є генетичний дефект ферменту фенілаланінгідроксилази, що в нормі перетворює фенілаланін на тирозин (рис. 73). За відсутності зазначеного ферменту окиснення фенілаланіну відбувається шляхом утворення фенілпіровиноградної і фенілмолочної кислот. Однак цей шлях має малу пропускну здатність, і тому фенілаланін накопичується у великій кількості в крові, тканинах і спинномозковій рідині, що в перші ж місяці життя веде до важкого ураження центральної нервової системи й невиліковного слабоумства.
Рис. 73. Блокада шляхів метаболізму фенілаланіну і тирозину: блок а - фенілкетонурія; б — тирозиноз; в — алкаптонурія; г - гіпотиреоз; д — альбінізм
22.9. У чому сутність і чим виявляють себе спадково обумовлені порушення обміну тирозину?
Залежно від рівня генетичних дефектів спадково обумовлені порушення обміну тирозину можуть виявлятися розвитком тирозинозу, алкаптонурії, альбінізму. Усі ці захворювання успадковуються аутосомно-рецесивно.
Тирозиноз виникає внаслідок генетичного дефекту ферменту - оксидази парагі-дроксифенілпіровиноградної кислоти. У результаті вона, будучи першим проміжним продуктом обміну тирозину, не перетворюється в гомогентизинову кислоту, накопичується в крові й разом з тирозином виводиться із сечею.
Алкаптонурія є наслідком порушення синтезу оксидази гомогентизинової кислоти, що перетворює останню в малеїлацетооцтову кислоту. У результаті в крові й сечі з'являється гомогентизинова кислота. Сеча при стоянні на повітрі, а також при додаванні до неї лугу стає чорною, що пояснюється окисненням гомогентизинової кислоти киснем повітря й утворенням алкаптону. Гомогентизинова кислота з крові проникає в тканини — хрящову, сухожилля, зв'язки, внутрішній шар стінки аорти, унаслідок чого з'являються темні плями в ділянці вух, носа, на склерах. Іноді розвиваються тяжкі зміни в суглобах.
Альбінізм обумовлений дефіцитом ферменту тирозинази. Унаслідок цього не утворюється пігмент шкіри й волосся — меланін. Організм, позбавлений пігменту, стає дуже чутливим до дії ультрафіолетового випромінювання.
Порушення пуринового та піримідинового обміну. Етіологія, патогенез подагри.
22.10. Що таке подагра?
Подагра - це хвороба, в основі якої лежить накопичення в організмі сечової кислоти - кінцевого продукту обміну пуринових основ, що входять до структури нуклеїнових кислот.
Для хворих на подагру характерне збільшення рівня сечової кислоти в крові (гіперурикемія). Гіперурикемія може супроводжуватися відкладенням солей сечової кислоти в суглобах і хрящах, де через слабке кровопостачання завжди є тенденція до зменшення рН (кисле середовище сприяє випаданню солей в осад). Відкладення солей викликає гостре подагричне запалення, що супроводжується болем, гарячкою і завершується утворенням подагричних вузлів і деформацією суглобів.
22.11. Що є факторами ризику подагри?
Факторами ризику подагри можуть бути:
1) надмірне надходження пуринів в організм (уживання в їжу великої кількості м'яса, особливо з вином і пивом);
2) надмірне надходження в організм молібдену, що входить до складу ксантинокси-дази, яка перетворює ксантин у гіпоксантин. Останній потім перетворюється в сечову кислоту;
3) стать (частіше хворіють чоловіки);
4) літній вік, для якого характерна вікова гіперурикемія;
5) спадкова схильність. Можливими є домінантно успадковуване підвищення рівня сечової кислоти в крові і зміни факторів, що підтримують сечову кислоту в розчиненому стані.
Гіпо- та гіпервітамінози: види, причини і механізми розвитку. Патогенез основних клінічних проявів. Принципи корекції вітамінної недостатності.
??????????????????????????????????
Порушення водно-сольового обміну. Форми гіпер- і гіпогідрії, їх етіологія, патогенез, наслідки. Порушення обміну натрію і калію: причини, механізми, клінічні прояви.
Водно електролітний обмін - це сукупність процесів надходження води і електролітів в організм, розподілу у внутрішньому середовищі та виведення з організму.
Вода складає 45-70% від маси тіла людини:
35-45 - інтрацелюлярна рідина
15-25 - екстрацилюлярна рідина( внутрішньоклітинна - 5%,міжклітинно - 12-15%, трансцелюлярно - 1-3% )
Регуляція ВЕО
Відбувається Нейрогуморальгим шляхом
Основним регуляторним органом є нирки.
МехАнізми затрмки води в організмі:
1) антидіуретичний механізм ( при подразненні осморецепторів гіпоталамуса при підвищ Р осм крові , подразн волюморецепторів у стінках серця)
Вазопресин - виробляється в суправентрикулярних ядрах гіпоталамуса, накопичується у нейрогіпофізі, накопичується у нейрогіпофізі , потім виділяється в кров. Забезпечує реабсорбцію води.
2) антинатрійуретичний механізм - реалізується при подраз рецепторів вас аференс у нирках а також при подр натрівих Рецепторів в ЮГА нирок > активація РААС ( ринін, ангіотензин, альдесторонова сист)
Ренін > L2макроглобулін крові ( ангіотензиноген), йде відщеплення ангіотензин 1 ( неактивна речовина)> АПФ> ангіотензин 2 при діЇ ангіотензинази > ангіотензин 3 > збільшення синтезу альдостерону ( реабсорбція Натрій +, секрецію і виведення К+ з сечею)
3) Стимуляція центру спраги у латеральній ділянці гіпоталамуса
виникає Внаслідок зменшення ОЦК, і дії ангіотензину
Механізми які сприяються виведенюю сечі
1) Реномедулярні простагландини ( ПгА та ПгЕ) ( артеріальний та гіпоталамічний) - збільшення діурезу та підвищ Натрійуреази з зниженням артеріального тиску
Стимули: гіпернатріємія, підвищений АТ, стимуляція альфоалдренорецепторів, та рецепторів вазопресину
Порушення ВОЕ
1) Гіпергідратація - накопичення позаклітинноі рідини ( позитивний водний баланс)
Прични: - збільшення надходження води в організм
- зменшення вивиедення води з організму
Види:
- ізоосмолярна( ізотонічна) - зберігається норм співвідношення між водою і іонами, Р осм не змінюється. Причини: внутрвенне введення великоі кількості інфузійних розчинів у пацієнтів в післяопераційних періодів, ниркова недостатність у стадію олігоанурії, введення великого обєму інфузійних розчинів у пацієнтів з нирковою недостатністю
- гіпоосмолярна (гіпотонічна)- водне отруєння - надмірне накопичення води без відповідноі затримки електролітів. Пр. цьому відбувається внутрішньоклітинний набряк. Причини: внтрішньовенне вливання великого обєму гіпотонічного розчину глюкози, неправильна корекція зневоднення, особливо у дітей, чистою водою, лікування нецукрового діабету препаратами вазопресину.
- гіперосмолярна( гіпертонічна) - надмірне накопичення електролітів і помірне накопичення води. При цьому виникає зневоднення клітин.Причини: інфузія гіпертонічних розчинів елеутролітів що перевищує іх можливість виведення нирками, вимушене пиття морскьоі води, алдостеронізм.
2) Дегідратація( негативний водний баланс) - розвивається тоді коли виведення води перевищує іі надходження в організм.
Причини:
-збільшення виділення води з оргінзму
-1зменшення надходження води в організм
Види:
- ізоосмолярна - розвивається при еквівалентній в раті води і електролітів. Причини: гостра крововтрата, нетривала поліурія.
- гіпоосмолярна - розвивається при переважноому виведенні солей. Виникає клітинна гіпергідратація. Причини: поліурія при цукровому діабеті.
- гіперосмолярна - розвивається коли втрата води перевищує втрату електролітів.( натрію )Причини: профузне потовиділення, слиновиділення, поліурія при нецукровому діабеті.
Ангідремія — це зменшення вмісту води в рідкій частині крові. Є крайнім проявом позаклітинного зневоднення й характеризується розвитком ангідремічного шоку
23.30. Назвіть причини гіпонатріємії, захисно-компенсаторні реакції, що виникають при цьому, і патогенетичне значення даного порушення.
Первинна (абсолютна) гіпонатріємія розвивається в результаті зменшення надходження натрію в організм (безсольова дієта, анорексія) або внаслідок збільшення виведення натрію з організму нирками (гіпофункція кори надниркових залоз, ниркова недостатність).
Причиною вторинної (відносної) гіпонатріємії є. надмірне надходження в організм води або її затримка - гіпонатріємія внаслідок розведення.
Зменшення концентрації іонів натрію в позаклітинній рідині викликає, з одного боку, посилення секреції альдостерону через ренін-ангіотензинний механізм, з другого - зменшення надходження в кров антидіуретичного гормону, оскільки зменшується імпульсація від осморецепторів. Наслідком цього є посилення реабсорбції іонів натрію й пригнічення реабсорбції води в нирках — осмотичний тиск позаклітинної рідини відновлюється.
Патогенетичне значення гіпонатріємії полягає в розвитку генералізованого набряку клітин (див. запит. 23.27).
23.31. Назвіть причини гіперкаліємії, захисно-компенсаторні реакції, що виникають при цьому, і патогенетичне значення даного порушення.
Гіперкаліємією називають збільшення вмісту іонів калію в плазмі крові понад 5,5 ммоль/л.
Причинами її розвитку можуть бути:
1) надмірне надходження калію в організм;
2) перехід іонів калію із внутрішньоклітинного в позаклітинний простір при масивному ушкодженні клітин, при збільшенні інтенсивності катаболічних процесів і ацидозі;
3) порушення виведення калію з організму (оліго- і анурія, недостатність функції кори надниркових залоз).
Збільшення концентрації іонів калію в крові безпосередньо активує клітини клу-бочкової зони кори надниркових залоз і викликає посилення секреції альдостерону. Останній збільшує секрецію іонів калію в ниркових нефронах і в такий спосіб відновлює їх концентрацію в крові.
Наслідками гіперкаліємії є:
1) порушення діяльності збудливих тканин (нервової і м'язової), у результаті чого розвиваються розлади функції ЦНС, серцево-судинної системи, скелетної мускулатури, гладких м'язів травного каналу;
2) розвиток негазового ацидозу (див. розд. 25).
23.32. Назвіть причини гіпокаліємії, захисно-компенсаторні реакції, що виникають при цьому, і патогенетичне значення даного порушення.
Гіпокшііємія — це зменшення концентрації іонів калію в плазмі крові нижче 3,5 ммоль/л.
Причинами її розвитку можуть бути:
1) недостатнє надходження калію в організм з їжею (тривале використання дієти, що не містить продуктів рослинного походження);
2) посилений перехід іонів калію з позаклітинного простору в клітини, що буває при посиленні анаболічних процесів і алкалозі;
3) втрата калію організмом (поліурія, гіперальдостеронізм, тривале використання сечогінних засобів).
При гіпокаліємії розвивається гіперполяризація мембран секреторних клітин і у зв'язку з цим зменшується секреція альдостерону корою надниркових залоз. Це викликає зменшення секреції іонів калію клітинами ниркового епітелію.
Патогенетичне значення гіпокаліємії полягає в тому, що:
а) збільшується поріг збудливості клітин і, як наслідок, з'являються загальна слабкість, метеоризм, гіпотонія скелетних м'язів, зменшується шкірна чутливість;
б) розвивається гіпокаліємічний алкалоз (див. розд. 25).
Набряк: визначення поняття, види, причини і механізми розвитку набряків (за Старлінгом).
23.18. Що таке набряки? Як їх класифікують?
Набряки — це накопичення рідини в тканинах організму і серозних порожнинах.
Розрізняють загальні й місцеві набряки. Загальні набряки є проявом позаклітинної гіпергідрії, місцеві — пов'язані з порушенням балансу рідини в обмеженій ділянці тканини або органа.
За етіологією виділяють набряки серцеві, ниркові, печінкові, кахектичні, запальні, алергічні, токсичні та ін.
Залежно від механізмів розвитку набряки можуть бути:
1) гідростатичними;
2) онкотичними;
3) мікседематозними.
Гідростатичні набряки виникають у результаті збільшення гідростатичного тиску в капілярах. Залежно від причин такого збільшення можна виділити:
а) гіперволемічні;
б) застійні;
в) мікроциркуляторні набряки.
Онкотичні набряки виникають унаслідок змін онкотичного тиску в капілярах або інтерстиціальній рідині. У цю групу входять:
а) гіпопротеїнемічні;
б) мембраногенні;
в) лімфогенні набряки.
23.19. У розвитку яких набряків провідна роль належить збільшенню гідростатичного тиску крові в капілярах?
Гідростатичні набряки можуть бути обумовлені такими механізмами: •
1) збільшенням об'єму крові (гіперволемічні набряки);
2) збільшенням венозного тиску (застійні набряки);
3) первинним порушенням мікроциркуляції - розширенням артеріол і спазмом ве-нул (мікроциркуляторні набряки). v^V-V
Гіперволемічнгши є набряки при позаклітинній гіпергідрії і набряки, пов'язані із затримкою в організмі іонів натрію, наприклад, при серцевій недостатності, вторинному гіперальдостеронізмі.
Причиною застійних набряків є порушення відтоку крові по венозних судинах (хронічна серцева недостатність, порушення венозних клапанів, тромбоз вен та ін.).
Розвиток набряків по мікроциркуляторному типу викликає гістамін, який одночасно розширює артеріоли і звужує вени.
23.20. У розвитку яких набряків провідна роль належить зменшенню онкотичного тиску крові?
Онкотичні набряки закономірно розвиваються при зменшенні вмісту білків (альбумінів) у плазмі крові. Це призводить до зменшення онкотичного тиску крові і переходу рідини з капілярів в інтерстиціальний простір.
За таким механізмом розвиваються, зокрема, набряки при голодуванні (кахек-тичні); нефротичні набряки, пов'язані із втратою білка (протеїнурією); печінкові набряки, що виникають унаслідок порушення синтезу альбумінів у печінці.
23.21. Які набряки відносять до мембраногенних?
Мембраногенні набряки виникають унаслідок підвищення проникності стінок судин (див. розд. 14). Збільшення проникності судин є причиною виходу білків плазми крові в міжклітинний простір, унаслідок цього зростає тканинний онкотичний тиск і вода переходить із кровоносних судин в інтерстицій.
Зазначений механізм є провідним у розвитку алергічних, запальних, токсичних набряків.
23.22. Що таке лімфогенні набряки?
Лімфогенні набряки виникають унаслідок порушень лімфоутворення і лімфо-відтоку. При цьому порушується виведення з лімфою білків, що в нормі фільтруються у тканину, і, як наслідок, збільшується тканинний онкотичний тиск.
Серед причин розвитку лімфогенних набряків варто виділити здавлення лімфатичних судин рубцем; збільшення центрального венозного тиску (недостатність серця), що перешкоджає припливу лімфи в систему кровообігу.
23.23. Що таке мікседематозні ("слизові") набряки?
Мікседематозні набряки - це особливий варіант набряків, в основі якого лежить збільшення гідрофільності тканинних колоїдів. При цьому в тканинах зростає кількість зв'язаної води.
Мікседематозні ("слизові") набряки характерні для гіпофункції щитоподібної залози.
23.24. Як відбувається накопичення води в тканинах при розвитку набряків?
У патогенезі набряків розрізняють дві стадії.
Перша стадія — накопичення зв'язаної води. Набрякова рідина зв'язується з тканинними колоїдами й накопичується в основному в гелеподібних структурах (колагенові волокна, основна речовина сполучної тканини). При цьому клінічні ознаки набряку незначні — трохи збільшується тургор тканини.
Друга стадія - накопичення вільної води. Коли маса зв'язаної води збільшується приблизно на ЗО %, а гідростатичний тиск у тканині досягає атмосферного, починає накопичуватися вільна незв'язана вода. Тоді з'являються виражені ознаки набряку: вільна вода переміщається відповідно до сили гравітації, з'являється симптом "ямки" при натискуванні на тканину.
23.25. Як впливає ацидоз на розвиток набряків?
Рис. 83. Вплив ацидозу на розвиток набряків
Ацидоз посилює розвиток набряків. Це пов'язане з тим, що при зміщенні рН у кислий бік зменшується гідрофільність структур сполучної тканини. Тому при надходженні води в тканину менша її кількість буде зв'язуватися з тканинними колоїдами і, отже, буде збільшуватися кількість вільної незв'язаної води. У зв'язку з цим за інших рівних умов клінічні ознаки набряку в кислому середовищі будуть більш вираженими (рис. 83).
23.26. У чому сутність дослідів Старлінга, присвячених вивченню патогенезу набряків?
Е. Старлінг (1896) у дослідах на жабах вивчав роль онкотичного тиску в розвитку набряків. Він показав, що при пер-фузії жаби фізіологічним розчином, що не містить білка, відбувається швидкий перехід рідини з суцин у тканину, внаслідок чого вода затримується в організмі й маса тварини зростає. Якщо потім перемкнути систему на перфузію сироваткою крові, то буде спостерігатися зворотний процес - рідина виходить із тканин у кровоносні судини й маса жаби поступово віднов-
люється до вихідного рівня. Дегідратація тканин відбувається значно повільніше, ніж розвиток набряків. Це пояснюється тим, що вода, яка надійшла в тканину, легко зв'язується із тканинними колоїдами й набагато важче переходить знову у вільний стан
Ацидоз: визначення поняття, класифікація, причини розвитку. Компенсаторні та патологічні реакції. Показники кислотно-основного стану при різних видах ацидозу, принципи корекції.
Ацидоз - це абсолютне або відносне накопичення кислот у крові.
Ацидоз і алкалоз можуть бути компенсованими (рН в межах норми) та декомпенсованими( рН виходить за межі норми)
За механізмом розвитку ацидози і алкалози поділяються на
- газові
- негазові
Газовий ацидоз - розвивається внаслідок надмірного накопичення в крові вугільноі кислоти внаслідок порушення виведення СО2 легенями
Причини:
- пригнічення дихального центру (передозування наркотиків, барбітуратами)
- різні захворювання, повязані з ураженням струкутр, що беруть участь в акті дихання (пневмонія, перелом ребер, пневмоторакс)
- дихання повітрям або штучними газовими сумішами з високою концентрацією СО2
Порушення функцій:
1) гіперкапнія причиняє спазм артеріол і підвищ артер тиску
2) спазм ниркових судин призвод до олігурії, виникає гіперкаліємія(оскільки клітини звязують іони водню а з клітин виходить калій) і гіперкальціємія
3) розширення судин головного болю та підвищ внутрішньочерепний біль
4) підвищ тонус блукаючого нерву і внаслідок чого можевиникати зупинка серцяі спазм бронхіол
5) гіпоксія выникає внаслідок зменшення здатності гемоглобіну звязувати кисень
6) клітинний алкалоз, який призведе до порушення конформаціі і властивостей струкутри і фунціі білків, а також білків ферментів
7) збільш гідрофільність клітинних колоїдів що призводить до набрякання клітин
8) в мітохондріях поруш процеси фосфорилювання та окислення
Негазовий ацидоз - розвивається внаслідок надмірного накопичення в крові нелетких кислот.
Види:
1) метаболічний ацидоз - виникає внаслІдок порушення обміну речовин. Є 2 види:
- кетоацидоз - повязаний з накопиченням кетонових тіл ( ацетооцтова кислота, ацетон). Причини: цукр діабет 1 типу, при голодуванні, в маленьких дітей на тлі інфекцейних захворювань
- лактатацидоз - повязаний з порушенням вуглеводного обміну і надмірним утворенням молочної кислоти. Причини: всі види гіпоксій, негативний ефект Пастера, фізичне навантаження( клепатура), рідко при цукр діабеті.
2) видільний ацидоз - розвив при порушенні фунціі органів виділення ( нирок, кишок) Види:
- азотемічний - при порушенні фільтраціі в клубочках нирок. Причини: гострий громерульнефрит, ниркова недостатНість.
- канальцевий - виникає при інгібуванні карбангідрази в нирках.
- гіперсалівація - слина втрачається і з нею зникають луги
- діарея - втрачання кишкового вмісту і як наслідок - ацидоз
3) Екзогенний ацидоз - розвивається внаслідок надмірного накопичення в організмі кислих продуктів ззовні (отруєння кислотами, саліцилатами)
Порушення функцій :
1) подразнення дихального центру іонами водню призведе до гіпервентиляції
2) виникає гіпокапнія, яка призведе до зниження артеріального тиску (розширення артеріол)
3) зменшення клубочкової фільтраціі
4) гіперкаліємія і гіперкальціємія
5) остеопороз
6) виникає гіпоксія внаслідок збільшення вмісту відновленого гемоглобіну
7) ацедотичне ушкодження клітин, може призвести до коми
8) пригнічення скоротливоії функціі серця внаслідок блокади іонами водню кальцієвих каналів
Показники КОС
Зі сторони крові:
SB - стандартний бікарбонат =24-28 ммоль/л
BB - сума всіх буферних основ крові = 44-54 ммоль/л
BE - надлишок буферних основ крові=(вираховується за формулою ВВ в нормі -- ВВ у хворого) = +_ 3 ммоль/л
принципи корекції порушень КОС
1) Відновити охідність дихальних шляхів
2) Негазовий ацидоз : введення натрію гідрокарбонату
3) Газовий алкалоз : відновити парціальний тиск СО2, спеціальні газові суміші(карбоген)
4) Негазовий алкалоз :залежить від його виду.
Якщо гіпохлоремічний - ввести розчин що містить хлор, але не містить калію (хлорид амонію)
Якщо гіпокаліємічний - ввести розчин що містить калій (калій хлор)
Якщо гіпернатріємічний - вивести надлишок натрія діуретиками
Алкалоз: визначення поняття, класифікація, причини розвитку. Компенсаторні та патологічні реакції. Показники кислотно-основного стану при різних видах алкалозу, принципи корекції.
Алкалоз - це абс або відн накопичення лугів в крові.
Ацидоз і алкалоз можуть бути компенсованими (рН в межах норми) та декомпенсованими( рН виходить за межі норми)
За механізмом розвитку ацидози і алкалози поділяються на
- газові
- негазові
Газовий алкалоз - це порушення КОС в основі якого лежить зниження рСО2 внаслідок гіпервентилації легень.
Причини:
1) стимуляція дихального центру (енцефаліт, епілепсія, пухлини)
2) рефлекторне збудження дихального центру при подразненні периферичних хеморецепторів (гірська звороба, гарячка)
3) неправильне проведення штучної вентиляції легень
Порушення функцій при газовому алкалозі:
1) гіперкапнія призводить до зниження артеріального тиску, внаслідок чого зменш клубочкава фільтрація і виникає поліурія
2) іони кальцію звязуються з плазмовими білками і виникає гіпокальціємія
3) судоми
4) гіпоксія, внаслідок збільшення спорідненості гемоглобіна до кисню ( більше оксигемоглобіну, менше відновленого)
5) всі прояви які супроводж клітинний алкалоз, який характерезуєтся набряканням клітини внаслідок збіль гідрофільності, і порушення окисного фосфорилювання в мітохондріях
Негазовий алкалоз - це таке порушення КОС в основі якого лежить введення лугів або виведення нелетких кислот. Види:
1) видільний (метаболічний)
- гіпохлоремічний, причинами є :блювання, гастростома.
- гіпокаліємічний - повязаний гіпокаліємією. Причини: альдостеронізм, аліментарний фактор ( дефіцит калію у їжі )
2) Екзогенний (гіпернатріємічний) - надмірне надходженяя натрію ззовні. Прични: неконтрольоване вживання лужних мінеральних вод, при неправильній корекції ацидозу (передозуванні бікарбонату), вживання антацидів системної дії( всмоктуються в шлунку), тривале введення натрієвих солей антибіотиків.
Порушення функцій при негазовому алкалозі:
1) алкалоз сприяє зменшенню активності дихального центру і гіповентиляції, що призведе до гіперкапнії> спазм артеріол > підвищ артеріального тиску
2) в судинах головного мозку відбув вазодилятація, підвищення внутрішньочерепного тиску і сильний головний біль
3) спазм ниркових судин веде до зменшення клубочкової фільтрації
4) підвищення тонусу блукаючого нерву призводить до зупинки серця, спазму бронхіол
5)гемічна гіпоксія внаслідок збільшення спорідненості гемоглобіну до кисню
6) зниження Росм позаклітинної рідини, що призводить до клітинної гіпергідратації
7) гіпокальціємія в крові, гіпокаліємія >тахиаритмія > зупинка серця в систолі
Показники КОС
Зі сторони крові:
SB - стандартний бікарбонат =24-28 ммоль/л
BB - сума всіх буферних основ крові = 44-54 ммоль/л
BE - надлишок буферних основ крові=(вираховується за формулою ВВ в нормі -- ВВ у хворого) = +_ 3 ммоль/л
принципи корекції порушень КОС
1) Відновити охідність дихальних шляхів
2) Негазовий ацидоз : введення натрію гідрокарбонату
3) Газовий алкалоз : відновити парціальний тиск СО2, спеціальні газові суміші(карбоген)
4) Негазовий алкалоз :залежить від його виду.
Якщо гіпохлоремічний - ввести розчин що містить хлор, але не містить калію (хлорид амонію)
Якщо гіпокаліємічний - ввести розчин що містить калій (калій хлор)
Якщо гіпернатріємічний - вивести надлишок натрія діуретиками
Порушення загального об'єму крові: класифікація, причини та механізми розвитку. Етіологія, патогенез крововтрати. Патогенез постгеморагічного шоку. Особливості у дітей.
26.3. Назвіть види порушень загального об'єму крові. Наведіть приклади.
Порушення об'єму крові виявляють себе гіповолемієїо або гіперволемією — зменшенням або збільшенням об'єму крові, якщо порівнювати з нормою (нормоволемією).
Гіпо- і гіперволемію поділяють на просту (зберігається нормальне співвідношення плазми і клітин крові), поліцитемічну (переважають клітини крові) і олігоци-темічну (переважає плазма).
Крім того, до порушень об'єму крові відносять зміни об'ємного співвідношення між клітинними елементами і плазмою при нормальному загальному об'ємі крові — оліго- і поліцитемічну нормоволемію (гемодилюція і гемоконцентрація). Показником об'ємного співвідношення є гематокрит, що визначає вміст клітинних елементів (переважно еритроцитів) у загальному об'ємі крові (у нормі 0,36-0,48, або 36-48 %).
Гіповолемія проста — зменшення об'єму крові без зміни гематокриту. Виникає відразу після гострої крововтрати і зберігається доти, доки рідина не перейде із тканини в кров.
Гіповолемія олігоцитемічна — зменшення об'єму крові з переважним зменшенням у ній клітин - еритроцитів. Спостерігається при гострій крововтраті в тих випадках, коли надходження крові й тканинної рідини в кровоносне русло не компенсує об'єм і особливо склад крові.
Гіповолемія поліцитемічна—зменшення об'єму крові внаслідок зменшення об'єму плазми при відносному збільшенні вмісту еритроцитів. Розвивається при зневодненні організму (пронос, блювота, посилене потовиділення, гіпервентиляція), шоку (вихід рідини в тканини внаслідок підвищення проникності стінок судин).
Гіперволемія проста— збільшення об'єму крові при збереженні нормального співвідношення між еритроцитами і плазмою. Виникає відразу ж після переливання великої кількості крові. Однак незабаром рідина виходить з кровоносного русла, а еритроцити залишаються, що веде до згущення крові. Проста гіперволемія при посиленій фізичній роботі обумовлена надходженням у загальний кровообіг крові з депо.
Гіперволемія олігоцитемічна — збільшення об'єму крові за рахунок плазми. Розвивається при затримці води в організмі у зв'язку із захворюваннями нирок, при вве-
денні кровозамінників. її можна моделювати в експерименті шляхом внутрішньовенного введення тваринам ізотонічного розчину натрію хлориду.
Гіперволемія поліцитемічна — збільшення об'єму крові за рахунок наростання кількості еритроцитів. Спостерігається при зниженні атмосферного тиску, а також при різних захворюваннях, пов'язаних з кисневим голодуванням (вади серця, емфізема), її розглядають як компенсаторне явище.
Однак при еритремії поліцитемічна гіперволемія є наслідком пухлинного розростання клітин еритроцитарного ряду кісткового мозку.
Олігоцитемічна нормоволемія виникає при анемії внаслідок крововтрати (об'єм крові нормалізувався за рахунок тканинної рідини, а кількість еритроцитів ще не відновилася), при гемолізі еритроцитів, порушеннях гемопоезу.
Поліцитемічна нормоволемія спостерігається при переливанні невеликих кількостей еритроцитарної маси.
26.4. Чим обумовлене патогенетичне значення гіпо- і гіперволемії?
Гіповолемія супроводжується порушенням дихальної й транспортної функцій еритроцитів; трофічної, екскреторної, захисної, регуляторної функцій крові. Це так чи інакше відбивається на сталості внутрішнього середовища організму (гомеостазі).
Гіперволемія обумовлює збільшення навантаження на серце. У випадку одночасного збільшення гематокриту (поліцитемічна гіперволемія) збільшується в'язкість крові, що може бути причиною порушень мікроциркуляції й провокувати утворення тромбів.
26.5. Що таке крововтрата? Які причини викликають крововтрату? Від чого залежать її перебіг і завершення?
Крововтрата - це патологічний процес, що виникає внаслідок кровотечі і характеризується складним комплексом патологічних порушень і компенсаторних реакцій, спрямованих проти зменшення об'єму циркулюючої крові й гіпоксії, обумовленої зниженням дихальної функції крові.
До етіологічних факторів, що викликають кровотечу, відносять:
1) порушення цілісності судин при пораненні або ураженні патологічним процесом (атеросклероз, пухлина, туберкульоз);
2) підвищення проникності судинної стінки (гостра променева хвороба);
3) зниження зсідання крові (геморагічний діатез).
Перебіг і результат крововтрати залежать від особливостей самої кровотечі (швидкості, величини, виду ушкодженої судини, механізму ушкодження); швидкості включення й вираженості компенсаторних реакцій організму; статі, віку, станів, що передують і супроводжують крововтрату (охолодження, травма, захворювання серця, глибокий наркоз). Серйозну небезпеку для життя людини являє втрата 50 % об'єму циркулюючої крові, смертельною є втрата крові понад 60 %.
26.6. Які стадії умовно виділяють у патогенезі гострої крововтрати?
І. Початкова стадія. Характеризується зменшенням об'єму циркулюючої крові -простою гіповолемією, зниженням артеріального тиску, гіпоксією переважно циркуляторного типу.
II. Компенсаторна стадія. Обумовлена здійсненням комплексу захисно-компенсаторних реакцій, спрямованих на ліквідацію наслідків втрати крові.
III. Термінальна стадія. Характеризується наростанням патологічних змін в організмі аж до настання смерті. Розвивається при недостатності компенсаторних реакцій, а також при інтенсивній і швидкій крововтраті, на тлі дії несприятливих факторів (охолодження, велика травма, серцево-судинні захворювання) і при відсутності лікувальних заходів.
26.7. Що є головною ланкою патогенезу гострої крововтрати?
У патогенезі гострої крововтрати центральне місце посідають порушення загальної гемодинаміки — зменшення об 'єму циркулюючої крові (гіповолемія) і обумовлене цим падіння артеріального тиску (гіпотензія) (рис. 94).
Зазначені порушення, з одного боку, є причиною розвитку власне патологічних змін в організмі (анемії, гіпоксії, ацидозу, інтоксикації), з другого- вмикають складний комплекс рефлекторних і гуморальних захисно-компенсаторних реакцій, спрямованих на відновлення й збереження гомеостазу.
Рис. 94. Головна ланка патогенезу гострої крововтрати
26.8. Які захисно-компенсаторні реакції закономірно розвиваються при крововтраті?
Залежно від строків виникнення захисні реакції організму поділяють на термінові і нетермінові.
Залежно від спрямованості виділяють такі групи механізмів компенсації: Іо4 спрямовані на зменшення об'єму судинного русла;
2) спрямовані на збільшення об'єму циркулюючої крові;
3) спрямовані на відновлення складу крові;
4) спрямовані на захист від гіпоксії-(див. розд. 19).
26.9. Які компенсаторні реакції організму спрямовані на зменшення об'єму судинного русла в початковій стадії крововтрати?
Мета цієї групи реакцій — привести у відповідність об'єм судинного русла зменшеному об'єму циркулюючої крові (централізація кровообігу). Це дає можливість на деякий час підтримати необхідний тиск крові й забезпечити кровопостачання життєво важливих органів.
Зазначені реакції- термінові, у їхньому розвитку провідне значення мають рефлекси.
Зменшення об'єму судинного русла при крововтраті обумовлюється такими змінами (рис. 95):
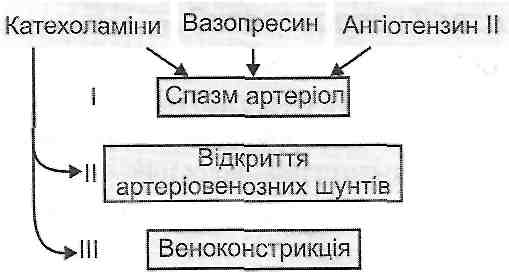
Рис. 95. Механізми централізації кровообігу при гострій крововтраті
1) спазм артеріол шкіри, м'язів, органів травної системи;
2) відкриття артеріовенозних анастомозів зазначених органів і тканин у результаті спазму прекапілярних сфінктерів;
3) веноконстрикція (скорочення гладких м'язів вен), що збільшує надходження крові до серця і зменшує ємність венозного відділу кровообігу.
В основі цих змін лежать такі механізми:
а) зниження артеріального тиску → збудження барорецепторів → активація симпа-тоадреналової системи → дія катехоламінів на а-адренорецептори гладких м'язів артерій, артеріол, прекапілярних сфінктерів і вен;
б) зменшення об'єму циркулюючої крові й артеріального тиску → збудження волю-мо- і барорецепторів → активація нейросекрсторних клітин гіпоталамуса, що продукують вазопресин → дія цього гормону на Vj-рецептори гладких м'язів судин з наступною вазоконстрикцією;
в) зменшення об'єму циркулюючої крові й активація симпатоадреналової системи → вивільнення клітинами юкстагломерулярного апарату нирок реніну → активація ренін-ангіотензинної системи з утворенням ангіотензину II → спазм гладких м'язів кровоносних судин.
26.10. У чому сутність компенсаторних реакцій організму, спрямованих на збільшення об'єму циркулюючої крові при крововтраті?
Реакції цієї групи здійснюються в період часу від кількох годин до кількох діб після кровотечі.
Збільшення об'єму циркулюючої крові в зазначений проміжок часу досягається за допомогою таких механізмів.
1. Перехід тканинної рідини в кровоносні судини. Основу цього процесу становить механізм, описаний Старлінгом. У результаті зменшення об'єму циркулюючої крові зменшується гідростатичний тиск у капілярах, що призводить до зменшення фільтрації води в артеріальній частині капілярів і збільшенню реабсорбції рідини у венозній. До зменшення гідростатичного тиску в капілярах призводить також розвиток спазму артеріол.
2. Посилення реабсорбції води та іонів натрію в нирках. Ця реакція, що запобігає втраті рідини із сечею, обумовлена такими механізмами:
а) дія вазопресину (антидіуретичного гормону) на У,-рецептори епітелію дистальних звивистих канальців і збірних трубок нирок, у результаті чого збільшується факультативна реабсорбція води;
б) активація ренін-ангіотензинної системи з наступним вивільненням альдостерону, що посилює реабсорбцію натрію в дистальних звивистих ка-нальцях нефронів;
в) активація симпатоадреналової системи, що призводить до перерозподілу течії крові між судинами кортикальних і юкстамедулярних нефронів, у результаті чого зростає площа й інтенсивність канальцевої реабсорбції води й натрію. 3. Вихід крові з депо в кровоносне русло. Основу цієї реакції становить активація симпатоадреналової системи й дія катехоламінів на судини печінки, селезінки, підшкірної жирової клітковини.
26.11. Які компенсаторні реакції забезпечують відновлення складу периферичної крові при крововтраті?
Відновлення складу периферичної крові забезпечується довгостроковими реакціями, які здійснюються в період часу від кількох діб до 1—2 тижнів після кровотечі. Вони виникають у відповідь на циркуляторну (зменшення об'єму циркулюючої крові) і гемічну (анемічну) гіпоксію. Основне значення надають кисневому голодуванню нирок, наслідком якого є утворення і надходження в кров великої кількості ниркових еритропоетинів. Останні діють на кровотворні клітини червоного кісткового мозку (еритропоетинчутливі клітини III класу) і стимулюють еритропоез - збільшується надходження молодих регенераторних форм еритроцитів у периферичну кров.
26.12. Які власне патологічні зміни можуть розвиватися при крововтраті?
1. Порушення системної' гемодинаміки (зменшення об 'єму циркулюючої крові, падіння артеріального тиску) і місцевого кровообігу (мікроциркуляції) аж до розвитку шоку.
2. Гостра постгеморагічна анемія (див. запит. 26.1.11).
3. Гіпоксія. Спочатку є циркуляторною, а потім гемічною (анемічною).
4. Негазовий ацидоз. Обумовлений гіпоксією й надходженням у кров молочної кислоти.
5. Порушення екскреторної функції нирок. При падінні артеріального тиску зменшується інтенсивність клубочкової фільтрації й розвиваються явища гострої ниркової недостатності: оліго- і анурія, інтоксикація (азотемія).
26.13. Що таке геморагічний шок? Назвіть головні патогенетичні ланки його розвитку.
Геморагічний шок — це шок, що виникає в результаті інтенсивної гострої крововтрати (рис. 96).
Провідним механізмом його розвитку є зменшення об 'єму циркулюючої крові, що викликає падіння
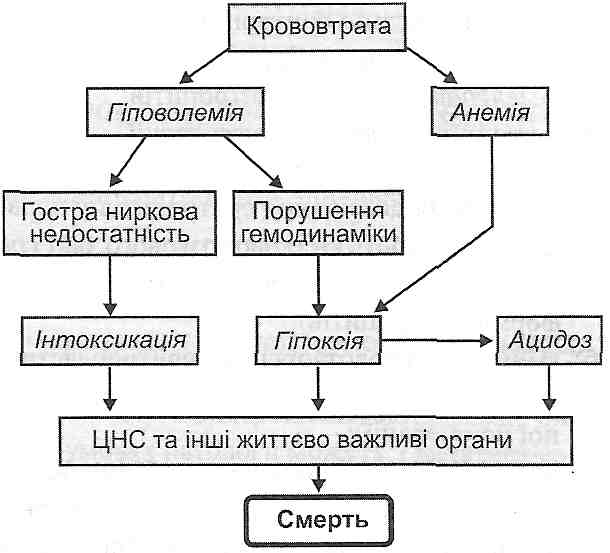
Рис. 96. Схема патогенеза геморагічного шоку
артеріального тиску, порушення мікроциркуляції, розлади кровопостачання життєво важливих органів (головного мозку, серця, нирок). Наслідком цього є розвиток гіпоксії, ацидозу й інтоксикації, що погіршує перебіг шоку, створює "зачаровані кола" у його патогенезі і в кінцевому підсумку веде до смерті.
Еритроцитози: визначення поняття, види, їх етіологія, патогенез.
Еритроцитоз — це збільшення в крові кількості еритроцитів понад б • 1012 віл і концентрації гемоглобіну понад 170 г/л.
Еритроцитоз поділяють на абсолютний і відносний.
Абсолютний еритроцитоз - це підвищення вмісту еритроцитів і гемоглобіну в одиниці об'єму крові внаслідок посилення еритропоезу. За етіологією виділяють набутий і спадковий абсолютний еритроцитоз.
Набутий абсолютний еритроцитоз виникає в результаті збільшення продукції еритропоетину переважно в нирках. Це може бути викликано такими причинами:
1) порушеннями нейрогуморальної регуляції- збудженням симпатичної частини нервової системи, гіперфункцією деяких ендокринних залоз. Катехоламіни, АКТГ, тиреоїдні гормони, глюкокортикоїди посилюють утилізацію кисню, що сприяє розвитку гіпоксії й утворенню еритропоетинів у нирках;
2) гіпоксичною, дихальною, циркуляторною гіпоксією - у випадку гірської хвороби, недостатності зовнішнього дихання й кровообігу (див. розд. 19);
3) місцевою гіпоксією нирок унаслідок їх ішемії (гідронефроз, стеноз ниркових артерій);
4) гіперпродукцією еритропоетинів деякими пухлинами (гіпернефрома, рак печінки та ін.).
Крім того, абсолютний еритроцитоз розвивається при істинній поліцитемії (еритремія, або хвороба Вакеза), що є різновидом хронічного лейкозу.
Причиною виникнення спадкового абсолютного еритроцитозу може бути генетично обумовлений дефект глобіну в молекулі гемоглобіну або дефіцит в еритроцитах 2,3-Дифосфогліцерату, що є регулятором оксигенації й дезоксигенації гемоглобіну. При цьому підвищується спорідненість гемоглобіну до кисню і зменшується віддача останнього тканинам (крива дисоціації оксигемоглобіну зміщена вліво). Розвивається гіпоксія, стимулюється продукція еритропоетинів, під впливом яких посилюється еритропоез.
Гіпоксичне, або дисрегуляторне посилення утворення еритропоетинів супроводжується підвищенням функціональної активності еритроцитарного паростка кісткового мозку, що виявляється збільшенням у крові вмісту еритроцитів, гемоглобіну, гематокриту. При цьому може збільшитися об'єм циркулюючої крові (поліцитемічна гіперволемія), її в'язкість, сповільнюється швидкість течії крові, порушується серцева діяльність. Артеріальний тиск підвищується, відзначається повнокрів'я внутрішніх органів, гіперемія шкіри й слизових оболонок, спочатку посилюється тромбоутворення, а потім виникають кровотечі (розвивається ДВЗ-синдром, див. розд. 26.3).
Зміни в крові при набутих абсолютних еритроцитозах часто носять компенсаторний характер, сприяють поліпшенню кисневого постачання тканин в умовах гіпоксії. Із припиненням дії етіологічного фактора відбувається нормалізація кількості еритроцитів і гемоглобіну. Саме в цьому полягає корінна відмінність абсолютного еритроцитозу, що характеризується підвищенням фізіологічної регенерації еритроцитарного паростка кісткового мозку, від еритремії (хвороба Вакеза),- при якій первинне й необоротне зростання кількості еритроцитів обумовлене гіперплазією еритроцитарного паростка пухлинної природи.
Відносний еритроцитоз — це збільшення вмісту еритроцитів і гемоглобіну в одиниці об'єму крові внаслідок зменшення об'єму плазми. Його розвиток пов'язаний з дією факторів, які обумовлюють зневоднення організму (див. розд. 23) або перерозподіл крові, що викликає поліцитемічну гіповолемію (наприклад, шок, опіки).
Анемії: визначення поняття, принципи класифікації. Регенеративні, дегенеративні, патологічні форми еритроцитів. Постгеморагічні анемії: види, причини, патогенез, картина крові. Особливості у дітей.
26.1.7. Дайте визначення поняття "анемія".
Анемія — це гематологічний синдром або самостійне захворювання, що характеризується зменшенням кількості еритроцитів і (або) вмісту гемоглобіну в одиниці об'єму крові, а також якісними змінами еритроцитів.
26.1.8. Якими гематологічними й загальними клінічними ознаками може виявляти себе анемія?
Гематологічні ознаки анемій поділяють на кількісні і якісні. До кількісних відносять:
1) зменшення вмісту еритроцитів в одиниці об'єму крові (у чоловіків нижче 4 • 1012, у жінок нижче 3,5 -1012 в 1 л крові);
2) зменшення концентрації гемоглобіну (у чоловіків нижче 130 г/л, у жінок нижче 120 г/л);
3) зменшення гематокриту (у чоловіків нижче 43 %, у жінок нижче 40 %);
4) зміни колірного показника (норма 0,85-1).
Якісними ознаками анемій є поява:
1) регенераторних форм еритроцитів (див. запит. 26.1.3);
2) дегенеративних змін у клітинах еритроцитарного ряду (див. запит. 26.1.4);
3) клітин патологічноїрегенерації(див. запит. 26.1.5).
Загальні клінічні прояви анемій:
1) гіпоксія - синдром, що виникає при будь-якому виді анемії (див. розд. 19);
2) синдроми, обумовлені особливістю патогенезу кожного окремого виду анемії (наприклад, при В12-фолієводефіцитній анемії- неврологічні розлади й ураження травної системи, при гемолітичній анемії- жовтяниця).
26.1.9. Як класифікують анемії? Наведіть приклади кожного виду анемій.
I. Патогенетична класифікація:
1) постгеморагічні анемії (наприклад, анемія після гострої крововтрати);
2) гемолітичні анемії (наприклад, серпоподібноклітинна);
3) анемії, обумовлені порушеннями еритропоезу (наприклад, залізодефіцитна).
II. За етіологією:
1) спадкові (наприклад, талассмія);
2) набуті (наприклад, хронічна постгеморагічна анемія).
III. За регенеративною здатністю червоного кісткового мозку:
1) регенераторні (наприклад, гостра постгеморагічна анемія);
2) гіперрегенераторні (наприклад, набута гемолітична анемія);
3) гіпорегенераторні (наприклад, залізодефіцитна анемія);
4) арегенераторні (наприклад, апластична анемія).
IV. За колірним показником (КП):
1) нормохромні (КП = 0,85-1; наприклад, гостра постгеморагічна анемія в перші кілька діб після крововтрати);
2) гіпохромні (КП < 0,85; наприклад, залізодефіцитна анемія);
3) гіперхромні (КП > 1; наприклад, ВІ2-фолієводефіцитна анемія).
V. За типом кровотворення:
1) анемії з еритробластичним типом кровотворення (наприклад, залізодефіцитна анемія);
2) анемії з мегалобластичним типом кровотворення (наприклад, В|2-фолієво-дефіцитна анемія).
VI. За клінічним перебігом:
1) гострі (наприклад, анемія після гемотрансфузійного шоку);
2) хронічні (наприклад, гіпопластична анемія).
26.1.10. Які ознаки свідчать про регенераторний характер анемії? »
Ознаками посиленої регенерації клітин еритроїдного ряду є:
1) з боку периферичної крові - збільшення вмісту ретикулоцитів і поліхроматофілів та поява нормобластів (регенераторні форми еритроцитів);
2) з боку червоного кісткового мозку -зміщення лейкоеритроїдного співвідношення від 3:1 до 1:1 і навіть до 1:2 і 1:3.
26.1.11. Що таке постгеморагічні анемії? Як їх класифікують?
Постгеморагічна анемія - це анемія, що розвивається в результаті крововтрати.
Залежно від характеру крововтрати виділяють два види анемій цієї групи: 1) гостру постгеморагічну і 2) хронічну постгеморагічну анемію.
Гостра постгеморагічна анемія виникає після швидкої масивної крововтрати при пораненні судин або їх ушкодженні патологічним процесом.
Хронічна постгеморагічна анемія розвивається внаслідок повторних, часто невеликих крововтрат, викликаних ураженням кровоносних судин при деяких хворобах (дисменорея, виразкова хвороба шлунка, геморой та ін.) і порушенням судин-но-тромбоцитарного і коагуляційного гемостазу (геморагічний діатез). Втрата заліза при частих кровотечах надає цій анемії залізодефіцитний характер.
26.1.12. Опишіть картину крові при гострій постгеморагічній анемії.
Картина крові при гострій постгеморагічній анемії зазнає змін залежно від часу, що пройшов після крововтрати. З урахуванням цього можна виділити три періоди, кожний з яких характеризується певною картиною периферичної крові (рис. 102).

Рис. 102. Картина периферичної крові в різні періоди після гострої крововтрати (N- норма)
I. Перші кілька годин після гострої крововтрати. У цей період часу зменшується загальний об'єм крові, а також загальна кількість еритроцитів в організмі. Однак в одиниці об'єму крові вміст еритроцитів і концентрація гемоглобіну не міняються. Це пояснюється тим, що відразу ж після крововтрати спрацьовують термінові компенсаторні реакції, спрямовані на зменшення об'єму судинного русла, і ще недостатньо виражені реакції, спрямовані на поповнення об'єму циркулюючої крові (перехід рідини із тканин у кров).
II. Період часу від кількох годин до кількох діб після гострої крововтрати. У результаті переходу рідини з інтерстиціального простору в кровоносні судини відбуваєть-
ся розведення крові (гемодшюі/ія). Як результат, зменшується кількість еритроцитів і гемоглобіну в одиниці об'єму крові, падає гематокрит. Колірний показник залишається без змін (нормохромна анемія). Якісні зміни еритроцитів у мазку крові ще не виявляються. III. Період часу від кількох діб до 1—2 тижнів після гострої крововтрати. Найбільш характерною рисою картини крові в цей період є поява великої кількості регенераторних форм еритроцитів (див. запит. 26.1.3), що пов'язане з посиленням еритропоезу в червоному кістковому мозку. Оскільки молоді незрілі еритроцити містять гемоглобіну менше в порівнянні зі зрілими клітинами, колірний показник зменшується й анемія стає гіпохромною.
26.1.13. Визначте місце гострої постгеморагічної анемії в різних класифікаціях анемій.
|
Класифікація |
Анемія після гострої крововтрати |
І. |
За патогенезом |
Постгеморагічна |
II. |
За етіологією |
Набута |
III. |
За регенераторною здатністю червоного кісткового мозку |
Регенераторна |
IV. |
За колірним показником |
Спочатку нормохромна, потім гіпохромна |
V. |
За типом кровотворення |
3 еритробластичним типом кровотворення |
VI. |
За клінічним перебігом |
Гостра |
26.1.14. Опишіть картину крові при хронічній постгеморагічній анемії.
У зв'язку із втратою заліза при частих кровотечах розвиваються гематологічні ознаки залізодефіцитної анемії: зменшується концентрація гемоглобіну й колірний показник, у мазку крові з'являються дегенеративні форми еритроцитів (мікро- і по-йкілоцитоз, гіпохромія). Кількість еритроцитів і гематокрит можуть залишатися без змін.
26.1.15. Визначте місце хронічної постгеморагічної анемії в різних класифікаціях анемій.
|
Класифікація |
Анемія після повторних крововтрат |
І. |
За патогенезом |
Постгеморагічна |
II. |
За етіологією |
Набута |
III. |
За регенераторною здатністю червоного кісткового мозку |
Гіпорегенераторна |
IV. |
За колірним показником |
Гіпохромна |
V |
За типом кровотворення |
3 еритробластичним типом кровотворення |
VI. |
За клінічним перебігом |
Хронічна |
Гемолітичні анемії, класифікація; причини та механізми гемолізу еритроцитів. Клінічна та гематологічна характеристика різних видів гемолітичних анемій.
26.1.16. Що таке гемолітичні анемії? Як їх класифікують?
Гемолітичними називають анемії, що виникають унаслідок руйнування (гемолізу) еритроцитів.
Класифікація. І. За походженням:
1) набуті;
2) спадково обумовлені. її. За причинами гемолізу:
1) анемії, обумовлені екзоеритроцитарними факторами (екстракорпускулярні);
2) анемії, обумовлені ендоеритроцитарними факторами (корпускулярні). НІ. За механізмами гемолізу:
1) анемії із внутрішньосудинним гемолізом;
2) анемії із внутрішньоклітинним гемолізом. IV. За клінічним перебігом:
1) гострі;
2) хронічні.
26.1.17. Які ендоеритроцитарні фактори можуть бути причиною розвитку гемолітичної анемії?
Розвиток гемолітичної анемії може бути пов'язаний з трьома групами дефектів еритроцитів:
1) дефекти мембрани (мембранопаті'і);
2) порушення ферментів (ферменто-, або ензимопатії);
3) зміни структури гемоглобіну (гемоглобінопатії).
26.1.18. Як визначити, які фактори (ендо- чи екзоеритроцитарні) є причиною гемолізу еритроцитів?
Із цією метою використовують пробу Моллісона у двох варіантах її постановки.
I. Еритроцити хворого з гемолітичною анемією вводять здоровій людині. Можливі результати:
а) якщо відбувся гемоліз цих еритроцитів, то анемія ендоеритроцитарна;
б) якщо гемолізу немає, то анемія екзоеритроцитарна.
II. Еритроцити здорової людини вводять хворому з гемолітичною анемією. Можливі результати:
а) якщо відбувається гемоліз цих еритроцитів, то анемія екзоеритроцитарна;
б) якщо гемолізу немає, то анемія ендоеритроцитарна.
26.1.19. Назвіть можливі причини й основні механізми внутрішньосудинного гемолізу еритроцитів.
Внутрішньосудиннш гемоліз виникає в кровоносних судинах унаслідок дії факторів, що ушкоджують еритроцити. Ці фактори отримали назву гемолітичних. До них відносять:
а) фактори фізичної природи (механічна травма, іонізуюча радіація, ультразвук, температура);
б) хімічні агенти (гемолітичні отрути);
в) біологічні фактори (збудники інфекційних захворювань, токсини, ферменти);
г) імунні фактори (антитіла).
Механізми внутрішньосудинного гемолізу.
I. Механічний гемоліз. Виникає внаслідок механічного руйнування мембран еритроцитів, наприклад, при роздавлюванні еритроцитів у судинах стопи (маршовий гемоліз).
II. Осмотичний гемоліз. Виникає тоді, коли осмотичний тиск усередині еритроцита більший, ніж осмотичний тиск плазми крові. У цьому випадку вода за законами осмосу надходить в еритроцит, об'єм його зростає, і в кінцевому підсумку відбувається розрив мембрани. Причиною осмотичного гемолізу може бути або зменшення осмотичного тиску середовища, у якому перебувають еритроцити (гіпотонічні розчини), або збільшення осмотичного тиску в самих еритроцитах. Останнє, як правило, пов'язане зі збільшенням концентрації іонів натрію усередині еритроцитів у результаті підвищення проникності їх мембрани або внаслідок порушення роботи Na-K-насосів.
III. Окисний гемоліз. Розвивається внаслідок вільнорадикального окиснення ліпідів і білків плазматичної мембрани еритроцитів. Результатом цього є збільшення проникності еритроцитарної мембрани, що потім веде до реалізації осмотичного механізму гемолізу.
IV. Детергентний гемоліз. Пов'язаний з розчиненням ліпідних компонентів мембрани еритроцитів речовинами-детергентами. Цей вид гемолізу викликають жовчні кислоти (холемічний синдром), жиророзчинні хімічні агенти, деякі токсини бактерій (лецитинази).
V. Комплементзалежний гемоліз. Обумовлений руйнуванням (перфорацією) мембрани еритроцитів активним комплементом. Цей механізм лежить в основі імунного гемолізу.
26.1.20. Які фактори можуть спричиняти окисний гемоліз еритроцитів?
Основу окисного гемолізу становлять реакції вільнорадикального окиснення, і зокрема, процеси пероксидного окиснення ліпідів еритроцитарної мембрани (див. розд. 11). Існує два механізми активації окисного гемолізу еритроцитів.
I. Посилене утворення вільних радикалів. Це буває при:
а) дії екзогенних речовин-окислювачів (деякі лікарські препарати, гемолітичні отрути, токсичні дози вітаміну D; продукти, що містяться в бобах (Vicia fava);
б) дії іонізуючої радіації;
в) гіпероксії.
II. Порушення діяльності антиоксидантних систем еритроцитів. Це може бути обумовлено:
а) спадковими або набутими порушеннями активності ферментів глюшатіоно-вої антиоксидантної системи (глютатіонпероксидази і глютатіонредуктази);
б) дефіцитом селену — мікроелемента, необхідного для функціонування глю-татіонпероксидази;
в) пригніченням реакцій пентозного циклу (наприклад, дефіцит глюкозо-6-фосфатдегідрогенази).
26.1.21. Які порушення розвиваються в організмі в результаті внутрішньосудинного гемолізу еритроцитів?
В нутрішньосу динний гемоліз супроводжується виходом гемоглобіну з клітин у плазму крові, де він частково з'єднується з білком гаптоглобіном (рис. 103). При цьому відбуваються такі процеси
1. Комплекс гемоглобін-гаптоглобін поглинається макрофагами і викликає утворення й вивільнення останніми макрофагальних еритропоетинів. Еритропоетини, впливаючи на червоний кістковий мозок, стимулюють еритропоез. У результаті в червоному кістковому мозку й периферичній крові з'являються ознаки посиленої регенерації клітин еритроїдного ряду.
2. Поглинений макрофагами гемоглобін зазнає біохімічних перетворень, у результаті яких білкова частина молекули розщеплюється до амінокислот, а з гема утворюється білірубін. Останній зв'язується з білками й надходить у кров (непрямий білірубін). У результаті розвивається синдром, відомий під назвою гемолітична жовтяниця (див. розд. 31).
З. Частина не зв 'язаного з гаптоглобіном гемоглобіну фільтрується в нирках. Це призводить, з одного боку, до появи гемоглобіну в сечі (гемоглобінурія), з другого - до "забивання" nop ниркового фільтра, що може бути причиною появи ознак гострої ниркової недостатності.
26.1.22. Що таке внутрішньоклітинний гемоліз еритроцитів? Чим він може бути обумовлений?
Внутрішньоклітинний гемоліз розвивається внаслідок поглинання і перетравлювання еритроцитів макрофагами (рис. 104).
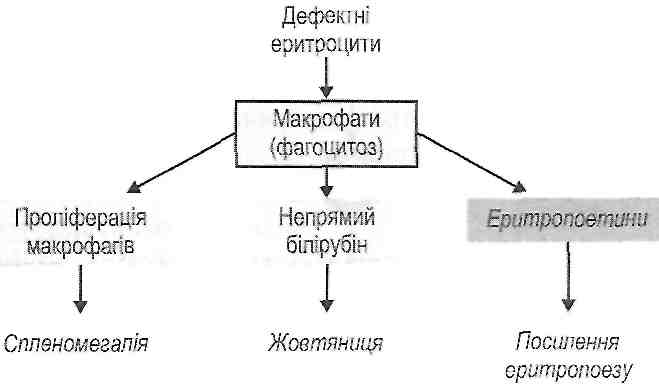
Рис. 104. Схема патогенезу порушень при внутрішньоклітинному гемолізі
У його основі можуть лежати такі причини:
а) поява дефектних еритроцитів. Зменшення пластичності еритроцитів, їхньої здатності до деформації, набряк призводять до того, що вони не можуть вільно проходити через міжендотеліальні щілини венозних синусів селезінки ("селезінковий фільтр") і надовго затримуються в червоній пульпі, контактуючи з макрофагами. Остання обставина і викликає поглинання дефектних еритроцитів макрофагами;
б) поява на поверхні еритроцитів хімічних груп, здатних специфічно взаємодіяти з рецепторами макрофагів. Такі групи виявляються при старінні еритроцитів (оголюються структури сіалових кислот еритроцитарної мембрани), а також при фіксації на їхній поверхні антитіл (з'являються Рс-фрагменти імуноглобулінів). В останньому випадку активується антитілозалежний фагоцитоз еритроцитів;
в) гіперспленізм - збільшення фагоцитарної активності макрофагів селезінки
26.1.23. Які порушення розвиваються в організмі в результаті внутрішньоклітинного гемолізу еритроцитів?
Посилений фагоцитоз еритроцитів викликає такі зміни:
а) утворення й вивільнення макрофагами epumponoemimie, у результаті чого посилюється еритропоез у червоному кістковому мозку і з'являється велика кількість регенераторних форм еритроцитів у периферичній крові;
б) утворення великої кількості білірубіну, що обумовлює розвиток жовтяниці;
в) проліферацію макрофагів, що призводить до збільшення селезінки (сплеиомегалїі).
26.1.24. Які причини можуть викликати розвиток набутої гемолітичної анемії?
Залежно від причин розвитку виділяють такі види набутої гемолітичної анемії.
I. Анемії, обумовлені механічним ушкодженням еритроцитів.
II. Імунні гемолітичні анемії.
III. Токсичні гемолітичні анемії.
IV. Інфекційні гемолітичні анемії.
V. Набуті мембранопатії.
26.1.25. Наведіть приклади анемій, обумовлених механічним ушкодженням еритроцитів.
1. Механічний гемоліз при протезуванні судин або клапанів серця.
2. "Маршова " гемоглобінурія — травматизація еритроцитів у капілярах стоп під час тривалого маршу.
3. Мікроангіопатична гемолітична анемія (хвороба Мошковича) — травматизація еритроцитів при зіткненні їх з нитками фібрину. Буває при ДВЗ-синдромі.
26.1.26. Що таке імунні гемолітичні анемії? Назвіть можливі причини.
Імунні гемолітичні анемії—це анемії, що виникають за участі специфічних імунних механізмів. Вони обумовлені взаємодією гуморальних антитіл з антигенами, фіксованими на поверхні еритроцитів, і тому є проявом II типу алергічних реакцій за класифікацією Кумбса і Джелла (див. розд. 10).
Залежно від причин розвитку виділяють такі види імунних гемолітичних анемій:
1. Алоімунні (ізоімунні) гемолітичні анемії. їх причиною можуть бути: а) надходження ззовні антитіл проти власних еритроцитів (гемолітична хвороба новонароджених) і б) надходження в організм еритроцитів, проти яких у плазмі є антитіла (переливання крові, не сумісної за групами АВО або Rh).
2. Аутоімунні гемолітичні анемії. Обумовлені утворенням в організмі антитіл проти власних еритроцитів. Це може бути пов'язано або з первинними змінами самих еритроцитів (поява аутоантигенів), або зі змінами в імунній системі (скасування імунологічної толерантності, поява "заборонених" клонів лімфоцитів).
3. Гетероімунні (гаптенові) гемолітичні анемії. Виникають при фіксації на поверхні еритроцитів чужорідних антигенів (гаптенів), зокрема, лікарських препаратів (пеніцилін, сульфаніламіди), вірусів
Гемолітичні анемії, класифікація. Клінічна та гематологічна характеристика різних видів гемолітичних анемій. Гемолітична хвороба новонародженого. АВО- конфлікт.
див вище!
26.1.27. Що таке гемолітична хвороба новонароджених?
Гемолітична хвороба новонароджених - це хвороба, що виникає в результаті гемолізу еритроцитів плода й новонародженого, викликаного антитілами матері.
Найчастіше бувають два варіанти гемолітичної хвороби новонароджених: резус-конфлікт і АВО-конфлікт.
Резус-конфлікт. Розвивається у випадку вагітності Шг-матері Ші+-плодом (найчастіше при повторній вагітності). Спочатку відбувається імунізація матері Rh+-epn-троцитами плода, які можуть потрапляти в організм матері під час пологів або при
дефектах плаценти. Найбільш імовірною є імунізація під час пологів, тому резус-конфлікт виникає найчастіше в умовах повторної вагітності Кп+-плодом.
У відповідь на надходження Ші+-еритроцитів в організмі матері синтезуються антитіла проти D-антигену. Ці антитіла (Ig G) здатні проникати через плаценту в організм плода й викликати гемоліз його еритроцитів.
АВО-конфлікт. Найчастіше виникає в ситуаціях, коли мати має групу крові 0(1), а плід - А(ІІ) або В(III). Нормальні ізоаглютиніни в системі АВО належать до класу IgM. Ці антитіла не проникають через плаценту й тому не можуть бути причиною АВ0-конфлікту. Однак у 10 % здорових людей, що мають групу крові 0(1), є антитіла проти аглютиногенів А і В, представлені IgG. Наявність цих антитіл не залежить від попередньої імунізації. Аглютиніни IgG проникають через плаценту і можуть викликати гемоліз еритроцитів плода з групами крові А(П), В(Ш). Серед дітей-первістків гемолітична анемія як результат АВО-конфлікту буває з такою ж частотою, як і у дітей, народжених після других, третіх і наступних пологів, на відміну від резус-конфлікту, при якому частота гемолітичної анемії збільшується зі збільшенням кількості пологів.
Залізодефіцитні анемії у дітей: причини і механізми розвитку, типові зміни периферичної крові, патогенез основних клінічних проявів. Залізорефрактерні анемії.
26.1.54. У чому полягає фізіологічне значення заліза?
В організмі дорослої людини масою 70 кг міститься 4,5 г заліза. Воно в гемовій і негемовій формах входить до складу цілого ряду білків.
I. Білки, які містять залізо в гемовій формі:
а) гемоглобін;
б) міоглобін;
в) цитохроми;
г) каталаза;
ґ) лактоферин.
II. Білки, які містять залізо в негемовій формі:
а) феритин;
б) гемосидерин;
в) трансферин;
г) ферменти: аконітаза, ксантиноксидаза, НАДН-дегідрогеназа.
26.1.55. Як відбувається обмін заліза в організмі?
Залізо надходить в організм у складі харчових продуктів (м'ясо, печінка, риба, рис, горох, курага). Найкраще всмоктується залізо, що входить до складу харчових білків у формі гема. Стінки кишок містять фермент гемоксигеназу, що розщеплює гем харчових продуктів на білірубін, оксид вуглецю (II) та іони заліза, останні і всмоктуються в тонкій кишці. Цьому процесу сприяють аскорбінова кислота, фруктоза, піровиноградна кислота. Системи транспорту заліза в кишках можуть забезпечити всмоктування максимум 2,5 мг заліза за добу, незалежно від потреби організму в цьому елементі.
Транспорт заліза в організмі здійснюється за допомогою білка трансферину, а депонування відбувається у формі іншого білка - феритину. Надходячи в клітини, залізо зв'язується з внутрішньоклітинним білком сидерохіліном, що транспортує залізо в мітохондрії, де синтезується гем.
Фізіологічні втрати заліза невеликі. У чоловіків вони становлять менш ніж 1 мг/ добу (втрати із сечею, потом, злущеним епітелієм шкіри). У жінок вони набагато більші і обумовлені менструаціями, вагітністю, пологами, лактацією.
26.1.56. Назвіть можливі причини розвитку залізодефіцитної анемії.
1. Недостатнє надходження заліза в організм:
а) аліментарна анемія в грудних дітей (вигодовування коров'ячим або козячим молоком);
б) порушення всмоктування заліза (резекція шлунка, кишок, гастрити, ентерити).
2. Крововтрати. Це найпоширеніша причина дефіциту заліза в організмі. Найчастіше буває при невеликих, але повторних кровотечах (хронічна постгеморагічна анемія).
3. Посилене використання заліза - вагітність, лактація.
26.1.57. Який патогенез порушень, що розвиваються в організмі у зв 'язку з дефіцитом заліза ?
Недостатність заліза в організмі призводить до порушення синтезу залізовмісних білків, а отже, до розладів функцій, у виконанні яких беруть участь ці білки. Найбільше значення мають такі лінії патогенезу:
1) дефіцит заліза → порушення синтезу гема і гемоглобіну → анемія;
2) дефіцит заліза → порушення синтезу гема → порушення утворення цитохро-мів → розлади клітинного дихання (порушення утилізації кисню) → тканинна гіпоксія;
3) дефіцит заліза → порушення синтезу гема → зменшення активності каталази → порушення функції антиоксидантних систем → активація вільнорадикального окиснення → ушкодження клітин → гемоліз еритроцитів і розвиток дистрофічних змін у клітинах;
4) дефіцит заліза → порушення синтезу гема → зменшення синтезу міоглобіну → зниження пристосувальних можливостей клітин щодо гіпоксії.
26.1.58. Дайте характеристику картини периферичної крові і червоного кісткового мозку при залізодефіцитній анемії.
Для залізодефіцитної анемії характерне зниження концентрації гемоглобіну в периферичній крові і зменшення колірного показника, що свідчить про зменшення насичення кожного окремого еритроцита гемоглобіном. Кількість еритроцитів в одиниці об'єму крові при цьому або трохи зменшується, або залишається без змін.
При біохімічних дослідженнях виявляється зниження вмісту заліза в сироватці крові, а також ступеня насичення залізом трансферину.
У мазку крові зменшується кількість регенераторних форм еритроцитів (ретику-лоцитів, поліхроматофілів) і з'являються дегенеративні форми. Характерні гіпохромія (з'являються анулоцити), анізоцитоз (мікроцитоз), пойкілоцитоз.
У червоному кістковому мозку зменшується вміст сидеробластів і сидероцитів (еритробластів і нормоцитів, що містять гранули заліза), частка яких у нормі становить 20-40 %. У той же час збільшується вміст базофільних і поліхроматофільних форм клітин еритроїдного ряду при одночасному зменшенні оксифільних. Зазначений феномен отримав назву "синього кісткового мозку ".
26.1.59. Визначте місце залізодефіцитної анемії в різних класифікаціях анемій.
|
Класифікація |
Залізодефіцитна анемія |
І. |
За патогенезом |
Анемія, пов'язана з порушеннями еритропоезу |
II. |
За етіологією |
Набута |
III. |
За регенераторною здатністю червоного кісткового мозку |
Гіпорегенераторна |
IV. |
За колірним показником |
Гіпохромна |
V. |
За типом кровотворення |
3 еритробластичним типом кровотворення |
VI. |
За клінічним перебігом |
Хронічна |
26.1.60. Якими синдромами виявляє себе залізодефіцитна анемія?
1. Гематологічний синдром. Охоплює порушення з боку периферичної крові і червоного кісткового мозку.
2. Гіпоксія. Виявляється загальною слабкістю, запамороченнями, серцебиттям, задишкою, непритомностями. Кисневе голодування в умовах дефіциту заліза має два механізми: кров'яний (зменшення кисневої ємності крові) і тканинний (порушення клітинного дихання й утилізації кисню).
3. Синдром трофічних порушень. Виявляється такими ознаками, як сухість і тріщини шкіри, тріщини в кутах рота, ангулярний стоматит, ураження нігтів (зміни форми, стоншення), атрофія сосочків язика (атрофічний глосит), атрофічний гастрит. Вважають, що розвиток зазначених порушень, з одного боку, пов'язаний з гіпоксичним і вільнорадикальним ушкодженням клітин, з другого — з розладами вторинних метаболічних шляхів, у здійсненні яких беруть участь ферменти, що містять залізо.
4. Сидеропенічний синдром. Це специфічний синдром, що виникає при дефіциті заліза. Він виявляється спотвореннями смаку й нюху. Хворі часто їдять крейду, зубний порошок, вугілля, глину, пісок, лід, сирі крупи, тісто, сирий м'ясний фарш. Мають пристрасть до запахів гасу, бензину, ацетону, вихлопних газів автомобілів. Патогенез зазначених порушень досі невідомий.
5. Синдром м'язової слабкості. М'язова слабість, що виникає при дефіциті заліза, завжди більш значна, ніж та, котрої варто було б очікувати, виходячи зі ступеня анемії. Вона виявляється слабкістю і стомлюваністю скелетних м'язів, слабкістю міокарда (міокардіопатія), порушеннями ковтання (дисфагія), порушеннями сечовипускання. Очевидно, що важливе значення в розвитку зазначених симптомів мають гіпоксія (кров'яна і тканинна), а також зменшення вмісту міоглобіну в м'язовій тканині.
26.1.61. Що таке залізорефрактерна анемія? Які її етіологія й патогенез?
Залізорефрактерна анемія-цс анемія, що виникає в результаті порушення включення заліза в гем при зниженні активності ферментів, що каталізують синтез порфіринів і гема.
Причинами розвитку цієї анемії можуть бути:
1) генетично обумовлене зниження активності декарбоксилази копропорфірииоге-ну - ферменту, що забезпечує один з кінцевих етапів синтезу гема (успадковується рецесивно, зчеплено з Х-хромосомою);
2) зменшення вмісту піридоксальфосфату - активної форми вітаміну В6, унаслідок чого залізо не вилучається з мітохондрій еритробластів і не включається в гем;
3) блокада свинцем сульфгідрильних груп ферментів, що беруть участь у синтезі гема, при побутовому й виробничому отруєнні свинцем.
Зменшення активності ферментів, що беруть участь в утворенні порфіринів і гема, призводить до зниження утилізації заліза й порушення синтезу гема гемоглобіну, що викликає недостатність еритропоезу і розвиток гіпохромної анемії з низьким вмістом гемоглобіну.
Порушення використання заліза супроводжується підвищенням вмісту сироваткового заліза, відкладенням його у внутрішніх органах з вторинним розростанням сполучної тканини (гемосидероз)
Причини виникнення і механізми розвитку недостатності вітаміну В,2 та фолієвої кислоти. Характеристика загальних порушень в організмі при дефіциті вітаміну В,2 та/або фолієвої кислоти. Гематологічна характеристика вітамін В12- та фолієводефіцитних анемій. Особливості у дітей.
26.1.47. Що таке металобластичні анемії? Наведіть приклади.
Мегалобласмичні анемії-це група анемій, в основі яких порушення синтезу нуклеїнових кислот у клітинах і, як наслідок, порушення розмноження останніх. До мегалобластичних відносять:
1) анемію, обумовлену дефіцитом вітаміну В і фолієвої кислоти, - Вп-фолієводефі-цитну анемію;
2) перніціозну анемію Аддісона-Бірмера (етіологію не встановлено);
3) анемію, пов'язану з порушеннями ферментів синтезу пуринових і піримідинових основ, — В 12-рефрактерну анемію (стійку до лікування вітаміном В|2).
За походженням мегалобластичні анемії можуть бути набутими (виявляються звичайно у дорослих і осіб літнього віку) і спадково обумовленими. В останньому випадку порушення можуть мати стосунок до системи транспорту речовин (усмоктування в травному каналі) і реакцій метаболізму (ферментопатії).
26.1.48. Яка роль вітаміну В12 і фолієвої кислоти в забезпеченні кровотворення?
Вітамін В (ціанокобаламін) надходить в організм у складі харчових продуктів (м'ясо, яйця, сир, печінка) у зв'язаному з білками вигляді. У результаті гідролізу білків вітамін В]2 вивільняється в шлунку, де утворюється комплекс вітаміну В,2 з гастромукопротеїном (внутрішнім фактором Кастла). 1 мг гастромукопротеїну зв'язує 25 мг вітаміну В]2. Комплекс, що утворився, всмоктується в середньому й нижньому відділах клубової кишки.
Транспорт вітаміну В12 кров'ю здійснюється за допомогою специфічних транспортних білків - транскобаламінів. Печінка є важливим депо вітаміну В12. Запаси його тут настільки великі, що потрібно 3-6 років, аби при порушеннях надходження вітаміну В в організмі виникли ознаки його дефіциту.
У клітинах з вітаміну В12 утворюється дві його коферментні форми: метилкоба-ламін і 5-дезоксиаденозилкобаламін.
Метилкобаламін необхідний для синтезу ДНК у клітинах, тому він істотно впливає на кровотворення (рис. 105).
Рис. 105. Вплив ціанокобаламіну на кровотворення
Цей кофермент входить до складу ферментного комплексу, що перетворює фолієву кислоту на її коферментну форму — тетрагідрофолієву кислоту. Тетрагідро-фолієва кислота, у свою чергу, є коферментом ферментного комплексу, що каталізує реакцію перетворення уридинмонофосфату на тимідинмонофосфат, що йде на побудову ланцюгів ДНК.
Друга коферментна форма вітаміну В]2 - 5-дезоксіаденозилкобаламін — необхідна для нормального обміну жирових кислот. Вона бере участь у реакціях перетворення токсичного продукту обміну - метилмалонової кислоти на сукцинат, котрий потім надходить у цикл Кребса.
26.1.49. Назвіть основні причини недостатності вітаміну В12в організмі.
1. Екзогенна (аліментарна) недостатність — недостатнє надходження вітаміну В12 в організм з продуктами харчування. Може розвиватися у маленьких дітей при вигодовуванні їх козяч им молоком або сухими молочними сумішами.
2. Порушення всмоктування вітаміну BJ2:
а) порушення утворення й секреції гастромукопротеїну (внутрішнього фактора Кастла). Буває при спадково обумовлених порушеннях, атрофії слизової оболонки шлунка, аутоімунних ушкодженнях парієтальних клітин слизової шлунка, після гастректомії або видалення понад 2/3 шлунка;
б) порушення функції тонкої кишки: хронічні проноси (целіакія, спру), резекція великих ділянок кишки;
в) конкурентне використання вітаміну В12 гельмінтами й мікрофлорою кишок (дифілоботріоз, синдром "сліпої петлі").
3. Порушення утворення транскобаламінів у печінці.
4. Порушення депонування вітаміну Впу печінці (наприклад, при цирозі).
5. Посилене використання вітаміну Вп (наприклад, при вагітності).
26.1.50. Який патогенез порушень, що розвиваються в організмі при дефіциті вітаміну В12?
Дефіцит вітаміну В12 призводить до розвитку розладів, пов'язаних з порушенням утворення двох коферментних форм цього вітаміну: метилкобаламіну та 5-дезоксіа-денозилкобаламіну.
Недостатнє утворення метилкобаламіну, як і дефіцит фолієвої кислоти, викликає порушення утворення тетрагідрофолієвої кислоти, у результаті чого не відбувається перетворення уридинмонофосфату в тимідинмонофосфат. Унаслідок дефіциту пуринових основ порушуються синтез ДНК і розмноження клітин, у першу чергу кровотворних і епітелію травного каналу.
У червоному кістковому мозку еритробластичний тип кровотворення змінюється на мегалобластичний, зростає неефективний еритропоез, стає коротшою тривалість життя еритроцитів. Розвивається анемія, при якій клітини патологічної регенерації й еритроцити з вираженими дегенеративними порушеннями з'являються не тільки в кістковому мозку, але й у крові.
Зміни в клітинах мієлоїдного й мегакаріоцитарного ряду виявляються зменшенням кількості лейкоцитів і тромбоцитів, вираженою атипією клітин (гігантські нейтрофіли, мегакаріоцити з дегенеративними змінами в ядрі).
Виникнення при дефіциті вітаміну В]2 і фолієвої кислоти атипового мітозу та поява гігантських клітин епітелію травного каналу призводять до розвитку запально-атрофічних процесів у слизовій оболонці його відділів (глосит, стоматит, езофагіт, ахі-лічний гастрит, ентерит). Це посилює первинні порушення секреції або всмоктування гастромукопротеїну, а отже, посилює дефіцит вітаміну Вр ("зачароване коло").
У результаті недостатності другої коферментної форми вітаміну В — 5-дезоксіа-денозилкобаламіну в організмі накопичуються пропіонова і метилмалонова кислоти, токсичні для нервових клітин. Крім того, у нервових волокнах синтезуються жирові кислоти зі зміненою структурою, що призводить до порушення утворення мієліну і ушкодження аксонів. Розвивається дегенерація задніх і бічних стовпів спинного мозку (фунікулярний мієлоз), уражаються черепні й периферичні нерви.
26.1.51. Дайте характеристику картини периферичної крові і червоного кісткового мозку при В 12-фолієводефіцитній анемії.
Найхарактернішою рисою цієї анемії є поява в крові й червоному кістковому мозку клітин патологічної регенерації — мегалобластів та їхніх без'ядерних форм-мегалоцитів.
На тлі істотного зменшення вмісту еритроцитів і концентрації гемоглобіну колірний показник збільшений, що пояснюється більшим діаметром еритроцитів. Характерне явище дегенерації еритроцитів: анізоцитоз (макроцитоз), пойкілоцитоз (поява клітин овальної форми), патологічні включення (тільця Жоллі, тільця Кебота).
У крові зменшений вміст гранулоцитів (особливо нейтрофілів) і тромбоцитів. Виявляються гігантські нейтрофіли з гіперсегментованими ядрами.
26.1.52. Визначте місце В 12-фолієводефіцитної анемії в різних класифікаціях анемій.
|
Класифікація |
В^-фолієводефіцитна анемія |
І. |
За патогенезом |
Анемія, пов'язана з порушеннями еритропоезу |
II. |
За етіологією |
Набута, є спадкові форми |
III. |
За регенераторною здатністю червоного кісткового мозку |
Гіпорегенераторна |
IV. |
За колірним показником |
Гіперхромна |
V. |
За типом кровотворення |
3 мегалобластичним типом кровотворення |
VI. |
За клінічним перебігом |
Хронічна |
26.1.53. Якими синдромами виявляє себе В 12-фолієводефіцитна анемія?
При В -фолієводефіцитній анемії спостерігають:
1. Гематологічний синдром:
а) анемія і пов'язана з нею гіпоксія;
б) лейкопенія (нейтропенія), що призводить до зниження резистентності організму до інфекцій;
в) тромбоцитопенія, що викликає розвиток геморагічного синдрому.
2. Ураження травного каналу, що виявляються розвитком запально-атрофічних змін у слизовій оболонці: глосит, стоматит, езофагіт, ахілічний гастрит, ентерит.
3. Ураження центральної й периферичної нервової системи: фунікулярний мієлоз, дегенерація периферичних нервів
Лейкоцитози: види, причини і механізми розвитку. Супутні ядерні зрушення нейтрофільних гранулоцитів. Лейкемоїдні реакції.
Лейкоцитоз - це збільшення кількості лейкоцитів в одиниці об'єму крові понад) 10 • 109/л. Лейкоцитоз не має самостійного значення, він є всього лише симптомом, що супроводжує розвиток багатьох хвороб.
Класифікація лейкоцитозів.
I. Залежно від причин розвитку виділяють фізіологічний і патологічний лейко-1 цитози.
II. Лейкоцитоз може бути абсолютним і відносним. Для абсолютного лейкоцитозу) характерне збільшення абсолютної кількості лейкоцитів в одиниці об'єму крові. Про відносний лейкоцитоз ідеться в тому випадку, коли зростає відносний вміси окремих форм лейкоцитів у периферичній крові.
III. За механізмом розвитку лейкоцитоз буває:
а) реактивним;
б) перерозподільним;
в) пухлинного походження.
IV. Залежно від видів лейкоцитів, вміст яких у крові збільшений, виділяють:
а) нейтрофільний лейкоцитоз (нейтрофільоз);
б) еозинофільний лейкоцитоз (еозинофілія);
в) базофільний лейкоцитоз;
г) лімфоцитарний лейкоцитоз (лімфоцитоз); ґ) моноцитарний лейкоцитоз (моноцитоз).
26.2.6. Наведіть приклади фізіологічного і патологічного лейкоцитозів.
Фізіологічний лейкоцитоз є фізіологічною реакцією організму на ті чи ті впливи. До його різновидів відносять:
а) емоціогенний лейкоцитоз — виникає під час сильних емоцій;
б) міогенний — розвивається під час інтенсивної фізичної роботи;
в) статичний — характерний для переходу людини з горизонтального у вертикальне положення;
г) аліментарний — розвивається після приймання їжі; ґ) лейкоцитоз у вагітних;
д) лейкоцитоз у новонароджених.
Патологічний лейкоцитоз пов'язаний з перебігом в організмі патологічних процесів. Він, як правило, розвивається при:
а) інфекційних захворюваннях;
б) запальних і алергічних процесах;
в) інтоксикації екзо- й ендогенного походження.
26.2.7. Що таке реактивний лейкоцитоз? Які механізми можуть лежати в основі його розвитку?
Реактивним називають лейкоцитоз, що виникає як реакція червоного кісткового мозку на патогенні впливи. Він закономірно розвивається при інфекційних захворюваннях, запаленні, дії низьких доз токсичних речовин.
В основі розвитку реактивного лейкоцитозу можуть лежати два механізми. І. Посилення проліферації і дозрівання лейкоцитів у червоному кістковому мозку. Це може бути пов'язано або зі збільшенням утворення в організмі лейкопоети-нів - речовин, що стимулюють утворення лейкоцитів, або зі зменшенням вмісту інгібіторів лейкопоезу. Серед лейкопоетинів найбільш вивченим нині є колонієс-тимулятивний фактор — речовина, що належить до групи цитокінів. Вона, будучи продуктом секреції активованими макрофагами, стимулює утворення гранулоцитів у червоному кістковому мозку.
На роль інгібіторів лейкопоезу претендують високомолекулярний інгібітор сироватки крові (ліпопротеїн), кейлони і лактоферин.
II. Збільшення переходу резервних лейкоцитів із червоного кісткового мозку в кров. Цьому сприяють інтерлейкін-1 і бактеріальні ендотоксини, що підвищують проникність стінки кровоносних судин червоного кісткового мозку.
26.2.8. Які особливості характерні для перерозподільного лейкоцитозу?
Перерозподільном називають лейкоцитоз, що виникає в результаті переходу лейкоцитів із пристінкового пулу в циркулюючий. Його особливостями є:
а) короткочасний характер зі швидким поверненням кількості лейкоцитів до норми після завершення дії причини;
б) збереження нормального кількісного співвідношення різних видів лейкоцитів (лейкоцитарна формула не міняється);
в) відсутність дегенеративних змін лейкоцитів. Більшість форм фізіологічного лейкоцитозу за механізмом свого розвитку є перерозподільними.
26.2.9. Наведіть приклади нейтрофільного, еозинофільного, базофільного, лімфоцитарного і моноцитарного лейкоцитозів.
Нейтрофільнш лейкоцитоз характерний для:
а) гнійно-запальних процесів, спричинених гноєтворними бактеріями (абсцеси, флегмони, сепсис);
б) важкого кисневого голодування (велика гостра крововтрата, гострий гемоліз);
в) ендогенної інтоксикації (уремія).
Еозинофільний лейкоцитоз розвивається при:
а) алергічних реакціях І типу за класифікацією Кумбса і Джелла;
б) гельмінтозах;
в) хронічному мієлолейкозі.
Базофільний лейкоцитоз буває дуже рідко. Він може супроводжувати розвиток:
а) хронічного мієлолейкозу;
б) гемофілії;
в) хвороби Вакеза (поліцитемії).
Лімфоцитарний лейкоцитоз часто буває при:
а) гострих інфекційних захворюваннях (кашлюк, вірусний гепатит);
б) деяких хронічних інфекційних хворобах (туберкульоз, сифіліс, бруцельоз);
в) хронічному лімфолейкозі.
Моноцитарний лейкоцитоз характерний для:
а) хронічних інфекцій (туберкульоз, бруцельоз);
б) інфекційного мононуклеозу;
в) інфекцій, збудниками яких є рикетсії та найпростіші (висипний тиф, малярія).
Лейкопенії: види, причини і механізми розвитку. Агранулоцитоз. І Ісйтропения. Супутні ядерні зрушення нейтрофільних гранулоцитів.
Лейкопенія - це зменшення кількості лейкоцитів в одиниці обєму крові менше 4*10^9
Класифікація за клітинним складом
-еозинофілія ( агранульоцитоз, лімфолейкоз)
-базофілопенія
-нейтропенія (причини: інфекц хвороби ( вірусний гепатит, інф мононуклеоз, тифи, снід), дія хімічних факторів, дія фізичних факторів, при мегалобластичних анеміях, білкове голодування, гіперсклонізм, анафілактичний шок, гемодіаліз)
-моноцитопенія (буває при сепсисі, опроміненні, агранульоцитозі)
-лімфопенія (буває и сепсисі, імунодифіцитних станах, при голодуванні, стресовиз ситуаціях, вживання імунодеприсантів)
Класифікація за походженням:
-первинні або спадкові - поділяються на постійні та періодичні ( спадкові імунодифіцити)
-вторинні або набуті - виникають при діі біологічних, фізичних, хімічних мієлотоксичних чинниках
Класифікація лейкопеній за патогенезом :
1) лейкопеніі внаслідок пригнічення лейкопоезу
причини:
-мієлотоксична дія фізични, хімічних, біологічних факторів
-зменшення кількості лейкопоетинів
-зменшенні вироблення цитокінів, що сприяють лейкопоезу
-недостатність вітаміну В12, фолієвоі кислоти, білкове голодування, мікроелементів
-пухлини кіскового мозку чи метастази у кісковий мозок
2) зменшення виходу лейкоцитів із кіскового мозку
синдром ледачих лейкоцитів (виникає при е
генетичному дефекті мембрани та цитосклелету лейкоцитів, а саме мікрофіламентів та мікротрубочок
3) внаслідок збільшення руйнування лейкоцитів
-імунний механізм
-ізоімунні - утв антитіл до лейкоцитів плода
-утв аутоантитіл до власних лейкоцитів які змінили свою АГ
структуру( наприклад приєднання гаптену до лейкоцита,
дія факторів зовн середовища)
-поява забороненого клону лімфоцитів, які утворюють аутоантитіла до
власних нормальних лейкоцитів
утвантитіл до чужих лейкоцитів при переливанні крові, які можуть руйнувати і власні лейкоцити
-неефективний лейкопоез
-гіперспленізм
-підвищений визід лейкоцитів за межі судин у тканини( запалення, алрегія)
-руйнування лейкоцитів при краш синдромі
-перерозподільний ( шок, ранні етапи запалення)
-вихід лейкоцитів назовні ( гнійні нориці, лімфорея, плазморея при опіках, піурія)
-перерозподільний при гемодилюції ( при гіперволемії) при переливанні кровозамінників
Класифікація лейкопеній за механізмом розвитку:
-абсолютні - пригнічення лейкопоезу, збільшення руйнування лейкоцитів
небезпечна тим що виникає імунодифіцитний стан
-відносні (перерозподільна)
Агранульоцитоз - це зменшення кількості чи відстуність гранульоцитів, нафонізменшення загальної кількості лейкоцитів менше 1*10^9/л
Види:
Мієлотоксичний агранульоцитоз
Причини
- цитостатики
- антитереоїдні препарати
- НПЗЗ ( німесулід, парацитамол)
- стероїдні протизапальні препарати
- нейролептики, протисудомні, транквілізатори
- антибіотики ( лінкоміцин)
- дія біологічних , фізичних, хімічних чинників
Одним із видів АЦ є аліментарно-токсична алейкія. Причиною ж токсини плісневих грибів таких як ерготоксин, спорофузагрин, які спричиняють панцитопенію.
Наслідком є некроз мяких тканин обличчя і рота
імунний
Причини:
-утворення антилейкоцитарних АТ (після застосування деяких лікарських препаратів)
Прояви агранульоцитозу
-виразково некротичне ураження
-некроз тканин обличчя і рота
-некроз слизових оболонок
Лейкопенія — це зменшення кількості лейкоцитів у периферичній крові нижче 4-109/л. Лейкопенія часто виступає симптомом якого-небудь захворювання. Однак є нозологічні одиниці, при яких саме лейкопенія є основним проявом хвороби, вона по суті визначає картину недуги та всі інші симптоми.
Класифікація лейкопепій.
І. За походженням лейкопенії бувають набутими і спадково обумовленими.
Набуті лейкопенії можуть виникати під впливом фізичних (іонізуюча радіація), хімічних (бензол, цитостатики, лікарські препарати), біологічних (віруси гепатиту, інфекційного мононуклеозу) та імунних факторів.
Прикладами спадкових лейкопеній є нейтропенія Костмана, спадкова нейтропе-нія аутосомно-домінантного типу, синдром "лінивих лейкоцитів", циклічна нейтропенія.
її. За видом лейкоцитів, кількість яких зменшена, виділяють:
а) нейтропенію;
б) лімфопенію;
в) еозинопенію.
III. За патогенезом розрізняють:
а) лейкопенії, обумовлені порушенням надходження лейкоцитів із червоного кісткового мозку в кров;
б) лейкопенії, пов'язані зі скороченням часу перебування лейкоцитів у периферичній крові;
в) перерозподільні лейкопенії.
IV. Виділяють цілий ряд клініко-гематологічних синдромів, для яких лейкопенія є провідною ознакою. Серед них агранулоцитоз, гіпопластична анемія, геморагічна алейкія.
26.2.11. Які механізми можуть лежати в основі розвитку лейкопеній, пов'язаних з порушеннями надходження лейкоцитів із червоного кісткового мозку в кров?
1. Ушкодження кровотворних клітин. При цьому розвивається так званамієлотоксична
лейкопенія. Виділяють три основних механізми ушкодження кровотворних клітин:
а) цитолітичний. Пов'язаний з дією на клітини іонізуючої радіації, цитоста-тичних препаратів, імунних факторів (антитіл, Т-лімфоцитів) та ін. Ступінь ураження червоного кісткового мозку при цьому залежить від дози й тривалості дії зазначених факторів;
б) антиметаболічний. У його основі лежить дія агентів, які втручаються в обмін пуринових і піримідинових основ, порушуючи при цьому процеси поділу стовбурових клітин. За таким принципом діють деякі протипухлинні препарати й антибіотики (левоміцетин).
в) ідіосинкразичний. Реалізується при повторному введенні лікарських препаратів, чутливість організму до яких підвищена (ідіосинкразія). Найчастіше це препарати, що містять у своїй структурі бензольні кільця. У випадку ідіосинкразії немає зв'язку між імовірністю розвитку лейкопенії і дозою, а також тривалістю дії лікарських засобів.
2. Порушення мітозу - неефективний лейкопоез. Найчастіше його причиною є:
а) дефіцит необхідних для клітинного поділу речовин, зокрема вітаміну В12 і фолієвої кислоти;
б) порушення регуляції мітозу — дефіцит лейкопоетинів.
3. Порушення дозрівання лейкоцитів. їхню основу можуть складати генетично обумовлені дефекти як самих кровотворних клітин (наприклад, нейтропенія Костмана), так і клітин "мікрооточення" (наприклад, лейкопенія у "сталевих" мишей лінії SL/SI/). При цьому дозрівання клітин крові досягає певної стадії (наприклад, промієлоцитів) і зупиняється.
4= Порушення виходу лейкоцитів із червоного кісткового мозку в кров. Подібні порушення часто пов'язані з генетичними дефектами лейкоцитів, що порушують основні їхні функції й властивості (наприклад, рухливість). Прикладами можуть бути синдром "лінивих лейкоцитів ", нейтропенія "єменських євреїв ".
5. Зменшення плацдарму лейкопоезу. Має місце при заміщенні кровотворної тканини лейкозними клітинами, метастазами пухлин та ін.
26.2.12. Які механізми можуть лежати в основі розвитку лейкопеній, пов 'язаних зі скороченням часу перебування лейкоцитів у
периферичній крові?
1. Деструкція (руйнування) лейкоцитів. Може бути обумовлена:
а) аутоімунними механізмами (ревматоїдний артрит, системний червоний вовчак);
б) гаптеновими механізмами (амідопіринова гостра нейтропенія);
в) гіперспленізмом (підвищенням фагоцитарної активності макрофагів селезінки).
2. Посилене використання лейкоцитів. Цьому передує прискорений вихід лейкоцитів із крові в тканини в умовах хронічного рецидивуючого запалення.
3. Посилене виведення лейкоцитів з організму. Виражена хронічна втрата нейтрофілів спостерігається у курців: під час ранкового кашлю з мокротинням втрачається від 0,5 до 2-Ю8 гранулоцитів і 0,8 - 1,6-108 макрофагів.
26.2.13. Що таке агранулоцитоз?
Агранулоцитоз - це клініко-гематологічний синдром, що характеризується різким зменшенням вмісту нейтрофілів нижче 0,75-109/л при зменшенні загальної кількості лейкоцитів нижче 1'109/л.
В основі розвитку агранулоцитозу можуть лежати два механізми:
а) мієлотоксичний — ураження червоного кісткового мозку;
б) імунний — руйнування клітин гранулоцитарного ряду антилейкоцитарними антитілами.
Агранулоцитоз супроводжується значним ослабленням реактивності організму у зв'язку з порушенням захисної функції лейкоцитів.
26.2.14. Що таке зміщення лейкоцитарної формули?
Зміщення лейкоцитарної формули (ядерне зміщення) - це порушення співвідношення між незрілими і зрілими формами нейтрофілів.
При дослідженні лейкограми встановлюють наявність ядерного зміщення ней-трофільних гранулоцитів уліво або вправо. Ця термінологія пов'язана з розташу-
ванням незрілих нейтрофільних гранулоцитів (мієлоцитів, метамієлоцитів, палич-коядерних нейтрофілів) у лівій частині лейкоцитарної формули (формули Арнета й Шилінга), тимчасом як зрілі, сегментоядерні нейтрофіли умовно поставлені в крайнє праве положенні. Збільшення вмісту в крові молодих форм нейтрофільних гранулоцитів свідчить про ядерне зміщення вліво, переважання зрілих нейтрофілів з великою кількістю сегментів (5-6) на тлі зникнення більш молодих клітин - про ядерне зміщення вправо.
26.2.15. Які виділяють різновиди зміщення лейкоцитарної формули вліво?
Розрізняють такі різновиди ядерного зміщення вліво.
1. Регенеративне зміщення є показником реактивної активації гранулоцитопоезу (на тлі помірного загального лейкоцитозу підвищений вміст паличкоядерних нейтрофілів та метамієлоцитів, можуть зустрічатися поодинокі мієлоцити).
2. Гіперрегенеративне зміщення відображає надмірну гіперплазію лейкопоетичної тканини з порушенням дозрівання клітин і вираженим омолодженням складу крові. При цьому різко зростає число паличкоядерних гранулоцитів та метамієлоцитів, з'являються мієлоцити, промієлоцити; загальна кількість лейкоцитів може бути збільшена, незмінена і навіть знижена внаслідок того, що розвивається виснаження мієлоїдного паростка після попередньої активізації.
3. Дегенеративне зміщення свідчить про пригнічення й глибокі порушення лейко-поезу, коли на тлі загальної лейкопенії в лейкограмі збільшується число паличкоядерних нейтрофільних гранулоцитів з дегенеративними змінами в їхній цитоплазмі та ядрі при зменшенні кількості сегментоядерних форм і відсутності метамієлоцитів.
4. Регенеративно-дегенеративне зміщення спостерігається при гіперпродукції в червоному кістковому мозку патологічно змінених лейкоцитів і порушенні їх дозрівання. У цьому випадку виявляють лейкоцитоз, а в мазку крові зростає число паличкоядерних гранулоцитів, метамієлоцитів та мієлоцитів з ознаками дегенерації.
26.2.16. Які дегенеративні зміни можуть бути характерні для лейкоцитів в умовах патології?
Дегенеративні зміни лейкоцитів можуть виявляти себе анізоцитозом, наявністю в цитоплазмі вакуоль, токсигенної зернистості, включень типу тілець Князько-ва—Деле (базофільно забарвлені грудочки цитоплазми); великою азурофільною зернистістю; зникненням звичайної зернистості, пікнозом або набряканням ядра, його гіпер- і гіпосегментацією, а також невідповідністю ступеня дозрівання ядра й цитоплазми, каріорексисом, цитолізом.
Дегенеративні зміни найчастіше спостерігають у нейтрофільних гранулоцитах і моноцитах. Причиною їх виникнення є утворення лейкоцитів з порушеним обміном речовин, що й обумовлює структурні аномалії (при лейкозі, спадковій ензимопатії), а також ушкодження лейкоцитів у кровотворних органах і крові під впливом різноманітних патогенних факторів (бактерій, вірусів, антитіл).
Лейкози: визначення поняття, принципи класифікації. Етіологія лейкозів. Аномалії генотипу і конституції як фактори ризику виникнення і розвитку лейкозів.
Гемобластози — це пухлинні захворювання системи крові. Інакше кажучи, це злоякісні пухлини, які ростуть із кровотворних клітин.
Залежно від того, чи уражується первинно червоний кістковий мозок, гемобластози поділяють на дві великі групи: лейкози і гемобластози, що мають первинну локалізацію поза червоним кістковим мозком. Найчастішим місцем локалізації останніх є лімфатичні вузли.
Лейкози - це злоякісні пухлини, що виникають із кровотворних клітин і первинно уражують червоний кістковий мозок.
26.2.18. Які існують докази того, що лейкози - це захворювання пухлинної природи?
1. При лейкозах, як і при злоякісних пухлинах, відбувається безмежний і нерегульо-ваний поділ клітин.
2. Лейкоз, як і будь-яка злоякісна пухлина, виникає з одної-єдиної первинно зміненої, трансформованої клітини. Усі лейкозні клітини, якими б різними вони не були, походять із однієї клітини.
3. Для лейкозів, як і для інших злоякісних пухлин, характерне явище анаплазії— поява морфологічних, біохімічних та інших властивостей, які наближають лейкозні клітини до ембріональних.
4. Для лейкозів, як і для інших злоякісних пухлин, характерна пухлинна прогресія, тобто набуття із часом лейкозними клітинами все більш і більш злоякісних властивостей.
5. Однаковість причин розвитку лейкозів і злоякісних пухлин. Одні й ті ж етіологічні фактори (фізичні, хімічні, біологічні) здатні викликати виникнення як лейкозів, так і інших злоякісних пухлин.
26.2.19. Чим лейкози відрізняються від інших злоякісних пухлин?
1. У випадку лейкозу неможливо встановити первинну локалізацію пухлини, а отже, радикально видалити її вже на ранніх етапах розвитку.
2. Лейкози - це пухлини, які з самого початку метастазують. Лейкозні клітини легко проникають у кров і розносяться нею по всьому організмі. Вони заселяють все нові й нові ділянки, на яких виникають вторинні лейкозні інфільтрати.
3. Для лейкозів характерна системність ураження. У зв'язку з раннім метастазуванням уражується вся система крові: червоний кістковий мозок, лімфатичні вузли, селезінка, печінка.
4. При лейкозах пригнічується нормальне кровотворення. Це пов'язано з витісненням нормальної кровотворної тканини лейкозними клітинами. З другого боку, має значення токсична дія продуктів лейкозних клітин на клітини гемопоезу.
5. Лейкози — це різновид пухлин, що вражають людину переважно в дитячому і молодому віці.
26.2.20. На які класи поділяють кровотворні клітини?
I клас — стовбурові поліпотентні клітини. Ці клітини дають початок усім клітинам крові.
II клас — частково детерміновані поліпотентні клітжи-попередники. Він складається з двох груп клітин: клітин-попередників лімфоцитарного ряду і клітин-по-передників мієлопоезу.
III клас — уніпотентні клітини-попередники. Клітини цього класу дають початок певному паростку крові. До них відносять клітини-попередники В-лімфоцитів, клітини-попередники Т-лімфоцитів, еритропоетинчутливі клітини, що утворюють колонії.
Клітини перших трьох класів неможливо розрізнити за допомогою відомих нині морфологічних і цитохімічних методів дослідження, тому їх об'єднують під загальною назвою - клітини, яких неможна морфологічно диференціювати.
IV клас — клітини, що проліферують. Клітини цього класу мають специфічні морфологічні й цитохімічні ознаки. До них належать бласти (плазмобласти, лімфо-бласти, монобласти, еритробласти, мегакаріобласти), проклітини (проплазмоцити, пролімфоцити, промієлоцити, пронормоцити, промегакаріоцити) і так звані цитні форми (мієлоцити, нормоцити, мегакаріоцити).
Уклас — клітини, що дозрівають. Ними є метамієлоцити, паличкоядерні гранулоцити, ретикулоцити. Загальна властивість цих клітин - втрата ними здатності до поділу.
VI клас — зрілі клітини крові.
26.2.21. Які кровотворні клітини можуть бути джерелом лейкозу?
Будь-яка пухлина може розвиватися тільки з тих клітин, що мають первісну здатність до поділу. Не є винятком і лейкози.
Джерелом лейкозів можуть бути клітини І—IV класів, тобто клітини, здатні до проліферації. Клітини V і VI класів (ті, що дозрівають, і зрілі) трансформуватися в лейкозні не можуть, оскільки втратили структури, необхідні для здійснення клітинного поділу.
26.2.22. Як класифікують лейкози?
I. Залежно від особливостей патогенезу і пов'язаної з ними гематологічної картини лейкози поділяють на гострі й хронічні.
II. Залежно від того, які кровотворні клітини втягуються в пухлинний процес, лейкози поділяють на лімфолейкози (уражується лімфоцитарний паросток), мієло-лейкози (уражується гранулоцитарний паросток), еритромієлози та ін.
III. Залежно від вмісту лейкоцитів у периферичній крові лейкози бувають лейкеміч-ними (виражений лейкоцитоз — від 20— 109/л до 100-109/л), сублейкемічними (помірний лейкоцитоз до 20-109/л), алейкемічними (вміст лейкоцитів не міняється), лейкопенічними (кількість лейкоцитів зменшується).
26.2.23. У чому полягає основна патогенетична відмінність між гострими і хронічними лейкозами?
Основною рисою патогенезу гострих лейкозів є те, що лейкозні клітини, набувши здатності до безмежного неконтрольованого росту, повністю втратили здатність дозрівати, тобто диференціюватися в наступні форми.
У той же час при хронічних лейкозах лейкозні клітини поряд зі здатністю до безмежного росту зберігають властивість дозрівати й давати наступні форми.
Таким чином, при гострих лейкозах пухлинні клітини тільки діляться і не дозрівають, при хронічних — діляться і дозрівають. З урахуванням цієї обставини гострі лейкози варто вважати більш злоякісним видом захворювання.
26.2.24. Які існують види гострих лейкозів?
Джерелом гострих лейкозів можуть бути кровотворні клітини перших чотирьох класів.
Якщо лейкоз розвивається з клітин І—III класів, що не мають специфічних морфологічних і цитохімічних ознак, то такий лейкоз називають недиферещійованим.
Якщо лейкоз розвивається з клітин IV класу, то за допомогою морфологічних і цитохімічних методів можна встановити клітину, від якої походить пухлина. Для цього використовують сім цитохімічних реакцій: 1) реакцію на пероксидазу; 2) реакцію із Суданом чорним (на ліпіди); 3) реакцію на кислу фосфатазу; 4) PAS-реакцію (на глікоген); 5) реакцію на а-нафтилацетатестеразу; 6) реакцію на хлорацетатестеразу; 7) реакцію на сульфатовані глікозаміноглікани.
Якщо джерелом лейкозних клітин є лімфобласт, то такий лейкоз називається гострим лімфобластним, якщо мієлобласт — гострим мієлобластним, якщо моно-бласт — гострим монобластним і т. д.
26.2.25. Дайте характеристику картини крові при гострих лейкозах.
Для гострих лейкозів характерні алейкемічний і лейкопенічний варіанти перебігу.
Як приклад розглянемо два види гострих лейкозів, що найчастіше зустрічаються: гострий мієлобластний і гострий лімфобластний.
Гострий мієлобластний лейкоз розвивається переважно у молодих людей і людей середнього віку. Назва лейкозу свідчить про те, що його джерелом є мієлобласт і що лейкозні клітини (мієлобласти) не здатні диференціюватися. Це визначає такі особливості картини крові (рис. 107; див. форзац).
1. Лейкозні мієлобласти постійно діляться і легко надходять у кров. Отже, у крові будуть виявлятися пухлинні клітини -мієлобласти.
2. Оскільки в червоному кістковому мозку зберігаються осередки нормального кровотворення, то вони будуть джерелом надходження у кров нормальних лейкоцитів, тобто тих, які мають бути в крові у нормі — метамієлоцитів, паличкоядерних і сегментоядерних нейтрофілів.
3. У крові відсутні перехідні форми лейкоцитів від мієлобластів до тих клітин, які виявляються в крові у нормі, тобто немає промієлоцитів~і мієлоцитів. Таке явище отримало назву лейкемічного провалу (hiatus leucemicus). Відсутність у крові пере-
хідних форм пояснюється тим, що лейкозні мієлобласти не здатні диференціюватися в зазначені клітини, а з другого боку, осередки нормального кровотворення не "випускають" у кров промієлоцитів і мієлоцитів (рис. 108).
Рис. 108. Походження
"лейкемічного провалу" при
гострому мієлобластному лейкозі
Гострий лімфооластнии лейкоз. Є типовим лейкозом дитячого віку. Лейкоз розвивається з лімфобласта. Пухлинні клітини втрачають здатність до дозрівання. Тому для картини крові будуть характерні лімфобласти (пухлинного походження), а також всі ті клітини, які мають бути в нормі (з осередків нормального кровотворення).
Картина червоного кісткового мозку при всіх гострих лейкозах має ряд особливостей. По-перше, у пунктаті кісткового мозку кількість бластних клітин значно зростає і перевищує 30 %. По-друге, лейкозні бластні клітини мають якісні відмінності, що є ознакою їх анаплазії.
26.2.26. Які існують види хронічних лейкозів?
Хронічні лейкози розвиваються з кровотворних клітин IV класу.
Номенклатура хронічних лейкозів ґрунтується на тому, який тип клітин становить переважну більшість у популяції лейкозних клітин. Назва лейкозу вказує на паросток кровотворення, що виявився ураженим.
Виділяють хронічний мієлоцитарний лейкоз (хронічний мієлолейкоз), хронічний лімфоцитарний лейкоз (хронічний лімфолейкоз), хронічний моноцитарний лейкоз, хронічний еритромієлоз. При цьому зазнають уражень відповідно гранулоцитарний, лімфоцитарний, моноцитарний та еритроїдний паростки крові.
26.2.27. Дайте характеристику картини крові при хронічних лейкозах.
Для хронічних лейкозів найчастіше характерні лейкемічний і сублейкемічний варіанти перебігу.
Розглянемо два види хронічних лейкозів, що найчастіше бувають: хронічний мієлоцитарний і хронічний лімфоцитарний.
Хронічний мієлоцитарний лейкоз. Найбільш імовірним джерелом розвитку цього лейкозу є мієлобласти (іноді можуть бути промієлоцити і мієлоцити). Оскільки лейкоз хронічний, то це означає, що лейкозні мієлобласти зберігають здатність до диференціювання у наступні форми. Тому з лейкозної тканини червоного кісткового мозку в кров у великій кількості виходять всі клітини, які походять від мієлобластів, а саме промієлоцити, мієлоцити, метамієлоцити, паличкоядерні І сегментоядернг гранулоцити.
У червоному кістковому мозку переважають клітинні елементи мієлоїдного ряду. Жирова тканина повністю витісняється кровотворною (рис. 109; див. форзац).
Хронічний лімфоцитарний лейкоз. Джерелом його розвитку елімфобласти, які зберегли здатність диференціюватися в наступні форми — пролімфоцити і лімфоцити, і
Тому основна маса лейкозних клітин крові представлена лімфоцитами. їхня 1 кількість у лейкоцитарній формулі становить 80-90 %. Крім лейкозних лімфоцитів, у крові можуть виявлятися пролімфоцити і поодинокі лімфобласти. Характерною є поява так званих тіней Гумпрехта - напівзруйнованих ядер лімфоцитів, що утворюються як артефакт при готуванні мазків крові.
Лімфоцити лейкозного клону (В-лімфоцити) можуть продукувати імуноглобу-ліни однієї специфічності (моноклональні), однак при цьому пригнічується антиті-лоутворення іншими нормальними клонами В-лімфоцитів і поступово розвивається імунологічна недостатність.
У червоному кістковому мозку відбувається майже тотальне заміщення кровотворної тканини лімфоцитами (рис. ПО; див. форзац).
26.2.28. Дайте порівняльну характеристику картини крові при гострому мієлобластному і хронічному мієлоцитарному лейкозах.
1. При гострому мієлобластному лейкозі пухлинні клітини представлено лейкозни-ми мієлобластами, у той час як при хронічному мієлолейкозі основну масу становлять похідні лейкозних мієлобластів: промієлоцити, мієлоцити, метамієлоци-ти, паличкоядерні і сегментоядерні клітини.
2. При гострому мієлобластному лейкозі виявляється феномен "лейкемічного провалу", у той час як при хронічному мієлолейкозі його немає - навпаки, у крові з'являються всі перехідні форми від мієлобласта до сегментоядерних гранулоцитів.
3. Хронічний мієлолейкоз на відміну від гострого мієлобластного характеризується гіперрегенеративним зміщенням лейкоцитарної формули вліво.
4. Для хронічного мієлолейкозу характерний гіперлейкоцитоз, тобто збільшення вмісту лейкоцитів крові понад 20- 109/л. У той же час при гострому мієлобластному лейкозі кількість лейкоцитів у периферичній крові або не міняється, або зменшується.
5. При хронічному мієлолейкозі часто виникає так звана базофільно-еозинофільна асоціація - збільшується частка базофілів і еозинофілів у периферичній крові.
26.2.29. Чи може гострий лейкоз із часом перейти в хронічний, і навпаки, хронічний - у гострий?
Перехід гострих лейкозів у хронічні неможливий. Таке перетворення означало б, що лейкозні клітини, які втратили здатність до дозрівання, знову набули цю властивість. Нині немає даних про те, що такий перебіг подій можливий, хоча б принципово } з урахуванням відомих механізмів розвитку пухлин.
З другого боку, хронічний лейкоз із часом може трансформуватися в гострий. Це означає, що лейкозні клітини, які ще зберігали здатність до диференціювання, таку властивість втрачають. При цьому лейкоз із менш злоякісної перетворюється у більш злоякісну форму. Подібне явище по суті відображає пухлинну прогресію — загальну властивість усіх злоякісних пухлин.
Гематологічним проявом переходу хронічного лейкозу в гострий є так званий "бластний криз", коли в крові й червоному кістковому мозку різко зростає кількість бластних клітин, а в крові поступово зникають перехідні форми.
26.2.30. Які фактори можуть бути причиною розвитку лейкозів?
Причиною лейкозів можуть бути всі фактори, що здатні викликати розвиток злоякісних пухлин узагалі. Такими є:
1) фізичні фактори (іонізуюча радіація);
2) хімічні агенти (хімічні канцерогени);
3) фактори біологічного походження (онкогенні віруси).
26.2.31. Які існують докази того, що іонізуюча радіація може бути причиною лейкозів?
Перші докази було отримано після вивчення наслідків атомного бомбардування японських міст Хіросіми й Нагасакі в 1945 р.
Було показано, що частота лейкозів серед тих, що вижили після бомбардування, різко зросла через кілька років і досягла максимуму через 6—7 років, зберігаючись підвищеною ще 25 років.
Установлено, що частота лейкозів залежить від дози поглиненої радіації. В осіб, що перебували ближче до епіцентру вибуху, частота виникнення лейкозів була вища.
Крім того, частота лейкозів залежить від виду іонізуючої радіації. Після опромінення нейтронами ріст числа лейкозів був значно вищий, ніж після у-опромінення.
Так, на Хіросіму скинули бомбу, вибух якої був джерелом нейтронів і у-випро-мінювання (уранова бомба), а на Нагасакі скинули плутонієву бомбу, після вибуху якої виділялося тільки у-випромінювання. У Хіросімі частота лейкозів істотно збільшилася вже при поглиненій дозі 0,5 Гр, у той час як у Нагасакі навіть при дозі 1 Гр частота лейкозів не зростала.
Нині всі ми є свідками й учасниками ще одного "експерименту" - чорнобильського. Отримана в ньому інформація, безумовно, розширить наші уявлення про роль іонізуючої радіації й зокрема радіонуклідів у виникненні лейкозів.
26.2.32. Що свідчить про роль хімічних агентів у виникненні лейкозів?
1. Індукція лейкозів у тварин (миші, щури, кури) за допомогою хімічних канцерогенів (поліциклічних ароматичних вуглеводнів, нітрозосполук).
2. Виникнення лейкозів у людей після тривалої інтоксикації бензолом і його похідними, після приймання цитостатичних засобів, що використовувалися з лікувальною метою.
26.2.33. Яке значення мають віруси у виникненні лейкозів?
У 1908 р. Елерман і Бат уперше отримали лейкоз у курей після введення їм безклітинних фільтратів лейкозних клітин. У 1950 р. Гросс аналогічним чином індукував лейкоз у ссавців (дитинчат мишей). Ці експерименти довели значення вірусів у виникненні лейкозів.
Нині відомо багато вірусів, здатних викликати трансформацію кровотворних !
клітин у пухлинні. їх можна розділити на дві групи.
І. Онкогешіі віруси тварин. Викликають лейкози у різних видів тварин - птахів, мишей, щурів, кішок, мавп. Більшість з них належить до РНК-вмісних вірусів -ретровірусів. Залежно від характеру онкогенної дії їх поділяють на:
а) гостротрансформуючі віруси - викликають розвиток пухлин після короткого латентного періоду (віруси гострих лейкозів тварин). Ці віруси містять j у своїй структурі онкоген;
б) повільнотрансформуючг віруси - викликають розвиток пухлин після тривалого латентного періоду (віруси хронічних лімфолейкозів). Геном цих вірусів не містить онкогена.
її. Онкогенні віруси людиіуі. Сьогодні виявлено два таких віруси.
1. Вірус Епштейна—Барр: ДНК-вмісний вірус сімейства герпес-вірусів. Викликає розвиток лімфами Беркітта у дітей віком від 2 до 14 років у деяких країнах Центральної Африки. Лімфома Беркітта не є в буквальному значенні слова лейкозом, оскільки протікає без первинного ураження червоного кісткового мозку. Джерелом цієї пухлини є підщелепні лімфатичні вузли. Пухлина росте дуже швидко, збільшуючись у масі в 2 рази через кожні 2 дні. Через 6-12 тижнів дитина гине.
2. Вірус Т-клітинної лімфоми—лейкемії людини. Є ретровірусом, належить до так званих Т-лімфотропних вірусів (HTLV-вірусів). Викликає розвиток Т-клітинного лейкозу в Японії, країнах басейну Карибського моря, у Південній Америці, на Алясці. За своїми властивостями подібний до вірусу імунодефіциту людини (ВІЛ), що викликає СНІД.
26.2.34. Які факти свідчать про роль спадкового фактора в розвитку лейкозів?
1. У людей із хромосомними хворобами істотно збільшується частота виникнення лейкозів, Так, в осіб з хворобою Дауна (трисомія по 21-й хромосомі) частота розвитку гострих лейкозів зростає в 18-20 разів. Таку ж закономірність виявлено й серед хворих на синдроми Клайнфельтера, Шерешевського-Тернера, Фанконі.
2. Майже при всіх видах лейкозів у пухлинних клітинах виявляють ознаки хромосомних аберацій: делеція, транслокація, інверсія. Дуже характерною ознакою хронічного мієлолейкозу є поява так званої "філадельфійської" хромосоми (вона виявляється у 92 % хворих з даним видом лейкозу). Ця хромосома являє собою одну з 22-ї пари хромосом, у якої відбулася делеція приблизно половини довгого плеча. Дуже часто цей фрагмент приєднується до 9-ї хромосоми.
3. Неодноразово в літературі описано випадки так званої сімейної форми хронічних лейкозів.
4. Виведено чисті лінії тварин (мишей), у яких спостерігається висока вразливість спонтанними лейкозами.
26.2.35. Як впливають на виникнення лейкозів імунні фактори?
Показано, що виникненню лейкозів сприяють стани імунологічної недостатності.
Так, при спадково обумовлених імунодефіцитних станах, пов'язаних з ураженням В- і Т-лімфоцитів (синдроми Брутона, Луї-Барр, Віскотта-Олдрича), значно зростає частота розвитку гострого лімфобластного лейкозу й лімфосарком.
Застосування цитостатиків, що викликають розвиток імунодепресивного стану (вторинних імунодефіцитів), супроводжується збільшенням частоти виникнення ракових пухлин у 2,5 рази, а лімфоцитарних пухлин — у 35 разів.
26.2.36. Опишіть патогенез лейкозів. Які стадії розвитку проходить лейкоз?
Під впливом онкогенних вірусів, іонізуючої радіації, хімічних речовин відбувається мутація генів або епігеномне порушення регуляції процесу розмноження й дозрівання кровотворних клітин. При цьому в кістковому мозку утворюється клон пухлинних клітин, для яких характерні безмежний ріст і знижена здатність до диференціювання. Швидкий ріст лейкозних клітин призводить до поширення (метастазування) їх по всій системі крові, включаючи кровотворні органи й кров. У лейкозних клітинах, що циркулюють у крові, виявляють однакові хромосомні маркери. Для хронічного мієлолейкозу таким маркером служить "філадельфійська" хромосома.
Нестабільність генотипу лейкозних клітин призводить до виникнення мутацій, як спонтанних, так і обумовлених тривалим впливом канцерогенних факторів, у результаті чого утворюються нові пухлинні клони.
Таким чином, лейкоз проходить дві стадії свого розвитку: 1) моноклонову (відносно більш доброякісну) і 2) поліклонову (злоякіснішу, термінальну). Перехід з першої стадії в другу є показником пухлинної прогресії — лейкозні клітини набувають більшої злоякісності. Вони стають такими, що їх неможна диференціювати ані морфологічними, ані цитохімічними методами, у кровотворних органах і крові збільшується кількість бластних клітин з дегенеративними змінами ядра і цитоплазми. Лейкозні клітини поширюються за межі кровотворних органів, утворюючи лейкозні інфільтрати в різних органах. Унаслідок добору знищуються клітини тих клонів, на які діяли імунна система й гормони організму, цитостатичні засоби (хімічні, гормональні, променеві). Домінують клони пухлинних клітин, найбільш стійких до цих впливів.
26.2.37. Якими клінічними синдромами можуть виявляти себе лейкози?
Усе різноманіття клінічних ознак лейкозів можна розділити на три групи. І. Гематологічні синдроми, пов'язані із заміщенням нормальної кровотворної тканини лейкозною і пригніченням у зв'язку з цим нормального кровотворення. 1. Панцитопенія ~ зменшення вмісту всіх формених елементів крові. Особливо виражена при гострих лейкозах.
2. Анемія. Основу її патогенезу становить порушення еритропоезу Однак при деяких видах лейкозів певне значення може мати імунний гемоліз еритроцитів (наприклад, при хронічному лімфолейкозі) і кровотечі (геморагічний синдром).
3. Геморагічний синдром. Обумовлений в основному тромбоцитопенією та лейкозними інфільтратами в стінках кровоносних судин.
4. Порушення неспецифічного протимікробного захисту, у зв 'язку з чим зменшується резистентність організму до інфекцій. Основною причиною цього є зменшення вмісту функціонально повноцінних гранулоцитів.
5. Імунологічна недостатність. Розвивається як наслідок лімфопенії (при гострих лейкозах і хронічному мієлолейкозі) або неповноцінності лейкозних лімфоцитів (хронічний лімфолейкоз).
II. Синдроми, пов'язані з особливостями функціонування лейкозних клітин.
1. Гарячка. Показано, що тільки у 7-8 % хворих на лейкози підвищення температури на початку захворювання пов'язане з інфекцією. У більшості ж випадків гарячка має неінфекційне походження.
2. Інтоксикація. Велика кількість лейкозних клітин гине й вивільняє у кров свій вміст. Багато компонентів загиблих клітин мають токсичну дію на центральну нервову систему. Звідси стомлюваність, загальна слабкість, нудота та ін.
3. Аутоімунні процеси. Пов'язані зі змінами, що відбуваються в лімфоци-тарному паростку крові, а саме з появою так званих "заборонених" клонів лімфоцитів, зі зменшенням кількості й функціональної активності Т-супресорів.
III. Синдроми, пов'язані з метастазуванням лейкозних клітин і розвитком лейкозних проліфератів у різних органах і тканинах.
1. Збільшення лімфатичних вузлів, печінки й селезінки.
2. Шкірний синдром. Обумовлений появою в шкірі проліфератів лейкозних клітин - лейкемідів.
3. Виразково-некротичні ураження слизових оболонок (виразково-некротичні стоматит, ангіна, ентеропатії).
4. Кістково-суглобовий синдром, що виявляється болем у кістках і суглобах.
5. Синдром нейролейкозу. Може виявлятися менінгіальним синдромом, синдромом підвищення внутрішньочерепного тиску, різноманітними неврологічними порушеннями: парезами, паралічами, парестезіями. В основі його розвитку — поява лейкозних проліфератів в оболонках головного й спинного мозку, речовині мозку, нервових стовбурах, вегетативних гангліях.
6. Лейкозний пневмоніт. Лейкозні проліферати порушують дихальну функцію легень - розвивається недостатність зовнішнього дихання.
7. Серцева недостатність. Може бути наслідком розмноження лейкозних клітин у м'язі серця.
Порушення клітинного складу кісткового мозку і периферичної крові при гострих і хронічних лейкозах. Патогенез лейкозів: прогресія, метастазування, системні порушення. Принципи діагностики і терапії лейкозів.
див вище
Лейкоз - це системне захворювання крові пухлинної природи , з обовязковим первинним враженнями кісткового мозку, що характерезується гуперплазією, метаплазією, та анаплазією кровотворно тканини.
Геперплазія - це розростання мієлоідного чи лімфоїдногоростка у кістковому мозку
Метаплазія - це заміщення одного ростка іншим у кістковому мозку
Анаплазія - це порушення дозрівання клітин.
Етіологія
- фізичні канцерогени (іонізуюче випромінювання)
- хімічні канцерогени (бензол, цитостатики, бутадіон)
- дія біологічних канцерогенів (онкогенні віруси, вірус Епштейна Бара)
- спадкова схильність (хромосомні аномалії)
Патогенез
є 2 стадії
1) Моноклонова - проліферація
2) Поліклонова - злоякісна, термінальна - феномен пухлинної прогресії.
Ознаки пухлинної прогресії:
1) пригнічення норм ростків кровотворення
2) заміщення диференційованих клітин бластними ( незрілими)
3) втрата ферментної специфічності клітинами крові
4) утворення екстрамедулярнрх вогних кровоутворення ( метастазування)
5) вислизання пухлинного процесу з під контролю цитостатиків : перехід від мононклонованості до поліклонованості
6) поява і прогресування клітинного атипізму
7) перехід від алейкемічної форми до лейкемічної
Класифікація лейкозів за морф картиною крові:
гострі
хронічні
Класифікація лейкозів за морф субстратом
мієлоїдний
лімфоїдний
моноцитарний
еритроцитарний
Класифікація лейкозів за кількістю лейкоцитів у переферичній крові:
лейкемічні форма - більше 50
сублейкемічні форма - 15-50
алейкемічна форма - 4-15
лейкопенічна - менше 4
Гострий лейкоз
Основні оЗнаки:
-виникає у дітей і молодих людей
-характерезується ураженням клтіин 2,3,4 класів які втратили здатність до диференціації
Гемограма:
-кількість бластів більше 30%
-лейкемічний провал - відсутність у крові дозріваючих клітин 5го класу
-анемія
-тромбоцитопенія
Хронічний мієлолейкоз
Ознаки:
-у дорослих осіб старше 30 років
-характерезується ураженням клітин 4,5,6 класів які зберегли здатність до диференцціації
-маркером хронічмієлолекозу є Філадельфійська хромоосма - це делеція половини довгого плеча 22 хромосоми і транслокація його на 9 ту хромосому
Гемограма:
-в лейкоцитарнній формулі клітини 4 класу мієлобластів не більше 5%
-велика кількість клітин 5 класу
-невелика кількість клітин 6го класу
-еозинофільно-базофільна асоціація
-анемія
-тромбоцитопенія
-відсутність моноцитів і лімфоцитів
Хронічний лімфолейкоз
Ознаки
-старше 30 років
- -характерезується ураженням клітин 4,5,6 класів які зберегли здатність до диференцціації
Гемограма:
лімфобластів до 5%
дозріваючих клітин 5 класу (пролімфоцитів) - небагато
багато зрілих клітин 6 класу (малих лімфоцитів) - 80-90%
тіні Боткіна-Гумпрехта
Основні клінічні прояви лейкозів:
1) геморагічний синдром - виникає при чищенні зубів
Виникає внаслідок:
тромбоцитопеніі (пригнічення мегакаріоцитарногоросткалейкозними клітинами)
ураження судин лейкозними інфільтратами
зниження згортання крові
2) тяжкі інфекційно-токсичні процеси, некротичні ураження органів ітканин внаслідок імунної недостатності
3) Порушення функцій органів і систем внаслідок метастазування
Порушення судинно-тромбоцитарного гемостазу. Етіологія і патогенез вазопатій, тромбоцитопеній, тромбоцитопатій.
Система гемостазу — це система, що забезпечує, з одного боку, збереження рідкого стану крові, з другого - зупинку кровотечі при ушкодженні кровоносних судин.
Існує два механізми гемостазу: судинно-тромбоцитарний і коагуляційний.
Судинно-тромбоцитарний (первинний, мікроциркуляторний) гемостаз забезпечує зупинку кровотечі з судин мікроциркуляторного русла, що мають діаметр до 100 мкм. Він обумовлений взаємодією судинної стінки з тромбоцитами. Унаслідок активації судинно-тромбоцитарного гемостазу утворюється білий тромбоцитарний тромб. Порушення цього механізму гемостазу є причиною майже 80 % кровотеч і 95 % випадків тромбоутворення.
26.3.11. Що таке геморагічні діатези? Як їх класифікують?
Геморагічний діатез — це схильність організму до повторних кровотеч і крововиливів, що виникають спонтанно або після незначних травм. Геморагічні діатези поділяють на дві великі групи (рис. 118).

Рис. 118. Класифікація геморагічних діатезів
I. Порушення судинно-тромбоцитарного гемостазу:
а) вазопатії)
б) тромбоцитопенії;
в) тромбоцитопатії.
II. Порушення коагуляційного гемостазу - коагулопатії.
26.3.12. Що таке вазопатії? Як їх класифікують?
Вазопатії—ц& спадково обумовлені або набуті геморагічні діатези, що виникають як наслідок первинних порушень судинної стінки.
Залежно від механізму розвитку вазопатії поділяють на дві групи:
1) запальні вазопатії— васкуліти;
2) диспластичні вазопатії— ураження судин, пов'язані з порушеннями їхньої сполучної тканини (неповноцінність судинної стінки).
26.3.13. Які етіологія і патогенез запальних вазопатій?
Залежно від причин виникнення запальні вазопатії поділяють на:
1) інфекційні васкуліти. Є проявом цілого ряду інфекційних захворювань (вірусних геморагічних гарячок, висипного тифу, сепсису);
2) імунні васкуліти. Розвиваються як наслідок імунокомплексних захворювань (алергічних реакцій III типу за класифікацією Кумбса і Джелла), наприклад, при системному червоному вовчаку, вузликовому періартеріїті, геморагічному васкуліті (хвороба Шенляйн-Геноха);
3) інфекційно-імунні васкуліти. Поєднують обидва попередніх механізми.
У патогенезі запальних вазопатій провідна роль належить ушкодженню ендотелію, що може бути обумовлено:
а) цитопатичною дією ендотеліотропних вірусів;
б) токсинами мікробів, наприклад веротоксином, що його виділяють бактерії кишкової групи; *
в) комплексами антиген-антитіло і комплементом.
Наслідком ушкодження ендотелію судин є:
1) діапедез еритроцитів, що клінічно виявляється точковими крововиливами (пете-хіями);
2) інтенсивне мікротромбоутворення, що викликає порушення мікроциркуляції і живлення тканин;
3) тромбоцитопенія споживання (результат утворення мікротромбів).
26.3.14. Які етіологія і патогенез диспластичних вазопатій?
В основі диспластичних вазопатій лежать набуті або спадково обумовлені порушення сполучної тканини стінки кровоносних судин. Прикладами таких порушень є:
1. Гіповітаміноз С. Аскорбінова кислота— необхідний компонент реакції гідро-ксилювання проліну, унаслідок якої він перетворюється в оксипролін. Ця реакція вважається однією з ключових в утворенні колагену. При гіповітамінозі С з урахуванням сказаного порушується утворення повноцінного колагену (ламкість судин, випадання зубів і т. д.).
2. Телеангіектазії. Це спадково обумовлені локальні дефекти сполучної тканини судин, що обумовлюють стоншення їх стінок і розширення просвіту. Телеангіектазії є джерелом небезпечних для життя кровотеч, особливо при локалізації у внутрішніх органах.
3. Гемангіоми. Це судинні пухлини, які часто кровоточать.
4. Синдром Елерса-Данло. Його основу складають генетично обумовлені дефекти ] колагену.
У патогенезі диспластичних вазопатій мають значення:
1) стоншення стінок мікросудин і розширення їхнього просвіту;
2) неповноцінний локальний гемостаз. Через недостатню кількість або неповноцінність колагену в субендотелії судин порушується адгезія тромбоцитів;
3) легка ранимість судин.
26.3.15. Що таке тромбоцитопенія? Які механізми можуть лежати в основі розвитку тромбоцитопенії?
Тромбоцитопенія — це зменшення вмісту тромбоцитів в одиниці об'єму периферичної крові нижче 150 109/л.
На думку багатьох авторів, геморагічні прояви тромбоцитопенії з'являються при зменшенні кількості тромбоцитів нижче 50 109/л.
За походженням тромбоцитопенії можуть бути спадково обумовленими і набутими.
За механізмом розвитку виділяють такі види тромбоцитопенії. І. Тромбоцитопенії, пов'язані з порушеннями утворення тромбоцитів:
а) мієлотоксичні тромбоцитопенії— виникають унаслідок ушкодження кровотворних клітин. Дуже часто поєднуються з анемією й лейкопенією. Причинами їх розвитку є ті самі фактори, які викликають розвиток гіпопластич-ної анемії;
б) дефіцитні тромбоцитопенії— обумовлені недостатністю вітаміну В12 або фолієвої кислоти;
в) дисрегуляторні тромбоцитопенії— пов'язані з порушенням утворення тромбоцитопоетинів — речовин, що стимулюють утворення тромбоцитів;
г) тромбоцитопенії, пов'язані зі зменшенням плацдарму кровотворення, — розвиваються при лейкозах і метастазах злоякісних пухлин.
її. Тромбоцитопенії, пов'язані з посиленим руйнуванням тромбоцитів. Причиною такого руйнування можуть бути:
а) імунне ушкодження, обумовлене антитромбоцитарними антитілами на власні компоненти кров'яних пластинок або на лікарські препарати, адсорбовані на тромбоцитах. Аутоімунне ушкодження вважають найбільш імовірним механізмом розвитку так званої ідіопатичноїтромбоцитопенічноїпурпури (хвороби Верльгофа);
б) ггперспленізм — гіперфункція селезінки, що супроводжується часто спле-номегалією. У результаті підвищення фагоцитарної активності макрофагів відбувається інтенсивне руйнування всіх формених елементів крові, у тому числі й тромбоцитів;
в) механічне ушкодження тромбоцитів. Часто виникає при гемангіомах і накладенні штучних клапанів серця;
г) набутімембранопатії(гемолітична анемія Маркіафави—Мікеллі). Соматичні мутації кровотворних клітин спричиняються до утворення пулів клітин
(еритроцитів, гранулоцитів, тромбоцитів) з дефектами мембрани. У результаті збільшується чутливість таких клітин до дії комплементу й відбувається їх руйнування. III. Тромбоцитопенії споживання. Виникають у результаті посиленого використання тромбоцитів на утворення тромбів (хвороба Шенляйн—Геноха, хвороба Мошковича, ДВЗ-синдром).
26.3.16. Який патогенез порушень гемостазу в умовах тромбоцитопенії?
У патогенезі геморагічного синдрому при тромбоцитопеніях мають значення:
1) порушення ангіотрофічної функції тромбоцитів, у результаті чого виникають дистрофічні зміни в ендотелії і збільшується ламкість мікросудин. Це веде до збільшення ранимості судин, діапедезу еритроцитів, крововиливів. Останні виявляють себе петехіями на шкірі, кровотечами з ясен і носа, крововиливами в головний мозок і сітківку ока;
2) порушення адгезії й агрегації тромбоцитів. Це викликає порушення формування тромбоцитарного тромбу й призводить до збільшення часу кровотечі (проба Дюка);
3) порушення вторинного спазму ушкоджених артеріол. При тромбоцитопеніях вивільняється мало біогенних амінів (катехоламінів, серотоніну), з дією яких пов'язане скорочення гладких м'язів судин;
4) порушення зсідання крові. Обумовлено недостатнім вивільненням фактора 3 пластинок і тромбостеніну. У результаті порушується І фаза зсідання крові і ретракція згустку.
26.3.17. Що таке тромбоцитопатії? Як їх класифікують?
Тромбоцитопатії—це порушення функціональних властивостей тромбоцитів, йґ якісна неповноцінність. При цьому кількість тромбоцитів може залишатися в нормі J
За походженням тромбоцитопатії бувають спадково обумовленими і набутими.
За характером якісних дефектів кров'яних пластинок тромбоцитопатії поділяють на ендо- і екзотромбоцитарні.
Ендотромбоцитарні тромбоцитопатії обумовлені порушеннями складових частин тромбоцитів. їх, у свою чергу, поділяють на мембранопатії, гранулопатії і фер-ментопатії. Мембранопатії виникають при спадкових аномаліях мембранних пгіко-протеїнів, що виконують функції клітинних рецепторів; при блокаді цих рецепторів аномальними білками плазми крові (парапротеїнами), при ушкодженні мембрани кров'яних пластинок патогенними факторами. Гранулопатії виявляються дефіцитом гранул І і II типів. В основі ферментопатії може лежати зменшення активності ферментів циклу Кребса, гліколізу, порушення функцій АТФ-аз, циклоксигенази і тром-боксансинтетази.
При екзотромбоцитарних тромбоцитопатіях причини порушення функцій тромбоцитів лежать поза кров'яними пластинками. У зв'язку з цим екзотромбоцитарні тромбоцитопатії можуть бути:
а) пов'язаними зі змінами плазми крові (дефіцит плазмових білків, що є плазмовими кофакторами агрегації тромбоцитів);
б) пов'язаними зі змінами в судинній стінці (порушення утворення фактора Вілле-бранда ендотелієм судин, розлади зовнішнього механізму зсідання крові). Залежно від сутності порушень гемостазу виділяють:
1) тромбоцитопатії з первинним порушенням адгезії тромбоцитів^
2) тромбоцитопатії з первинними порушеннями агрегації тромбоцитів;
3) тромбоцитопатії з первинним порушенням реакцій вивільнення вмісту тромбоцитів',
4) тромбоцитопатії, пов'язані з дефіцитом або зменшенням доступності фактора З тромбоцитів.
26.3.18. Наведіть приклади тромбоцитопатій з різними механізмами порушень гемостазу.
1. Тромбоцитопатії з первинним порушенням адгезії тромбоцитів:
а) хвороба Віллебранда — ангіогемофілія. Обумовлена генетичними порушеннями синтезу фактора Віллебранда ендотеліальними клітинами (тип спадкування аутосомно-домінантний);
б) хвороба Бернара—Сульє — макротромбоцитодистрофія. Причиною є спадково обумовлений дефект глікопротеїнів тромбоцитарної мембрани (ГШЬ), що взаємодіють з фактором Віллебранда. При цьому тромбоцити набувають гігантських розмірів. Тип спадкування аутосомно-рецесивний.
2. Тромбоцитопатії з первинними порушеннями агрегації тромбоцитів - диза-грегаційні. Найчастіше буває тромбастенія Гланцмана, що виникає як наслідок дефектів мембранних глікопротеїнів І і II типів, що беруть участь в агрегації. При цьому адгезія тромбоцитів і вивільнення їхніх гранул відбуваються, а агрегація -ні, незважаючи на дію таких потужних агрегантів, як АДФ, адреналін, тромбін. Тип спадкування аутосомно-рецесивний.
3. Тромбоцитопатії з первинним порушенням реакцій вивільнення вмісту тромбоцитів:
а) порушення дегрануляції тромбоцитів — "парез реакції вивільнення ". Виникає, зокрема, при порушенні утворення тромбоксану А2 при дії ацетилсаліцилової кислоти. Показано, одо одноразове приймання аспірину необоротно збільшує час вивільнення гранул від 3,5 до 6 діб, поки не з'являться нові тромбоцити;
б) недостатність накопичення й збереження вмісту гранул тромбоцитів. Виникає, як правило, при генетично обумовлених порушеннях.
Розлади реакцій вивільнення гранул викликають порушення другої хвилі агрегації тромбоцитів. Початкова агрегація кров'яних пластинок закінчується їх дезагрегацією.
4. Тромбоцитопатії, пов'язані з дефіцитом або зменшенням доступності фактора 3 пластинок. У їхній основі можуть лежати або генетично обумовлені дефекти структури цього фактора, або порушення його вивільнення з ушкоджених тромбоцитів. При цьому порушується зсідання крові (коагуляційний гемостаз). Адгезив-но-агрегаційні властивості тромбоцитів не міняються.
Недостатність коагуляційного гемостазу. Причини та механізми порушень окремих стадій згортання крові.
Система гемостазу — це система, що забезпечує, з одного боку, збереження рідкого стану крові, з другого - зупинку кровотечі при ушкодженні кровоносних судин.
Коагуляційний (вторинний, макроциркуляторний) гемостаз є продовженням судинно-тромбоцитарного і здійснюється на його основі. Коагуляційний гемостаз забезпечує зупинку кровотеч із судин, діаметр яких перевищує 100 мкм. У результаті його активації утворюється червоний тромб, що складається з фібрину і формених елементів крові
26.3.19. Чим можуть бути обумовлені порушення коагуляційного гемостазу - коагулопатії?
В основі розвитку коагулопатій можуть бути:
1) зменшення активності системи зсідання крові;
2) підвищенням активності антикоагулянтної системи;
3) збільшенням активності фібринолітичної системи.
26.3.20. Що може бути причиною безпосереднього порушення І фази зсідання крові?
Залежно від характеру порушень І фази зсідання крові виділяють три групи розладів.
I. Ізольовані порушення зовнішнього механізму активації зсідання. Виникають при дефіциті ф. VII - гіпопроконвертинемії. Цей дефіцит може бути спадково обумовленим (тип спадкування аутосомно-рецесивний) або набутим (гіповітаміноз К, ураження печінки).
II. Ізольовані порушення внутрішнього механізму активації зсідання:
а) дефіцит ф.УШ- гемофілія А. Найчастіше виникає як генетичний дефект коагулянтної частини ф.УШ (тип спадкування зчеплений з Х-хромосомою). Можливе утворення аутоантитіл проти білкових компонентів цього фактора зсідання;
б) дефіцит ф. IX- гемофілія В. Причиною розвитку є спадкова патологія (тип спадкування зчеплений з Х-хромосомою), дефіцит вітаміну К або ураження печінки, антитіла проти ф. IX;
в) дефіцит ф. XI— гемофілія С. Виникає при генетичних порушеннях (тип спадкування аутосомно-рецесивний) або ураженнях печінки;
г) дефіцит ф. ХП. Спадкова патологія, що буває дуже рідко. Завдяки калікре-їн-кініновій системі цей дефект добре компенсується, оскільки запуск внутрішнього механізму зсідання відбувається через зовнішній;
ґ) дефіцит ф. З тромбоцитів. Є наслідком тромбоцитопенії або певних видів тромбоцитопатій.
III. Поєднані порушення зовнішнього і внутрішнього механізмів зсідання. Розвиваються при дефіциті ф. X, що може бути спадково обумовленим (тип спадкування аутосомно-рецесивний) або набутим (гіповітаміноз К, ураження печінки).
26.3.21. Що може бути причиною безпосереднього порушення II фази зсідання крові?
1. Дефіцит ф.ІІ— гіпопротромбінемія. Найчастіше має набутий характер і розвивається внаслідок гіповітамінозу К або уражень печінки.
2. Дефіцит ф. V— парагемофілія. Порушення утворення проакцелерину може бути обумовлено ураженнями печінки або аутоантитілами проти ф.У
26.3.22. Що може бути причиною безпосереднього порушення III фази зсідання крові?
1. Дефіцит фібриногену:
а) афібриногенемія - повна відсутність фібриногену (спадкове захворювання з аутосомно-рецесивним типом спадкування);
б) гіпофібрииогенемія - зменшення синтезу фібриногену в печінці при її ураженнях.
2. Дисфібршшгенемії - якісні порушення фібриногену. Розвиваються як наслідок генетичних дефектів (тип спадкування аутосомно-домінантний). Виявляються утворенням аномального фібрину.
3. Порушення полімеризації фібрину. Розвиваються б результаті утворення комплексів фібриногену з фібрином-мономером і проміжними продуктами, від яких ще повністю не відщепились пептиди А і В. При цьому утворюється так званий "заблокований фібриноген" ("тромбінрезистентний фібриноген"), що не піддається дії тромбіну.
4. Дефіцит ф. ХШ. Виникає як наслідок спадкових порушень (тип спадкування ау-тосомно-рецесивний). виявляється порушеннями перетворення розчинного фібрину (фібрину S) у нерозчинний (фібрин І).
26.3.23. Які речовини складають антикоатулянтну систему крові?
І. Первинні антикоагулянти. Постійно синтезуються в організмі й тому завжди містяться в плазмі крові. До них відносять:
1) антитромбін ПІ- основний універсальний антикоагулянт, що є інгібітором протеаз. Синтезується ендотелієм судин. Пригнічує активність усіх протеолітичних ферментів крові, у тому числі тромбіну, калікреїну, плазміну, ф. ХІІа, ф. ХІа, ф. Ха, ф. ІХа, ф.УІІа;
2) гепарин (антитромбін II) — глікозаміноглікан, що вивільняється тканинними базофілами і базофілами крові при їх дегрануляції. Антикоагулянтні властивості має не сам гепарин, а комплекс гепарину з антитромбіном III. Завдяки гепарину антитромбін III фіксується на поверхні ендотелію судин, де його антикоагулянтні властивості в багато разів зростають;
3) а -антитрипсин, а2-макроглобулін, інгібітор СІ-компонента комплементу - усі вони є неспецифічними інгібіторами протеаз, у тому числі й факторів зсідання крові.
її. Вторинні антикоагулянти. У плазмі крові в нормі не містяться. Утворюються в процесі зсідання крові й фібринолізу. До них відносять:
1) антитромбін І— фібрин, що адсорбує і в такий спосіб інактивує велику кількість тромбіну;
2) продукти фібринолізу. Перешкоджають процесам полімеризації фібрину і утворенню фібринових структур.
26.3.24. Чим може бути обумовлене підвищення активності антикоагулянтної системи крові?
Підвищення активності антикоагулянтної системи крові закономірно виникає при:
1) збільшенні вмісту гепарину в крові — гіпергепаринеміг. Це може бути обумовлено посиленою дегрануляцією тканинних базофілів і базофілів крові, зокрема, при алергічних реакціях І типу за класифікацією Кумбса і Джелла, руйнуванням базо-фільних лейкоцитів при лейкозах, введенням гепарину ззовні;
2) появою "патологічних " антикоагулянтів, до яких відносять антитромбін V, що порушує полімеризацію фібрину-мономеру; "вовчаковий" антикоагулянт, що порушує утворення протромбіназного комплексу; парапротеїни, що унеможливлюють полімеризацію фібрину.
26.3.25. Що входить у поняття "фібринолітична система"?
Фібриноліпшчна система — це система, яка забезпечує лізис (протеоліз) фібрину в кровоносному руслі. У такий спосіб вона бере участь у підтримці рідкого стану крові й у відновленні кровообігу в тромбованих судинах.
До складу системи фібринолізу входять (рис. 119):
Рис. 119. Схема фібринолізу
1) плазміноген (профібринолізин) — неактивний протеолітичний фермент, що завжди міститься в плазмі крові;
2) плазмін (фібринолізин) — активна форма плазміногену. Утворюється в результаті дії активних протеаз на плазміноген і відщеплення від його молекули пептиду, який "закриває" активний центр;
3) активатори фібринолізу — велика група речовин, які або самі є протеазами і здатні перетворювати плазміноген у плазмін, або викликають появу таких протеаз;
4) інгібітори фібринолізу. До них відносять інгібітори протеаз, серед яких найбільше значення має а2-антиплазмін.
Розрізняють внутрішній і зовнішній механізми активації фібринолізу.
Внутрішній механізм обумовлений активацією фактора XII зсідання крові й утворенням калікреїну, унаслідок чого в крові з'являється велика кількість активаторів фібринолізу.
Зовнішній механізм пов'язаний з надходженням у кров готових активаторів фібринолізу: ендотеліального, клітинного, тканинного (урокіназа), бактеріального (стрептокіназа).
26.3.26. Які фактори викликають підвищення активності фібриполітичної системи крові?
1. Посилене утворення й надходження в кров активаторів фібринолізу. Відбувається при великих ушкодженнях тканин: великі травми, ушкодження клітин токсинами, операційні втручання, лейкози та ін.
2. Зменшення вмісту в крові інгібіторів протеолізу. Має місце при недостатньому їх утворенні або посиленому використанні.
26.3.27. Якими клінічними ознаками виявляють себе порушення коагуляційного гемостазу?
На відміну від порушень судинно-тромбоцитарного гемостазу для коагулопатій характерні не капілярні (точкові) кровотечі, а кровотечі з великих судин — артерій і вен. Такі кровотечі клінічно виявляють себе:
а) гематомами — великими крововиливами в м'язи, під шкіру, у порожнину суглобів (гемартрози);
б) тривалими кровотечами після операційних втручань (видалення зуба та ін.).
Синдром дисемінованого внутрішньосудинного згортання крові, принципи класифікації, етіологія, патогенез, клінічні прояви. Роль в патології.
26.3.28. Що таке ДВЗ-синдром?
Синдром дисемінованого внутрішньосудинного зсідання крові (ДВЗ-син-дром) — це генералізоване зсідання крові всередині судин, що викликає утворення великої кількості мікрозгустків і агрегатів клітин, які порушують мікроциркуляцію в органах і тканинах.
Цей синдром часто характеризують як катастрофу для організму.
26.3.29. Що може бути причиною ДВЗ-синдрому?
Залежно від причин розвитку виділяють такі різновиди ДВЗ-синдрому:
1) інфекційно-септичний (розвивається при сепсисі);
2) посттравматичний (при краш-синдромі, опіковій хворобі, множинних переломах кісток);
3) шокогенний (при всіх видах шоку);
4) хірургічний (після операцій з великою травматизацією тканин);
5) акушерський (при передчасному відшаруванні плаценти, надходженні в кров навколоплідних вод);
6) токсигенний (після укусу змії);
7) пухлинний (при злоякісному пухлинному рості);
8) алергічний (при імунному ушкодженні тканин) та ін.
26.3.30. Що є патогенетичною основою розвитку ДВЗ-синдрому?
В основі патогенезу ДВЗ-синдрому лежить так званий "гуморальний протеазний вибух ", тобто одночасна активація всіх протеолітичних ферментів плазми крові, що входять до складу чотирьох позаклітинних біохімічних систем (рис. 120):
а) системи зсідання крові;
б) фібринолітичної системи;
в) калікреїн-кінінової системи;
г) системи комплементу.
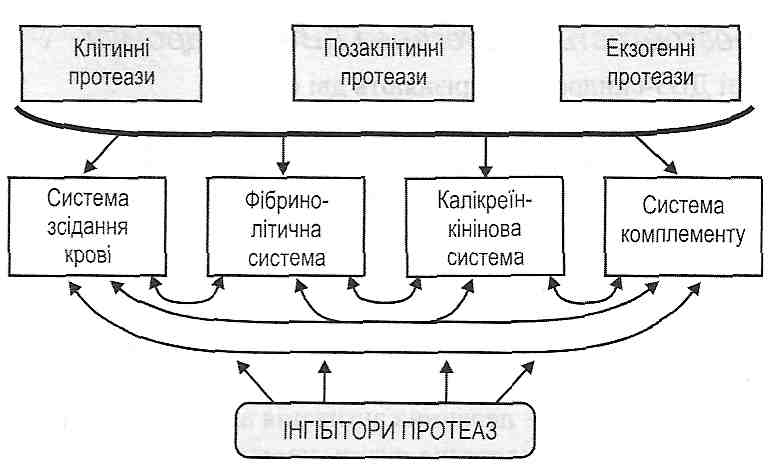
Рис. 120. "Гуморальний протеазний вибух"
Основний принцип активації позаклітинних протеаз — відщеплення пептидів, що закривають їхні активні центри. Утворення активних протеолітичних ферментів крові має свої особливості:
а) можлива самоактивація ферментів — активний фермент, впливаючи на неактивну форму, переводить її в активну;
б) одні активні протеази здатні активувати інші (перехресна активація)',
в) ланцюговий характер активації. Теоретично поява навіть однієї молекули активної протеази може викликати активацію всіх наявних протеаз крові.
Однак у нормі реакції активації протеолітичних ферментів мають обмежений характер, що пояснюється існуванням великої групи інгібіторів протеаз.
При патології, коли у кров надходять великі кількості активних протеаз, потужність існуючих інгібіторів може виявитися недостатньою. Отоді й виявить себе ланцюговий характер активації протеолітичних систем плазми крові. Така активація набуває генералізованого характеру, втягує всі протеази крові - відбувається "гуморальний протеазний вибух".
26.3.31. Назвіть основні джерела надходження в кров активних протеаз при ДВЗ- синдромі.
Існує три основних джерела надходження протеаз у кров.
I. Ушкоджені клітини. Має значення гостре ушкодження великої кількості клітин, з яких у позаклітинний простір і кров надходять лізосомні протеази, тканинний тромбопластин.
Запалення як місцевий процес, що виникає при ушкодженні клітин, обмежує надходження продуктів розпаду в кров, локалізуючи ушкодження і в такий спосіб попереджаючи розвиток ДВЗ-синдрому.
II. Надходження в кров великої кількості позаклітинних протеаз; наприклад трипсину при гострому панкреатиті, ферментів, що містяться в навколоплідних водах.
III. Екзогенні протеази. їхніми джерелами можуть бути бактеріальні клітини при сепсисі, зміїна отрута та ін.
26.3.32. Як розгортається патогенез ДВЗ-синдрому?
У патогенезі ДВЗ-синдрому розрізняють дві фази.
I фаза - фаза гіперкоагуляції і агрегації тромбоцитів. Основу цієї фази становить генералізована активація системи зсідання крові, тобто утворення тромбіну (тромбінемія), що призводить до утворення фібрину і агрегатів тромбоцитів.
Існує три механізми запуску цієї фази:
1) ферментативний механізм - надходження в кров великої кількості активних протеаз і тканинного тромбопластину;
2) контактний механізм - активація ф. XII при контакті його з чужорідними поверхнями (екстракорпоральний кровообіг, гемодіаліз, штучні клапани серця);
3) тромбоцитарний механізм - первинна активація агрегації тромбоцитів при гене-ралізованому ушкодженні ендотелію судин, порушеннях реологічних властивостей крові, гострому внутрішньосудинному гемолізі еритроцитів.
У результаті реалізації зазначених механізмів утворюється велика кількість мі-крозгустків і агрегатів клітин, що призводить до розладів мікроциркуляції, розвитку сладж-синдрому (див. розд. 13).
Усе це веде до появи таких клінічних проявів, як гіпоксія, ацидоз, інтоксикація продуктами розпаду, гостра недостатність зовнішнього дихання (мікрозгустками закупорюються капіляри легень), гостра ниркова недостатність (забиваються капіляри клубочків), порушення мозкового кровообігу.
II фаза - фаза гіпокоагуляції (геморагічний синдром). Ця фаза розвивається як наслідок виснаження механізмів судинно-тромбоцитарного і коагуляційного гемостазу.
У її виникненні мають значення:
а) зменшення активності системи зсідання крові (споживання факторів І, V, VIII);
б) активація фібринолітичної системи (надходження в кров великої кількості активаторів фібринолізу);
в) підвищення антикоагулянтної активності крові за рахунок утворення продуктів фібринолізу;
г) розвиток тромбоцитопенії споживання;
ґ) підвищення проникності стінки судин (має значення утворення великих кількостей кінінів). Фаза гіпокоагуляції клінічно виявляє себе масивними кровотечами, що їх важко зупинити.
26.3.33. Що таке тромбофільні діатези? Що може лежати в основі їх розвитку?
Тромбофільні діатези - це захворювання і синдроми, при яких схильність до утворення тромбів обумовлена первинними порушеннями механізмів гемостазу.
Патогенетичну основу тромбофілій можуть становити: 1) ендотеліальні механізми, що обумовлюють зменшення тромборезистентності стінки судин;
2) тромбоцитарнімеханізми, пов'язані зі збільшенням агрегаційної здатності тромбоцитів;
3) зменшення антикоагулянтної активності крові (зменшення утворення антитромбіну III);
4) зниження активності фібринолітичної системи (зменшення утворення ендоте-ліального активатора плазміногену, поява потужних інгібіторів плазміну, дефіцит плазміногену);
5) порушення системи зсідання крові (дисфібриногенемії).
Клінічно тромбофільні діатези виявляють себе тромбозами і тромбоемболіями венозних і артеріальних судин у різних органах і тканинах.
Недостатність кровообігу у дітей: визначення поняття, принципи класифікації, причини і механізми розвитку її різних типів. Патогенез основних клінічних проявів недостатності кровообігу.
Недостатність кровообігу - це стан, при якому серцево-судинна система не може забезпечити органи й тканини організму необхідною кількістю крові. Є найчастішим проявом різних порушень функцій системи кровообігу.
Недостатність кровообігу може бути обумовлена:
1) недостатністю серця;
2) недостатністю кровоносних судин;
3) серцево-судинною недостатністю, тобто одночасною недостатністю серця і судин.
27.2. Які наслідки для органів, тканин і організму в цілому має недостатність кровообігу?
1. Порушення трофічного забезпечення органів і тканин. Розвиваються як наслідок:
а) зменшення доставки кисню (циркуляторпа гіпоксія, див. розд. 19);
б) зменшення доставки поживних речовин.
2. Порушення видалення з органів і тканин кінцевих продуктів обміну речовин. Наслідком цього є розвиток:
а) негазового ацидозу;
б) інтоксикації.
Недостатність серця: визначення поняття, принципи класифікації. Причини перевантаження серця об'ємом та опором. Механізми негайної та довготривалої адаптації серця до надмірного навантаження. Гіпертрофія серця, її патогенез (за Ф. Меерсоном). Особливості гіпертрофованого міокарда.
27.3. Що таке недостатність серця?
Недостатність серця - це патологічний стан, обумовлений нездатністю серця забезпечити кровопостачання органів і тканин відповідно до їхніх потреб.
Це стан, при якому навантаження на серце перевищує його здатність виконувати роботу. Він виявляє себе тим, що серце не здатне переміщати в артеріальне русло всю кров, що надходить до нього по венах.
27.4. Як класифікують недостатність серця?
I. Залежно від клінічного перебігу розрізняють гостру і хронічну недостатність серця.
II. За вираженістю клінічних проявів недостатність серця може бути прихованою (компенсованою) і явною (декомпенсованою).
III. Залежно від порушення функції переважно того чи того відділу серця розрізняють лівоишуночкову, правошлуночкову і тотальну недостатність серця.
IV. За патогенезом виділяють:
а) недостатність серця від перевантаження;
б) міокардіальну недостатність серця;
в) позаміокардіальну недостатність.
27.5. Дайте коротку характеристику різних патогенетичних варіантів недостатності серця.
I. Недостатність серця від перевантаження розвивається в результаті дії на здорове серце великих навантажень опором або об'ємом, тобто коли збільшується опір серцевому виштовху або приплив крові до певного відділу серця. Це буває при вадах серця, гіпертензії великого або малого кола кровообігу, артеріовенозних фістулах або при виконанні дуже важкої фізичної роботи. При цьому до серця з нормальною скорочувальною здатністю висуваються надмірні вимоги.
II. Міокардіальна недостатність серця розвивається в результаті первинного ураження міокарда. Вона може бути пов'язана з: а) ушкодженням провідникової системи серця (аритмічна) і б) ушкодженням волокон робочого міокарда (міо-кардіопатична). Причиною її розвитку часто є інфекції, інтоксикація, гіпоксія, авітамінози, порушення вінцевого кровообігу, деякі спадкові дефекти обміну речовин. При цьому недостатність розвивається навіть при нормальному або зниженому навантаженні на серце.
III. Позаміокардіальна недостатність серця розвивається в результаті дії причин, не пов'язаних безпосередньо з міокардом. її виникнення можуть обумовлювати зменшення припливу крові до серця (гіповолемія, колапс) або перешкоди здійсненню діастоли, у результаті чого серце не може прийняти всю кров, що притікає до нього (накопичення ексудату або транссудату в порожнині перикарда, гостра тампонада серця).
27.6. Які типи перевантажень серця можуть бути причиною розвитку його недостатності?
Виділяють два типи перевантажень серця.
1. Перевантаження об'ємом виникає тоді, коли до серця або до окремих його порожнин притікає збільшений об'єм крові. У цих умовах серце або його відділ, що зазнає перевантаження, мають переміщати збільшений об'єм крові в артеріальну систему. Це досягається збільшенням хвилинного об'єму серця відповідно до збільшеного венозного повернення.
Перевантаження об'ємом виникає при:
а) збільшенні венозного повернення крові до серця, зокрема при збільшенні об'єму циркулюючої крові (гіперволемія) або збільшенні тонусу венозних судин (зменшення ємності венозної системи);
б) вадах серця — недостатності його клапанів. Так, при недостатності аортального і двостулкового клапанів розвивається перевантаження лівого шлуночка, при недостатності клапана легеневої артерії і тристулкового клапана - перевантаження правого шлуночка.
2. Перевантаження опором виникає тоді, коли серце або окремі його відділи змушені виконувати роботу проти збільшеного опору, що перешкоджає переміщенню всієї крові в артеріальну систему. При перевантаженні опором серце має зберегти свій хвилинний об'єм, незважаючи на збільшений опір вигнанню крові.
Перевантаження опором розвивається при:
а) збільшенні артеріального тиску (збільшенні периферичного судинного опору). При гіпертензії великого кола кровообігу перевантаження опором діє на лівий шлуночок, а при гіпертензії малого кола - на правий шлуночок;
б) вадах серця — стенозах клапанних отворів. 'Так, при стенозі отвору аорти розвивається перевантаження лівого шлуночка, при стенозі отвору двостулкового клапана - лівого передсердя, при стенозі отвору легеневої артерії - правого шлуночка, при стенозі отвору тристулкового клапана - правого передсердя.
27.7. Які механізми можуть забезпечувати компенсацію серця при дії на нього збільшених навантажень?
При дії на серце навантажень об'ємом і опором збільшення роботи серця забезпечується двома типами компенсаторних механізмів.
I. Негайні механізми компенсації серця. До них відносять:
а) гетерюметричтш механізм;
б) гомеометричний механізм;
в) хроноінотропний механізм;.
г) інотропна дія катехоламінів.
II. Механізми довгострокової адаптації серця - гіпертрофія міокарда.
27.8. У чому сутність тетерометричното механізму компенсації серця?
Гетерометричпиіі механізм є одним із негайних механізмів компенсації серця до дії навантажень об'ємом.
Його сутність полягає в збільшенні сили серцевих скорочень в умовах надходження до серця збільшеного об'єму крові.
Основу гетерометричного механізму становить закон Франка—Старлінга. Він має два формулювання: для окремих м'язових волокон і для серця в цілому. У першому варіанті його сутність виражено таким положенням: що більша вихідна довжина м 'язового волокна (у певних межах), то більша сила його скорочень. Для серця в цілому уживаним є формулювання: що більший кінцеводіастолічний об'єм шлуночків серця, то більший їхній ударний об'єм.
Основними проявами гетерометричного механізму компенсації є збільшення кін-цеводіастолічного тиску за рахунок збільшення надходження крові в порожнини шлуночків і збільшення ударного, а отже, і хвилинного об'ємів серця за рахунок збільшення сили серцевих скорочень. Напруга м'язових волокон міокарда при цьому не міняється. Змінюється тільки їх довжина, звідси назва механізму - гетерометричний.
27.9. У чому сутність томеометричното механізму компенсації серця?
Гомеометричний механізм є негайним механізмом компенсації серця до дії навантажень опором. Його сутність полягає в збільшенні сили серцевих скорочень в умовах збільшення опору вигнанню крові.
Нині показано, що в основі цього механізму, як і гетерометричного, закон Фран-ка-Старлінга, тобто збільшення вихідної довжини волокон міокарда і пов'язане з цим збільшення кінцеводіастолічного тиску.
Становлення гомеометричного механізму відбувається в такій послідовності. При збільшенні опору вигнанню крові різко падає ударний об'єм, унаслідок чого збільшується кінцевосистолічний об'єм шлуночків. Оскільки надходження крові в шлуночки продовжує залишатися попереднім, то в наступному циклі скорочень серця збільшується кінцеводіастолічний об'єм. А це за законом Франка-Старлінга веде до збільшення сили скорочень серця.
Для гомеометричного механізму характерні такі зміни показників кардіодинаміки:
а) збільшення кінцевосистолічного об'єму;
б) збільшення кінцеводіастолічного об'єму за рахунок первинного підвищення попереднього показника, а не за рахунок збільшення припливу крові, як при гетеро-метричному механізмі;
в) ударний, а отже, і хвилинний об'єми за рахунок збільшення сили серцевих скорочень залишаються на попередньому рівні, незважаючи на збільшення опору вигнанню крові.
При гомеометричному механізмі збільшується напруга м'язових волокон міокарда, тимчасом як їхня довжина не міняється. Звідси назва механізму компенсації — го-меометричний.
27.10. Чим характеризується хроноінотропний механізм компенсації серця?
Хроноінотропний механізм (феномен "сходинок", феномен Боудича) є одним з негайних механізмів компенсації серця до дії підвищених навантажень. Його сутність полягає в тому, що при збільшенні частоти скорочень серця збільшується сила його скорочень. При цьому одночасно зменшується час розслаблення міокарда, що сприяє швидкому наповненню шлуночків серця кров'ю.
Нині вважають, що основу хроноінотропного механізму становить збільшення надходження іонів кальцію в саркоплазму кардіоміоцитів під час потенціалів дії, сумарна тривалість яких при тахікардії зростає. Підвищення концентрації іонів кальцію в саркоплазмі призводить до збільшення кількості утворюваних кальцій-тропонінових комплексів і, як наслідок, до збільшення сили скорочень м'язових волокон.
Для хроноінотропного механізму характерні такі зміни показників кардіодинаміки:
а) ударний об'єм збільшується (при навантаженні об'ємом) або залишається постійним (при навантаженні опором). У результаті збільшення частоти серцевих скорочень зростає хвилинний об'єм серця;
б) кінцеводіастолічний об'єм зменшується, якщо приплив крові до серця постійний, або залишається без змін, якщо приплив крові зростає;
в) кінцевосистолічний об'єм зменшується.
27.11. Яка роль катехоламінів у здійсненні механізмів негайної компенсації серця?
Участь катехоламінів у здійсненні негайної адаптації серця до підвищених навантажень пов'язана зі здатністю адреналіну і норадреналіну безпосередньо збільшувати силу серцевих скорочень - з позитивним інотропним ефектом.
Установлено, що під впливом катехоламінів збільшується кількість Са-каналів сарколеми, здатних відкриватися під час потенціалу дії (катехоламіни через цАМФ-опосередкований механізм викликають фосфорування білків Са-каналів). У результаті цих процесів збільшується надходження іонів кальцію в саркоплазму, де підвищується їхня концентрація, і, як наслідок, збільшується сила скорочень кардіоміоци-тів, оскільки зростає кількість утворюваних кальцій-тропонінових комплексів.
Інотропна дія катехоламінів (а не закон Франка-Старлінга) є провідним механізмом адаптації серця до фізичних навантажень. При цьому показники кардіодинаміки змінюються таким чином:
а) збільшується ударний об'єм;
б) збільшується хвилинний об'єм серця як за рахунок збільшення ударного об'єму, так і за рахунок збільшення частоти серцевих скорочень (позитивний хронотроп-ний ефект катехоламінів);
в) зменшується кінцеводіастолічний об'єм (при рентгенівському дослідженні визначається зменшення об'єму серця);
г) зменшується кінцево систолічний об'єм.
27.12. Назвіть варіанти довгострокової адаптації серця до дії навантажень.
За Ф. Меєрсоном, виділяють три варіанти довгострокової адаптації серця.
1. Гіпертрофія серця у спортсменів ("адаптоване" серце). Розвивається при періодичних навантаженнях, інтенсивність яких поступово зростає, тобто в умовах тренувань. Є збалансованою гіпертрофією, при якій рівномірно збільшуються всі складові компоненти серця. Завдяки такій гіпертрофії істотно збільшуються функціональні резерви серця.
2. Компенсаторна гіпертрофія серця ("переадаптоване " серце). Є наслідком патологічних процесів, що мають стосунок до серця. Розрізняють два види компенсаторної гіпертрофії:
а) гіпертрофію від перевантажень (розвивається при вадах серця, артеріальній гіпертензії);
б) гіпертрофію від ушкодження (характерна для атеросклеротичних уражень, міокардіопатії).
Розвиток компенсаторної гіпертрофії серця характеризується такими особливостями:
1) патогенний фактор, що викликає гіпертрофію, діє постійно;
2) компенсаторна гіпертрофія, на відміну від гіпертрофії серця у спортсменів, є незбалансованою;
3) при компенсаторній гіпертрофії згодом розвивається недостатність серця.
3. Атрофія міокарда ("deadагітоване" серце). Характеризується зменшенням маси серця в результаті тривалої гіпокінезії і зменшення навантажень на серце.
27.13. Які механізми лежать в основі розвитку гіпертрофії серця?
При тривалому підвищенні навантаження на серце розвивається його гіперфункція, що згодом викликає структурні зміни в серці - гіпертрофію міокарда.
Найбільш доказовою теорією, що пояснює механізми переходу гіперфункції серця в його гіпертрофію, є концепція Ф. Меєрсона (рис. 121).
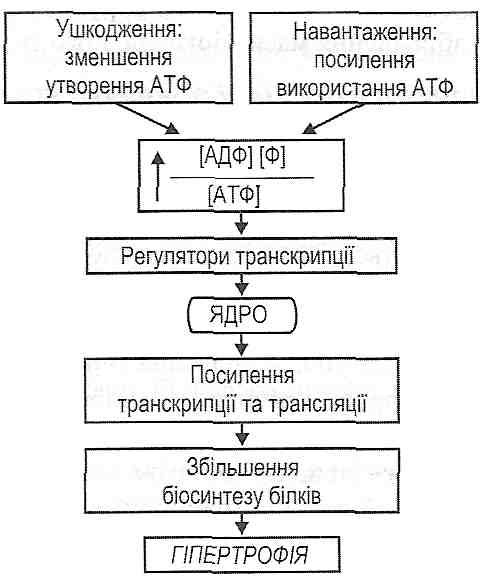
Рис. 121. Механізми компенсаторної гіпертрофії міокарда
Відповідно до цієї концепції, головною ланкою, що зв'язує підвищення функції клітини з роботою її генетичного апарату, є збільшення потенціалу фосфорування (ПФ):
![]()
де [АДФ], [Ф], [АТФ] - концентрації в цитоплазмі клітин відповідно АДФ, неорганічного фосфату і АТФ.
ПФ закономірно збільшується у двох випадках:
а) при посиленому використанні АТФ, що завжди спостерігається при збільшенні функціонального навантаження на клітини (при їхній гіперфункції);
б) при порушеннях утворення АТФ, що характерно для різних видів ушкодження клітин.
Збільшення показника ПФ викликає появу в клітинах речовин - регуляторів транскрипції, які, впливаючи на геном клітини, посилюють синтез інформаційної РНК на матриці генів, що кодують структуру функціонально важливих білків клі-
тини, у тому числі скорочувальних білків і ферментів. На роль речовин - регуляторів транскрипції претендує цілий ряд метаболітів, серед яких цАМФ, креатин, іони Mg2+, поліаміни (спермін, спермідин) та ін.
Таким чином, розвиток гіпертрофії серця можна описати такою послідовністю процесів: збільшення навантаження на серце (гіперфункція) → посилене використання АТФ, що перевищує інтенсивність його ресинтезу → збільшення потенціалу фосфорування → поява або збільшення концентрації в клітинах речовин - регуляторів транскрипції → зростання інтенсивності синтезу інформаційної РНК і процесів трансляції в рибосомах → посилення біосинтезу структурних, функціональних білків і білків-ферментів → збільшення маси міокарда, його гіпертрофія.
27.14. Які стадії виділяють у процесі розвитку компенсаторної гіпертрофії серця? Дайте їх характеристику.
За динамікою змін обміну, структури й функції міокарда в розвитку компенсаторної гіпертрофії серця виділяють три основні стадії (Ф. Мсєрсон).
1. Аварійна стадія. Розвивається безпосередньо після підвищення навантаження, характеризується поєднанням патологічних змін у міокарді (зникнення глікогену, зниження рівня креатинфосфату, зменшення вмісту внутрішньоклітинного калію і підвищення вмісту натрію, мобілізація гліколізу, накопичення лактату) з мобілізацією резервів міокарда і організму в цілому. У цій стадії підвищується навантаження на одиницю м'язової маси, а отже, інтенсивність функціонування структур (ІФС), відбувається швидке, протягом тижнів, збільшення маси серця за рахунок посиленого синтезу білків і потовщення м'язових волокон.
2. Стадія завершеної гіпертрофії і відносно стійкої гіперфункції. У цій стадії процес гіпертрофії завершений, маса міокарда збільшена на 100-120 % і далі не зростає, ІФС нормалізується. Патологічні зміни в обміні речовин і структурі міокарда не виявляються, споживання кисню, утворення енергії, вміст макроергічних сполук не відрізняються від норми. Нормалізуються гемодинамічні порушення. Гіпертрофоване серце пристосувалося до нових умов навантаження і протягом тривалого часу компенсує їх.
3. Стадія поступового виснаження і прогресуючого кардіосклерозу. Характеризується глибокими метаболічними і структурними змінами, які поволі накопичуються в енергоутворюючих і скорочувальних елементах клітин міокарда. Частина м'язових волокон гине й заміщається сполучною тканиною, ІФС знову зростає. Порушується регуляторний апарат серця. Прогресуюче виснаження компенсаторних резервів призводить до виникнення хронічної недостатності серця, а надалі - до недостатності кровообігу.
27.15. Які особливості гіпертрофованого серця є передумовою розвитку його декомпенсації?
Гіпертрофоване серце відрізняється від нормального низкою обмінних, функціональних і структурних ознак, які, з одного боку, дають йому можливість тривалий час долати підвищене навантаження, а з другого - створюють передумови для виникнення патологічних змін.
1. Збільшення маси серця відбувається за рахунок потовщення кожного м'язового волокна, що супроводжується зміною співвідношення внутрішньоклітинних структур. Об'єм клітини при цьому збільшується пропорційно кубу лінійних розмірів, а поверхня — пропорційно їх квадрату, що призводить до зменшення клітинної поверхні на одиницю маси клітини. Відомо, що через поверхню клітини відбувається її обмін з позаклітинною рідиною — поглинання кисню, поживних речовин, виведення продуктів метаболізму, обмін води і електролітів. Зазначені вище зміни створюють умови для погіршення постачання м 'язового волокна, особливо його центральних відділів.
2. Клітинна мембрана відіграє важливу роль у проведенні збудження і спряженні процесів збудження і скорочення, що здійснюється через тубулярну систему і сар-коплазматичний ретикулум. Оскільки ріст цих утворень при гіпертрофії м'язового волокна також відстає, то створюються передумови для порушення процесів скорочення і розслаблення кардіоміоцитів: унаслідок уповільнення виходу іонів кальцію в саркоплазму погіршується скорочення, а в результаті утруднення зворотного транспорту іонів кальцію в саркоплазматичний ретикулум - розслаблення, іноді можуть виникати локальні контрактури окремих кардіоміоцитів.
3. При гіпертрофії об'єм клітини зростає у більшій мірі, ніж об'єм ядра. Оскільки роль ядра полягає в забезпеченні білкового синтезу, а отже, і процесів відновлення внутрішньоклітинних структур, то відносне зменшення об'єму ядра (якщо порівнювати з цитоплазмою) може призводити до порушення синтезу білків і погіршення пластичного забезпечення клітини.
4. У процесі розвитку гіпертрофії маса мітохондрій спочатку збільшується швидше, ніж маса скорочувальних білків, створюючи умови для достатнього енергетичного забезпечення і належної компенсації функції серця. Однак надалі, у міру посилення процесу, збільшення маси мітохондрій починає відставати від росту маси цитоплазми. Мітохондрії починають працювати з граничним навантаженням, у них розвиваються деструктивні зміни, знижується ефективність їхньої роботи, порушується окисне фосфорування. Це веде до погіршення енергетичного забезпечення гіпертрофованої клітини.
5. Збільшення маси м'язових волокон найчастіше не супроводжується адекватним збільшенням капілярної мережі, особливо у випадках швидкого розвитку недостатності серця. Великі вінцеві артерії також не можуть забезпечити необхідне пристосування. Тому під час навантаження погіршується судинне забезпечення гіпертрофованого міокарда.
6. При розвитку гіпертрофії міокарда в процес обов'язково втягується нервовий апарат серця. Спостерігається посилене функціонування внутрішньосерцевих і екстракардіальних нервових елементів. Однак ріст нервових закінчень відстає від збільшення маси скорочувального міокарда. Відбувається виснаження нервових клітин, порушуються трофічні впливи, зменшується вміст норадреналіну в міокарді, що веде до погіршення його скорочувальних властивостей, утруднення мобілізації його резервів. Отже, порушується (регуляторне забезпечення серця.
7. Гіпертрофоване серце за рахунок збільшення маси його скорочувального апарату і систем енергозабезпечення здатне тривалий час виконувати значно більшу роботу, ніж серце нормальне, зберігаючи при цьому нормальний метаболізм. Однак здатність пристосовуватися до навантажень, що змінюються, діапазон адаптаційних можливостей у гіпертрофованого серця обмежені, зменшений функціональний резерв. Це робить гіпертрофоване серце через зазначену вище незбалансованість внутрішньоклітинних і тканинних структур більш уразливим у різних несприятливих обставинах.
Міокардіальна недостатність серця. Етіологія, патогенез некоронарогенних ушкоджень міокарду. Експериментальне моделювання. Особливості у дітей.
Міокардіальна недостатність серця розвивається внаслідок первинних уражень міокарда. Оскільки міокард складається з двох типів м'язових волокон - атипових, що становлять провідникову систему серця, і скорочувальних клітин робочого міокарда— то розвиток недостатності серця може бути пов'язаний з ушкодженням як перших, так і других. Звідси можна виділити два варіанти міокардіальної недостатності серця:
а) обумовлену ураженням провідникової системи серця — аритмічну;
б) обумовлену ушкодженням робочого міокарда. Іноді її називають міокардіопатичною
Кардіоміопатії у дітей: визначення поняття, принципи класифікації; етіологія, патогенез. Особливості у дітей.
Кардіоміопатія (застаріла назва - міокардіолістрофія) та її різновиди поділяються на три основних види: дилатаційна кардіоміопатія, гіпертрофічна кардіоміопатія, рестріківная кардіоміопатія. Кожен з трьох видів кардіоміопатії має свої «окремі випадки» - кардіоміопатії, зумовлені конкретними факторами. Причини кардіоміопатії різні - від генетичної схильності до зловживання алкоголем. У разі, якщо причини кардіоміопатії встановлені, кардіоміопатія вважається вторинною. При неможливості встановлення точної причини кардіоміопатії прийнято говорити про первинну кардіоміопатії. Всі різновиди кардіоміопатії мають схожі симптоми: задишка, набряки (переважно нижніх кінцівок), тахікардія, збільшення маси тіла, підвищений показник артеріального тиску, неможливість займатися фізичною активністю, серцева недостатність. Нерідко саме розвинулася серцева недостатність дозволяє встановити наявність кардіоміопатії. Кардіоміопатія і її різновиди має свої характерні особливості впливу на серцевий м'яз. Дилатаційна кардіоміопатія призводить до збільшення обсягу всіх камер серця (шлуночків і передсердь), стінки камер при цьому стають тонкими і в'ялими. Істотно знижується скорочувальна здатність серцевого м'яза. В результаті гіпертрофічної кардіоміопатії спостерігається потовщення стінок серця, порожнину лівого шлуночка поглинається, порушується кровообіг. Рестриктивна кардіоміопатія зустрічається найбільш рідко і характеризується жорсткістю стінок серця, поганою здатністю переходити у фазу розслаблення. В результаті ускладнюється доставка крові і кисню в лівий шлуночок серця, порушується кровообіг у всьому організмі. У ряді випадків спостерігається рестриктивная кардіоміопатія у дітей, обумовлена спадковими факторами. Окремими випадками дилатаційною кардіоміопатії є алкогольна кардіоміопатія, токсична кардіоміопатія, ішемічна кардіоміопатія, ідіопатична кардіоміопатія. Алкогольна кардіоміопатія і токсична кардіоміопатія провокуються надмірним вживанням алкоголю і токсичними отруєннями (наркотичними, хімічними та ін.) Ішемічна кардіоміопатія характеризується надмірним пошкодженням судин серця атеросклеротичними бляшками, тобто, причина виникнення ішемічної кардіоміопатії - атеросклероз судин. Для ішемічної кардіоміопатії властиво наявність хронічної серцевої недостатності. Причиною ідіопатичною дилатаційною кардіоміопатії, як правило, є спадковий фактор. Ідіопатична кардіоміопатія здатна тривалий час протікати безсимптомно, і починає проявлятися з скарг пацієнтів на задишку, тахікардію, занепад сил. Метаболічна кардіоміопатія (дисметаболічна кардіоміопатія) виникає внаслідок порушення обміну речовин в серцевому м'язі і призводить до дистрофії стінок і порушень скорочувальної здатності серцевого м'яза. Метаболічна кардіоміопатія є вторинною, тобто, причини її виникнення відомі, і вони породжують «підклас» метаболічних кардіоміопатій. До числа метаболічних кардіоміопатій відносять такі види. Клімактерична кардіоміопатія або дисгормональная кардіоміопатія викликана порушенням вироблення гормонів і взаємного споживання вироблюваних гормонів внутрішніми органами. У період клімаксу організму властива «гормональна перебудова», тому виникає ймовірність розвитку клімактеричнийкардіоміопатії. Захворювання, що тягнуть гормональні збої, завідомо здатні спровокувати дисгормональну кардиомиопатию. Причинами виникнення метаболічної кардіоміопатії також можуть стати ожиріння, недостатнє вживання вітамінів, захворювання ендокринної системи, шлунково-кишкового тракту, порушення обміну речовин та ін Деякі різновиди метаболічної кардіоміопатії у дітей мають вроджений характер захворювання. Діабетична кардіоміопатія є окремим випадком гіпертрофічної кардіоміопатії з аналогічним механізмом впливу на серцевий м'яз: товщають (гіпертрофуються) стінки серця, ускладнюється робота лівого шлуночка, порушується кровообіг. Цукровий діабет - кардіоміопатія являють собою причинно-наслідковий зв'язок виникнення діабетичної кардіоміопатії. Тобто, у ряді випадків діабетична кардіоміопатія діагностується у дитини внаслідок наявності у матері цукрового діабету. Діабетична кардіоміопатія у дітей може тривалий час протікати безсимптомно. Крім вродженої форми, діабетична кардіоміопатія може бути спровокована розвинувся цукровий діабет. Цукровий діабет сприяє закупорці судин атеросклеротичними бляшками, що ускладнює доставку крові до серця. Схильність до ожиріння при цукровому діабеті створює додаткове навантаження на серцевий м'яз. Діабетична кардіоміопатія, причиною виникнення якої є цукровий діабет, може протікати без конкретної симптоматики, що ускладнює її своєчасну діагностику. Аритмогенну кардіоміопатія або, користуючись прийнятої в медицині термінологією, аритмогенна правошлуночкова кардіоміопатія, відрізняється неухильним заміщенням м'язової серцевої тканини правого шлуночка серця на жирову і фіброзну (сполучну) тканини. Тобто, порушується скорочувальна функція серцевого м'яза за рахунок «сторонніх» серцю тканин, не здатних скорочуватися. Сучасна медицина схильна вважати, що аритмогенного кардіоміопатія має спадковий характер виникнення. Аритмогенну кардіоміопатія може існувати в організмі поряд з міокардитом, який буде вторинним і виникати внаслідок вразливості серцевого м'яза вірусів і інфекцій. Аритмогенну кардіоміопатія зустрічається у людей різних віків, аж до глибокої старості. У ряді випадків аритмогенного кардіоміопатія діагностується у спортсменів. Симптоми аритмогенну кардіоміопатії, як правило, починають проявлятися до досягнення пацієнтом 40-річного віку і виражаються в аритмії, тахікардії, запамороченнях, симптомах серцевої недостатності.
Недостатність вінцевого кровообігу: визначення поняття, причини і механізми розвитку, клінічні прояви. Механізми ішемічного і реперфузійного пошкодження кардіоміоцитів.
1. Високий рівень екстракції кисню в капілярах серця. У серці екстрагується 70—75 % 02, що надходить з артеріальною кров'ю, тимчасом як у тканинах головного мозку — 25 %, у скелетних м'язах і печінці — близько 20 %, у нирках — 10 %. Високий рівень вилучення 02 у серці пояснюється значною довжиною його капілярів і у зв'язку з цим — більшим часом контакту крові зі стінкою капілярів.
При збільшенні потреби серця в кисні вона не може бути задоволена збільшенням екстракції 02 (як у скелетних м'язах), оскільки остання і так є максимально можливою в стані спокою. Тому для забезпечення збільшених енергетичних потреб серця залишається тільки один шлях — збільшення вінцевого кровообігу.
2. Високий базальний тонус вінцевих судин. Він дає можливість у стані спокою забезпечувати вінцевий кровообіг на рівні 250-300 мл/хв, що становить близько 5 % хвилинного об'єму крові.
Високий базальний тонус вінцевих судин обумовлює високий резерв вінцевого кровообігу. Так, при зменшенні базального тонусу судин серця Інтенсивність кровообігу у них може зростати в 7—10 разів.
3. Фазний характер вінцевого кровообігу, пов'язаний з періодами серцевого циклу. Під час систоли відбувається здавлювання інтрамуральних судин - кровообіг мінімальний і становить близько 15 % загального вінцевого кровообігу. Під час діастоли здавлювання судин припиняється і кровообіг стає максимальним (близько 85 % загальної величини).
Фазність вінцевого кровообігу найбільш виражена в субендокардіальній зоні міокарда (найбільше здавлювання судин) і найменш виражена - у субепікардіальній зоні.
4. Підпорядкованість вінцевого кровообігу метаболічним потребам серця і відносна незалежність його від нервових регуляторних впливів. В умовах патології ця підпорядкованість часто порушується і збільшується чутливість вінцевих судин до нервових імпульсів.
5. Винятково висока чутливість вінцевих судин до зменшення напруги кисню в крові. Зменшення р02 артеріальної крові всього лише на 5 % істотно збільшує інтенсивність вінцевого кровообігу.
6. Недостатній розвиток колатеральних судин. При несприятливих умовах колате-ралі в серці не можуть компенсувати порушення течії крові у вінцевих судинах, тому колатеральний кровообіг тут функціонально неповноцінний.
27.46. Як здійснюється регуляція вінцевого кровообігу?
У регуляції вінцевого кровообігу розрізняють міогенну ауторегуляцію, метаболічну і нервову регуляцію.
1. Міогенна ауторегуляція. її основу становить закон Бейліса, відповідно до якого при розтягненні гладких м 'язів кровоносних судин збільшується сила їх скорочення. Міогенна ауторегуляція забезпечує сталість вінцевого кровообігу і відносну незалежність його від змін артеріального тиску. Так, при збільшенні тиску крові в аорті збільшується розтягування гладком'язових клітин вінцевих артерій, що веде до їх скорочення, підвищення тонусу артерій і збереження сталості кровообігу. При зменшенні артеріального тиску вінцевий кровообіг підтримується на постійному рівні завдяки розслабленню гладких м'язів і розширенню артерій.
Нині показано, що при розтягуванні гладком'язових клітин судин збільшується проникність їхньої плазматичної мембрани до іонів кальцію. Останні проникають у клітини і викликають їх скорочення.
2. Метаболічна регуляція. Підпорядковує вінцевий кровообіг метаболічним потребам серця. Здійснюється за допомогою цілого ряду іонів і метаболітів, серед яких іони водню, калію, молочна кислота, простагландини. Однак найбільше значення мають два фактори: зменшення напруги 02 в артеріальній крові і аденозин. Останній утворюється в результаті гідролізу аденінових нуклеотидів при гіпоксії і при посиленій роботі серця. Будучи природним блокатором Са-каналів, аденозин зменшує надходження іонів Са2+ у цитоплазму гладком'язових клітин вінцевих судин, унаслідок чого зменшується ступінь їхнього скорочення і падає базальний тонус - вінцеві судини розширюються, коронарний кровообіг зростає (рис. 129).
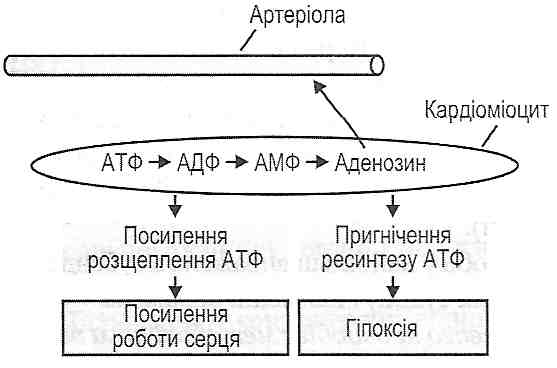
Рис. 129. Один з механізмів метаболічної регуляції вінцевого кровообігу
3. Нервова регуляція. Нейрогенний тонус вінцевих судин незначний, про що свідчить майже повна відсутність змін вінцевого кровообігу після повної денерва-ції судин серця. Набагато більше значення має опосередкований вплив нервової системи на коронарний кровообіг. Він здійснюється через зміни роботи серця та інтенсивності обміну речовин у ньому.
В експерименті показано можливість і безпосереднього впливу нервів на тонус вінцевих судин. Так, при подразненні парасимпатичних нервів і введенні ацетилхоліну відбувається незначне розширення вінцевих артерій. Медіатори симпатичної нервової системи (катехоламіни) при дії на а-адренорецептори викликають звуження судин, а впливаючи на р-адренорецептори — розширення. Оскільки в нормі у вінцевих судинах переважають β-адренорецептори, то загальний ефект симпатичних впливів — незначне розширення судин серця.
27.47. Що таке недостатність вінцевого кровообігу? Чим відрізняються відносна і абсолютна коронарна недостатність?
Недостатність вінцевого кровообігу - це патологічний стан, що характеризується нездатністю вінцевих судин забезпечувати кровопостачання серця відповідно до його енергетичних потреб. Інакше кажучи, виникає невідповідність між енергетичними потребами серця і доставкою кисню і поживних речовин вінцевими судинами.
Недостатність вінцевого кровообігу може бути відносною і абсолютною.
Відносна коронарна недостатність виникає у випадку первинного збільшення енергетичних потреб серця (збільшення навантаження на серце при фізичній роботі, артеріальній гіпертензії). За таких обставин інтенсивність вінцевого кровообігу може зростати, але цього буває замало, щоб задовольнити потреби серця.
Абсолютна коронарна недостатність виникає у випадку первинного порушення вінцевого кровообігу, у результаті чого зменшується доставка кисню і поживних речовин міокарду як у стані спокою, так і при збільшенні енергетичних потреб серця.
27.48. Які патогенетичні фактори можуть обумовлювати розвиток абсолютної недостатності вінцевого кровообігу?
Інтенсивність вінцевого кровообігу визначається формулою
![]()
де Q — об'ємна швидкість течії крові; Ра і Рвсн — тиск крові відповідно на початку і в кінці системи вінцевих судин; (Ра - Р ) - перфузійний тиск; R - опір вінцевих судин.
Оскільки абсолютна коронарна недостатність характеризується зменшенням інтенсивності вінцевого кровообігу (зменшенням Q), можливими є два патогенетичних варіанти її розвитку.
І. Зменшення перфузійного тиску. При цьому розвивається коронарна недостатність центрального походження. її причинами можуть бути:
а) артеріальна гіпотензія (зменшення Ра п), наприклад, при всіх видах шоку, колапсі, недостатності аортальних клапанів. При зменшенні артеріального тиску спрацьовують механізми міогенної ауторегуляції вінцевого кровообігу, що підтримують його на постійному рівні. Однак, коли артеріальний тиск падає нижче 70 мм рт. ст., ці механізми виявляються неспроможними;
б) порушення венозного відтоку (збільшення Рвен). В експерименті моделюють перев'язуванням вен серця. Може мати значення при декомпенсованій недостатності правого шлуночка, коли збільшується центральний венозний і кінцеводіастолічний тиск.
II. Збільшення опору вінцевих судин. При цьому розвивається коронарна недостатність місцевого походження. Оскільки
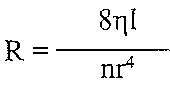
де tj — в'язкість крові; 1 — довжина судин; г — радіус судин, то збільшення опору може бути обумовлене:
а) збільшенням в 'язкості крові при порушенні її реологічних властивостей (зневоднення, поліцитемія, ДВЗ-синдром). При цьому розвиваються порушення мікроциркуляції, аж до сладж-синдрому та істинного капілярного стазу; : б) зменшенням радіуса судин. Це основний фактор розвитку абсолютної коронарної недостатності. Він викликає ішемію серця.
27.49. Які механізми можуть лежати в основі розвитку ішемії міокарда?
I. Обтураційний механізм — зменшення просвіту вінцевих артерій. Його причинами можуть бути:
а) стенозуючш атеросклероз (є причиною ішемії міокарда у 90 % випадків);
б) тромбоз вінцевих артерій (найчастіше є наслідком атеросклерозу);
в) емболія вінцевих артерій;
г) запальні процеси в стінці судин серця — коронаріїти. Бувають при ревматизмі, сифілісі.
Для розвитку клінічних ознак ішемії міокарда має значення величина "критичного стенозу ". Це мінімальне зменшення просвіту судин, при якому виникає ішемія. У людини цей показник становить 75 % (у свиней - менше, у собак - більше).
II. Ангіоспастичний механізм- спазм вінцевих судин. Його причинами можуть бути:
а) збудження а-адренорецепторів на тлі блокади р-адренорецепторів;
б) вазопресин;
в) ангіотензин II;
г) тромбоксан А^;
ґ) гіпокапнія;
д) ендотелій - біологічно активна речовина ендотеліального походження. Існує клінічна форма коронарного ангіоспазму — стенокардія Принцметала.
III. Компресійний механізм — здавлювання вінцевих судин. Може мати місце при тахікардії (збільшується загальна тривалість періоду здавлювання вінцевих судин під час систоли серця). Іноді його причиною є рубці і пухлини. В експерименті використовують для моделювання ішемії й інфаркту міокарда шляхом накладення лігатури на вінцеві артерії.
27.50. Які патогенетичні фактори впливають на міокард в умовах коронарної недостатності?
1. Гіпоксія.
2. Ацидоз. Розвивається в результаті накопичення кислих продуктів обміну речовин унаслідок порушення відтоку крові і активації гліколізу.
3. Збільшення позаклітинної концентрації іонів калію в зоні ішемії. Виявляють від самого початку порушень вінцевого кровообігу (через кілька хвилин), коли ще немає ушкодження кардіоміоцитів. У цей період з невідомих ще причин відбувається пасивний вихід іонів калію з клітин (можливо, збільшується проникність К-каналів), і його позаклітинна концентрація зростає до 8 ммоль/л і більше. Через декілька годин "витікання" іонів К+ із клітин може бути обумовлене порушенням роботи Na-K-насосів (дефіцит АТФ) та збільшенням проникності ушкоджених мембран.
27.51. Які наслідки для міокарда може мати недостатність вінцевого кровообігу?
1. Порушення скорочувальної здатності міокарда з розвитком недостатності серця.
2. Поява аномальної електричної активності — електрична нестабільність серця, розвиток аритмій.
3. Ушкодження кардіоміоцитів, обумовлене ішемією (див. розд. 11).
4. Реперфузійний синдром.
27.52. Які порушення скорочувальної функції міокарда можуть виникати при його ішемії?
При ішемії міокарда розрізняють ранні і пізні порушення його скорочувальної функції.
Ранні порушення виникають дуже рано, вже при незначному дефіциті АТФ. Вони є відображенням гіпокальцієвого варіанта порушень скорочувальної функції серцевого м'яза і розвиваються в результаті блокади Са-каналів сарколеми кардіоміоцитів. Порушення провідності Са-каналів в умовах ішемії має щонайменше два механізми:
а) дефіцит АТФ → порушення фосфорування білків Са-каналів;
б) активація гліколізу → накопичення в клітинах іонів Н+ → безпосередня блокада Са-каналів.
Наслідком зазначених порушень є зменшення надходження іонів Са2+ у саркоплазму м'язових волокон, розлади електромеханічного спряження, зменшення сили скорочень кардіоміоцитів.
Пізні порушення скорочувальної функції виникають при тривалих розладах вінцевого кровообігу - понад ЗО хв. їх відносять до гіперкальцієвого (контрактурного) типу порушень. Концентрація іонів кальцію в саркоплазмі кардіоміоцитів збільшується через дві основні причини:
а) порушення видалення іонів Са2+ із саркоплазми внаслідок дефіциту АТФ;
б) збільшення надходження іонів Са2+ у м'язові волокна через ушкоджену плазматичну мембрану клітин.
У результаті збільшення вмісту Са2+ порушуються процеси розслаблення кардіоміоцитів, настає контрактура міофібрил, порушується скорочувальна здатність серця.
Ішемічна хвороба серця: види, етіологія, патогенез, клінічні прояви. Патогенез проявів та ускладнень інфаркту міокарда.
Ішемічна хвороба серця — це хвороба, що розвивається в результаті абсолютної недостатності вінцевого кровообігу й виявляється ушкодженнями міокарда різного ступеня тяжкості.
її основними клінічними формами є:
1) стенокардія — напади короткочасної (до 20 хв.) гострої коронарної недостатності, які супроводжуються больовим синдромом, відчуттям страху і пов'язаними з цим вегетативними реакціями.
Розрізняють стенокардію напруги, стенокардію спокою і стенокардію Принцме-тала (спазм вінцевих артерій);
2) передінфарктний стан (проміжний коронарний синдром, або гостра вогнищева дистрофія міокарда) — розвивається при тривалості'ішемії міокарда від 20 до 40 хв.;
3) інфаркт міокарда— некроз серцевого м'яза, обумовлений порушеннями вінцевого кровообігу. Виникає при оборотній (транзиторній) ішемії, що триває понад 40-60 хв.? або при необоротних порушеннях коронарного кровообігу;
4) кардіосклероз — склеротичні зміни серцевого м'яза. Можуть бути дифузними (атеросклеротичний кардіосклероз) і вогнищевими (постінфарктний кардіосклероз).
27.56. Назвіть основні причини розвитку інфаркту міокарда.
1. Атеросклероз вінцевих артерій (див. розд. 28). Його розвиток супроводжується порушенням постачання міокарда киснем.
2. Збільшення навантаоїсення на серце (фізична напруга, артеріальна гіпертензія). При цьому збільшується потреба серця в кисні.
3. Стрес (див. розд. 33).
27.57. Яка роль катехоламінів у розвитку інфаркту міокарда?
У розвитку інфаркту міокарда можуть мати значення такі ефекти катехоламінів.
1. Порушення вінцевого кровообігу. Це пов'язане з тим, що катехоламіни:
а) сприяють розвитку атеросклерозу (стрес і катехоламіни, що беруть участь у його реалізації, є фактором ризику цієї хвороби);
б) викликають контрактурний спазм гладких м'язів вінцевих артерій (катехо-ламінове ушкодження клітин судинної стінки);
в) активують тромбоутворення і зсідання крові.
2. Збільшення потреби серця в кисні. Енергетичні потреби серця зростають у результаті реалізації позитивного іно- і хронотропного ефектів катехоламінів та
збільшення загального периферичного опору, внаслідок чого збільшується навантаження на серце. 3. Некоронарогенні катехоламіновіушкодження кардіоміоцитів. Вони не пов'язані з порушенням вінцевого кровообігу. Виникають у результаті того, що великі дози катехоламінів активують ліпідні і кальцієві механізми ушкодження клітин (див. розд. 11).
27.58. Які клінічні синдроми характерні для інфаркту міокарда ?
1. Больовий синдром.
2. Гостра серцева недостатність. Розвивається при уражені великих ділянок міокарда. Може виявляти себе синдромом серцевої астми і набряку легень або карді-огенним шоком.
3. Аритмічний синдром. Можливий розвиток усіх видів аритмій. Найнебезпечнішою є поява фібриляції шлуночків.
4. Резорбційно-некротичний синдром.
27.59. Які механізми розвитку і значення больового синдрому при інфаркті міокарда?
У розвитку больового синдрому при некрозі серцевого м'яза мають значення:
а) хімічні фактори, що з'являються в тканинах при ушкодженні клітин. Серед них іони Н+, К+, простагландини, лізосомні ферменти;
б) зміни скорочувальних властивостей ішемізованої ділянки міокарда, у результаті чого відбувається патологічне розтягування (пролабування) стінки серця при його скороченні. Це веде до подразнення механорецепторів серця і розвитку болю.
Патогенетичне значення больового синдрому в розвитку інфаркту міокарда полягає в тому, що:
1) біль є потужним чинником ініціації стресу і активації симпатоадреналової системи. Великі дози катехоламінів, що вивільняються при цьому, сприяють ушкодженню міокарда (див. запит. 27.57);
2) сильний біль викликає спочатку збудження, а потім і перезбудження життєво важливих центрів головного мозку (дихального, серцево-судинного). Ця обставина є важливим чинником розвитку кардіогенного шоку
27.60. Що таке кардіотенний шок? У яких формах він може виявляти себе?
Кардіогенний шок — це шок, що виникає в результаті різкого падіння нагнітальної (насосної) функції серця. Це - найнебезпечніше ускладнення інфаркту міокарда, що часто призводить до смерті.
Розрізняють 4 форми кардіогенного шоку. 1. Рефлекторна форма (больовий шок). Основним механізмом її розвитку є тривалий біль, що викликає активацію симпатоадреналової системи, яка переходить у гальмування. Це призводить до депресії скорочувальної функції серця, брадикардії, зменшення тонусу периферичних судин і падіння артеріального тиску.
2. Гіпокінетична форма (істинний кардіогенний шок). Основним фактором її розвитку є різке зменшення скорочувальної функції серця в результаті ішемічного ушкодження кардіоміоцитів. Істинний кардіогенний шок розвивається, коли площа ураженого міокарда перевищує 40 %.
3. Дискінетична форма. Виникає в результаті асинергії (неузгодженості) скорочень міокарда. Причиною такої асинергії є грубі ушкодження серця - аневризми, розрив міжшлуночкової перегородки, відрив хорд клапанів.
4. Аритмічна форма. Є наслідком важких аритмій.
27.61. Який патогенез кардіогенного шоку?
У патогенезі кардіогенного шоку розрізняють кілька етапів.
I етап - первинне падіння артеріального тиску. Всі патогенетичні фактори кардіогенного шоку (рефлекторна депресія, збільшення площі ушкодженого міокарда, асинергія серцевих скорочень, аритмії) викликають зменшення серцевого ви-штовху. Це, за законами гемодинаміки, призводить до зменшення хвилинного об'єму серця і падіння артеріального тиску.
II етап - компенсаторний спазм артеріол. Характеризується активацією сим-патоадреналової системи, надходженням у кров катехоламінів, вазопресину, глкжо-кортикоїдів, утворенням ангіотензину II. Вивільнення потужних судинозвужувальних факторів викликає генералізований спазм артеріол, у результаті чого збільшується загальний периферичний опір. Зазначена реакція є компенсаторною і спрямована на попередження подальшого падіння артеріального тиску.
III етап - вторинне падіння артеріального тиску. Тривалий спазм артеріол у периферичних тканинах викликає порушення мікроциркуляції і гіпоксію. Наслідком кисневого голодування є:
а) ацидоз, що викликає депресію скорочувальної функції міокарда;
б) розширення артеріол, що виникає в результаті накопичення в тканинах метаболі-тів-вазодилататорів ( "метаболічний симпатоліз ");
в) надходження у кров із тканин так званих ішемічних токсинів. Серед них велике патогенетичне значення має фактор депресії міокарда, що вивільняється з підшлункової залози.
Усі зазначені зміни, погіршуючи скорочувальну функцію серця й "знімаючи" компенсаторний спазм артеріол, викликають подальше падіння артеріального тиску.
IV етап - термінальні зміни. У результаті істотного падіння артеріального тиску (нижче 40 мм рт. ст.):
а) ще більше порушується коронарний кровообіг і збільшується ішемія міокарда -зменшення скорочувальної функції міокарда прогресує;
б) розвивається гостра ниркова недостатність (повністю припиняється клубочко-ва фільтрація, виникають анурія, інтоксикація);
в) порушується мозковий кровообіг, розвивається гіпоксія головного мозку, виникають розлади функції життєво важливих центрів.
Сукупність зазначених змін призводить до смерті.
27.62. У чому сутність резорбційно-некротичного синдрому, що розвивається при інфаркті міокарда?
Резорбційію-некротіїчний синдром при інфаркті міокарда є наслідком надходження в кров продуктів розпаду змертвілої тканини серця. Він виявляє себе такими ознаками:
а) гарячкою (див. розд. 15);
б) нейтрофільним лейкоцитозом;
в) збільшенням швидкості осідання еритроцитів (ШОЕ);
г) ферментемією — появою в крові ферментів, що надходять із ушкоджених кардіоміо-цитів (креатинкіназа, аспартатамінотрансфераза, лактатдегідрогеназа І типу та ін.);
j) аутоімунним синдромом (синдромом Дреслера). Розвивається в результаті кон-формаційних змін білків міокарда. Виявляється запаленням серозних оболонок організму - полісерозитом (перикардитом, плевритом, перитонітом).
27.63. Що таке некоронарогенні некрози серця? Як їх моделюють в експерименті?
Некоронарогеншіми називають некрози серця, що виникають не в результаті недостатності вінцевого кровообігу, а через інші причини.
Існує кілька експериментальних моделей некрозу серцевого м'яза, причина виникнення якого не пов'язана з патологією вінцевих судин. Ці моделі певною мірою відбивають ситуацію, що спостерігається в природних умовах.
1. "Гіпоксичний некроз міокарда. Може бути відтворений за допомогою різних видів гіпоксії: гіпоксичної, гемічної. При цьому на тлі загальної недостатності кисню в організмі, що сама по собі веде до підвищення навантаження на систему кровообігу, розвивається некротичне ушкодження м'язових волокон серця. Розвитку некрозу сприяє фіксація тварини в незручній позі, наприклад, розтягування у верстаті, або додаткове навантаження — біг у тредбані.
2. Електролітно-стероїдна кардіопатія з некрозом. За спостереженнями Сельє, при введенні щурам значної кількості солей натрію разом з деякими аніонами (сульфатними, фосфатними) у серці з'являються осередки ушкодження дегенеративно-некротичного типу, що часто супроводжуються гіалінозом судин інших органів. Ці ушкодження стають більшими або виникають при введенні меншої кількості солей, якщо одночасно вводити деякі стероїдні гормони надниркових залоз. На цьому тлі легше розвиваються і мають тяжчий перебіг ушкодження серця, викликані іншими причинами. Так, введення навіть невеликих доз норадреналіну, похідних кальциферолу, гіпоксія, м'язова напруга або, навпаки, значне обмеження рухливості ведуть до розвитку великого некрозу міокарда. Солі калію і магнію при цьому мають захисну дію.
3. Імунні ушкодження серця. Можливі при введенні в організм експериментальної тварини гетерогенної сироватки, що містить антитіла проти білків серця тварини даного виду (кардіоцитотоксини). Доведено також, що в організмі за певних умов можуть виникати антитіла і сенсибілізовані лімфоцити, які діють на тканини власного серця і спричиняють його ушкодження. Цьому сприяє проникнення в
кров денатурованих компонентів некротизованих кардіоміоцитів. В експерименті аналогічний процес можна викликати введенням тварині суспензії міокарда зі стимулятором імунологічної реакції (ад'ювантом Фрейнда). Серце може бути ушкоджене і циркулюючими імунними комплексами антиген-антитіло, а також при фіксації на його структурах цитофільних антитіл типу IgE з наступною їх реакцією з антигеном. 4. Нейрогенні ураження серця. Дистрофічні зміни і некроз міокарда можна відтворити гострим або хронічним подразненням шийно-грудного вузла симпатичного стовбура, блукаючого нерва, гіпоталамуса, мозкового стовбура або інших відділів головного мозку. Введення в кров великих доз адреналіну або норадреналіну також веде до ураження серця. В основі механізму нейрогенних ушкоджень лежить невідповідність між рівнями функції, метаболізму і кровопостачання. Подразнення серцевих симпатичних нервів супроводжується значним збільшенням споживання кисню міокардом. При цьому збільшення вінцевого кровообігу є недостатнім (відносна коронарна недостатність), а тому розвивається гіпоксія міокарда. При склерозуванні вінцевих артерій невідповідність інтенсивності кровообігу рівневі обміну речовин виявляється ще в більшій мірі, що може виявитися катастрофічним як для серця, так і для організму в цілому.
Аритмії серця. Експериментальне моделювання. Причини, механізми порушень автоматизму, збудливості, провідності, типові електрокардіографічні прояви.
27.17. Що таке аритмії серця? Як їх класифікують?
Аритміями серця називають порушення частоти, ритму, узгодженості й послідовності його скорочень.
Аритмія – порушення ЧСС, ритмічності скорочень, локалізації водія ритму чи провідності (Мурашко В.В., Електрокардіографія).
Розвиток аритмій може бути пов'язаний з порушеннями основних функцій провідникової системи серця: автоматизму, збудливості і провідності. На цьому ґрунтується класифікація аритмій, відповідно до якої виділяють:
I. Аритмії, обумовлені порушеннями автоматизму.
II. Аритмії, пов'язані з порушеннями збудливості.
III. Аритмії, обумовлені порушеннями провідності.
IV. Аритмії, пов'язані з поєднаними порушеннями збудливості і провідності.
27.18. Які аритмії серця можуть виникати в результаті порушення функції автоматизму?
Розрізняють дві групи аритмій, пов'язаних з порушенням автоматизму серця.
I. Номотопні аритмії. Генерація імпульсів до скорочення, як і в нормі, відбувається в синусно-передсердному вузлі. До цієї групи відносять:
а) синусну тахікардію - збільшення частоти серцевих скорочень;
б) синусну брадикардію - зменшення частоти серцевих скорочень;
в) синусну (дихальну) аритмію - зміну частоти серцевих скорочень у різні фази дихального циклу (прискорення при вдиху і уповільнення при видиху).
II. Гетеротопні аритмії - синдром слабкості синусно-передсердного вузла. Генерація імпульсів до скорочення відбувається не в синусно-передсердному вузлі, а в інших структурах провідникової системи, що є водіями ритму II і III порядку.
Синдром розвивається в результаті зменшення активності або припинення діяльності синусно-передсердного вузла при ушкодженні його клітин або первинних функціональних порушеннях. При цьому можуть розвиватися такі види патологічних ритмів серця:
а) передсердний повільний ритм — водій ритму міститься в структурах лівого передсердя, частота серцевих скорочень менше 70/хв.;
б) атріовентрикулярний ритм — джерелом імпульсів є водії ритму II порядку (верхня, середня або нижня частина атріовентрикулярного вузла), частота серцевих скорочень залежно від місця генерації імпульсів зменшується від 70 до 40/хв;
в) ідіовентрикулярний шлуночковий ритм — генерація імпульсів відбувається у водіях ритму III порядку (пучок Гісса або його ніжки), частота скорочень серця менше 40/хв.
27.19. Які причини і механізми розвитку синусної тахі- і брадикардії?
Синусні тахікардія і брадикардія належать до групи номотопних аритмій, пов'язаних з порушеннями функції автоматизму.
Здатність до автоматичного утворення імпульсів залежить від клітин, розташованих у провідниковій системі серця (β-клітини), у яких відбувається спонтанна повільна деполяризація клітинної мембрани в період діастоли (рис. 122). У результаті, при досягненні певного критичного рівня, виникає потенціал дії. Частота генерації імпульсів залежить від трьох чинників: а) максимального діастолічного потенціалу цих клітин; б) рівня критичного потенціалу, після досягнення якого виникає потенціал дії; і в) швидкості діастолічної деполяризації.
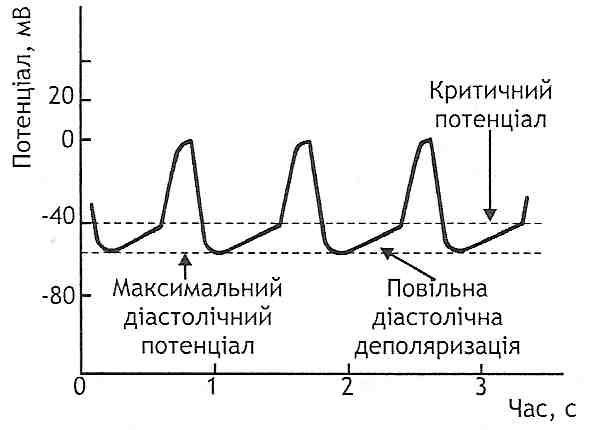
Рис. 122. Спонтанна генерація потенціалів дії в клітинах синусно-передсердного вузла
Зміна рівня максимального діастолічного потенціалу, критичного потенціалу або швидкості діастолічної деполяризації в той чи той бік веде до зміни частоти генерації імпульсів або до появи інших джерел імпульсації, якщо ці зміни виникають в інших, здатних до збудження ділянках серця і призводять до появи в них потенціалів дії. При зменшенні рівня максимального діастолічного потенціалу клітин синусно-передсердного вузла, при наближенні до нього критичного потенціалу або збільшенні швидкості повільної діастолічної деполяризації імпульси генеруються частіше, розвивається тахікардія. Це відбувається під впливом підвищеної температури тіла, при розтягненні ділянки синусно-передсердного вузла, при збудженні симпатичних нервів і дії катехоламінів.
Навпаки, зменшення швидкості повільної діастолічної деполяризації, гіперпо-ляризація в діастолі й віддалення критичного потенціалу від рівня максимального діастолічного, як це спостерігається при подразненні блукаючого нерва, супроводжуються уповільненням генерації імпульсів, а отже, і скорочень серця — брадикардією. Періодичні зміни тонусу блукаючого нерва під час акту дихання можуть викликати дихальну аритмію (прискорення серцебиття при вдиху, уповільнення — при видиху). Дихальна аритмія в нормі буває у дітей, але зрідка може спостерігатися і у дорослих.
27.20. Які аритмії виникають у результаті порушення збудливості міокарда? Який механізм їх розвитку?
В основі аритмій, пов'язаних з порушеннями функції збудливості, лежить поява розташованих поза синусно-передсердним вузлом так званих ектопічних осередків збудження, що генерують позачергові імпульси до скорочення.
У патологічних умовах може виявитися власний автоматизм нижчерозташова-них відділів провідникової системи серця (потенційних водіїв ритму). Такі умови можуть виникнути при зниженні автоматизму синусно-передсердного вузла або при підвищенні здатності до генерації імпульсів в інших ділянках міокарда. У цих випадках частота імпульсів, що генеруються нормальним водієм ритму, виявляється недостатньою для пригнічення автоматизму інших відділів, що призводить до появи додаткових імпульсів з ектопічно розташованих осередків збудження.
Іншим механізмом, що призводить до появи ектопічних осередків збудження, може бути виникнення різниці потенціалів між розташованими поруч міоцитами внаслідок, наприклад, неодночасного закінчення реполяризації в них, що може викликати збудження у волокнах, які вже вийшли з фази рефрактерності. Це явище спостерігається при локальній ішемії міокарда і при отруєнні серцевими глікозидами.
Серед аритмій цієї групи найчастіше бувають екстрасистолія і пароксизмальна тахікардія.
27.21. Що таке екстрасистолія? Назвіть види екстрасистол і їх основні електрокардіографічні характеристики.
Екстрасистолія - це вид аритмій, обумовлених порушеннями функції збудливості, що виявляє себе виникненням позачергових скорочень серця або тільки шлуночків. Такі позачергові скорочення отримали назву екстрасистол.
Залежно від локалізації осередку, з якого виходить позачерговий імпульс, розрізняють кілька видів екстрасистол: синусну (або номотопну), передсердну, передсерд-но-шлуночкову і шлуночкову (рис. 123). Оскільки хвиля збудження, що виникла в незвичному місці, поширюється в зміненому напрямку, це відображається на структурі електричного поля серця й знаходить відображення на електрокардіограмі. Кожний
вид екстрасистоли має свою електрокардіографічну картину, що дає можливість визначити місце ектопічного осередку збудження.
Синусна екстрасистола виникає внаслідок передчасного збудження частини клітин синусно-передсердного вузла. Електрокардіографічно вона не відрізняється від нормального скорочення, за винятком укорочення діасто-лічного інтервалу Т-Р. Унаслідок укорочення діастоли й зменшення наповнення шлуночків пульсова хвиля при екстрасистолі зменшена.
Передсердна екстрасистола спостерігається при наявності осередку ектопічного збудження в різних ділянках передсердь. Характеризується зміною форми зубця Р (знижений, двофазний, негативний) при збереженому комплексі QRS і деякому подовженні діастолічного інтервалу після екстрасистоли. Це обумовлено тим, що, ідучи ретроградним шляхом, збудження передчасно розряджає нормальний синусний імпульс, який збігається у часі зі збудженням шлуночків. Наступний передсердний імпульс, що виникає через нормальний інтервал, виявляється трохи віддаленим у часі від моменту закінчення збудження шлуночків - неповна компенсаторна пауза.
Передсердно-шлуночкова екстрасистола спостерігається при виникненні додаткового імпульсу в передсердно-шлуночковому вузлі. Хвиля збудження, що виходить з верхньої і середньої частин вузла, поширюється у двох напрямках: у шлуночках — у нормальному, у передсердях — у ретроградному. При цьому негативний зубець Р може накладатися на комплекс QRS. Діастолічний інтервал після екстрасистоли трохи подовжений. Екстрасистола може супроводжуватися також одночасним скороченням передсердь і шлуночків. При передсердно-шлуночковій екстрасистолі, що виходить із нижньої частини вузла, виникає компенсаторна пауза, така ж, як і при шлуночковій екстрасистолі.
Для шлуночкової екстрасистоли характерна наявність повної компенсаторної паузи після позачергового скорочення. Вона виникає внаслідок того, що збудження, яке охопило шлуночки, не передається через передсердно-шлуночковий вузол назад на передсердя, водночас черговий нормальний імпульс збудження, що йде від синусно-передсердного вузла, не поширюється на шлуночки, бо вони перебувають у фазі рефрактерності. Наступне скорочення шлуночків виникає тільки після приходу до них чергового нормального імпульсу. Тому тривалість компенсаторної паузи, разом з інтервалом, що передує екстрасистолі, дорівнює тривалості двох нормальних діасто-лічних пауз. Однак якщо скорочення серця настільки рідкі, що до моменту приходу чергового нормального імпульсу шлуночки встигають вийти зі стану рефрактерності, то компенсаторної паузи не буває. Позачергове скорочення потрапляє в інтервал між двома нормальними і в цьому випадку воно має назву вставної екстрасистолії. Оскільки хвиля збудження при шлуночковій екстрасистолії поширюється по шлуночках як у прямому, так і в ретроградному напрямках, це супроводжується значними змінами форми комплексу QRS, його деформацією.
Рис. 123. Екстрасистоли: а — передсердна, б — шлуночкова
27.22. Що таке пароксизмальна тахікардія? Чим вона характеризується?
Пароксизмальна тахікардія - це аритмія, обумовлена порушеннями функції збудливості, яка виявляється виникненням групи екстрасистол, що швидко повторюються і повністю пригнічують фізіологічний ритм.
При пароксизмальній тахікардії нормальний ритм серця раптово переривається нападом скорочень частотою від 140 до 250 ударів за хвилину. Тривалість нападу може бути різною - від кількох секунд до кількох хвилин, після чого він так само раптово припиняється і установлюється нормальний ритм.
Найчастіше спостерігається передсердна форма пароксизмальної тахікардії. А оскільки тривалість потенціалів дії і тривалість рефрактерних періодів збільшується по ходу провідникової системи, то дистально розташовані ділянки її не завжди здатні відтворити частоту імпульсації, що надходить з проксимальних відділів. Тому більша частина імпульсів при передсердній тахікардії не може проводитися передсердно-шлуночковим вузлом. Оскільки тривалість рефрактерних періодів і потенціалів дії у волокнах правої ніжки пучка Гісса більші, ніж у лівій, при високій частоті імпульсів частіше порушується проведення збудження саме до правого шлуночка.
27.23. Які аритмії виникають у результаті порушень функції провідності міокарда ?
Виділяють дві групи аритмій, пов'язаних з порушеннями провідності.
1. Блокади серця. Це аритмії, обумовлені уповільненням або повним припиненням проведення імпульсів по провідниковій системі.
Причиною блокади може бути ушкодження провідникових шляхів, що веде до подовження рефрактерного періоду і уповільнення або повного припиненням проведення імпульсу.
Порушення провідності можуть виникати між синусно-передсердним вузлом і передсердями, усередині передсердь, між передсердями і шлуночками, а також в одній із ніжок пучка Гісса. Тому виділяють такі види блокад:
а) внутрішньопередсердну;
б) передсердно-шлуночкову;
в) внутрішньошлуночкову.
2. Прискорене проведення імпульсів- синдром Вольфа-Паркінсона-Уайта (див. запит. 27.1.25).
27.24. Дайте характеристику передсердно-шлуночкової блокади серця.
Передсердно-шлуночкова, або поперечна, блокада серця може бути повною і неповною. У неповній блокаді серця розрізняють три ступені (рис. 124).
Передсердно-шлуночкова блокада І ступеня характеризується збільшенням часу проведення імпульсу від передсердь до шлуночків, що супроводжується подовженням інтервалу P-Q (0,2-0,5 с).
Блокада II ступеня (періоди Венкебаха) характеризується прогресуючим збільшенням інтервалу P-Q доти, доки одне із збуджень, найчастіше восьме-десяте, не проводиться. Після випадіння скорочення шлуночка інтервал P-Q відновлюється, поступово подовжуючись із кожним наступним скороченням серця. Вважають, що цей феномен пов'язаний з прогресуючим утрудненням проведення імпульсів через вузол.
При блокаді III ступеня спостерігають випадання кожного другого-третього скорочення або, навпаки, проводиться тільки кожне друге, третє або четверте збудження передсердь.
При повній передсердно-шлуночковій блокаді передсердя і шлуночки скорочуються незалежно одне від одного, кожне у своєму ритмі: передсердя з частотою близько 70, шлуночки — близько 35 скорочень в 1 хв. (ідіовентрикулярний ритм).
Особливе значення має момент переходу неповної блокади в повну, коли до шлуночків не надходять імпульси від передсердь. Повільна діастолічна деполяризація в потенційних водіях ритму виникає тільки через якийсь час після припинення надходження імпульсів від синусно-передсердного вузла. Цей період носить назву пре-автоматичної паузи, під час якої спостерігається асистолія шлуночків. При цьому внаслідок припинення надходження крові до головного мозку виникає втрата свідомості, судоми (синдром Морганьї—Едемса—Стокса). Можлива смерть, але найчастіше при поновленні скорочень шлуночків зазначені явища проходять. Синдром може повторюватися багаторазово.
Рис. 124. Передсердно-шлуночкова
блокада: а — І ступеня; б—II ступеня;
в — III ступеня; г - повна поперечна
блокада
27.25. Чим може бути обумовлений розвиток синдрому Вольфа-Паркінсона-Уайта ?
Синдром Вольфа—Паркінсона—Уайта характеризується прискореним проведенням імпульсів від передсердь до шлуночків, у результаті чого відбувається передчасне збудження останніх, розвивається тахікардія, зменшується інтервал P-Q на електрокардіограмі.
Причиною розвитку цього синдрому вважають існування додаткових шляхів проведення імпульсів. До таких шляхів відносять (рис. 125):
а) пучок Паладіно—Кента: видозмінена міокардіальна тканина, локалізована в зоні атріовентрикулярного кільця. Може існувати два пучки (лівий і правий), що містяться відповідно у кільцях двостулкового і тристулкового клапанів. Зазначені пучки проводять імпульси від передсердь до шлуночків, минаючи перед-сердно-шлуночковий вузол;
Рис. 125. Додаткові провідникові шляхи серця:
1 — пучок Паладіно— Кента;
2 — пучок Махайма;
3 — пучок Джеймса
б) пучок Махайма. З'єднує верхню частину пучка Гісса зі шлуночками;
в) пучок Джеймса. Зв'язує передсердя з нижньою частиною передсердно-шлуночкового вузла або з пучком Гісса.
По додаткових провідникових шляхах імпульси проводяться швидше і досягають шлуночків раніше імпульсів, які йдуть звичайним чином через передсердно-шлуноч-ковий вузол. Це призводить до передчасної активації частини шлуночків, інша їх частина збуджується пізніше імпульсами, що проходять через передсердно-шлуноч-ковий вузол. ,
27.26. Які аритмії виникають у результаті одночасного порушення функцій збудливості й провідності?
1. Тріпотіння передсердь (частота скорочень передсердь — 250-400/хв.).
2. Миготіння передсердь (частота імпульсів, що виникають у передсердях, становить 400-600/хв.).
Тріпотіння й миготіння передсердь мають однакові причини розвитку і можуть переходити одне в одне. Тому ці два види порушень ритму серця поєднують одним поняттям — миготлива аритмія.
3. Тріпотіння шлуночків (частота скорочень шлуночків — 150—300/хв.).
4. Миготіння (фібриляція) шлуночків (частота імпульсів у шлуночках- 300-500/хв, серце не скорочується).
27.27. Якими ознаками виявляє себе миготлива аритмія? Які механізми її розвитку?
При наявності численних ектопічних осередків збудження і такої зміни проведення імпульсу, при якій порушується швидкість його проведення по різних ділянках міокарда або відбувається поширення імпульсу тільки в одному напрямку, створюються умови для
тривалої циркуляції хвилі збудження в певному відділі серця, виникають розлади ритму —миготлива аритмія, що може виявлятися тріпотінням [миготінням передсердь (рис. 126).
Рис. 126. Миготлива аритмія
При тріпотінні передсердь частота їхніх скорочень досягає 250—400/хв. При цьому внаслідок нездатності шлуночків відтворювати високий ритм передсердь розвивається відносна серцева блокада; шлуночки відповідають скороченням на кожне друге, третє або четверте скорочення передсердь, тому що інші хвилі збудження потрапляють у фазу рефрактерності. Скорочення шлуночків можуть виникати раніше, ніж настане достатнє наповнення їх кров'ю - це викликає важкі порушення кровообігу.
Якщо кількість імпульсів у передсердях досягає 400-600/хв, говорять про миготіння, або фібриляцію передсердь. При цьому скорочуються лише окремі м'язові волокна, а все передсердя перебуває в стані неповного скорочення, його участь у перекачуванні крові припиняється. Імпульси, що безладно приходять до передсерд-но-шлуночкового вузла по окремих м'язових волокнах передсердя, здебільшого нездатні викликати його збудження, тому що застають вузол у стані рефрактерності або не досягають критичного рівня. Тому передсердно-шлуночковий вузол збуджується нерегулярно, і скорочення шлуночків носять випадковий характер. Як правило, число скорочень шлуночків за хвилину перевищує нормальне. Нерідко скорочення шлуночків відбуваються до їх наповнення кров'ю й не супроводжуються пульсовою хвилею. Тому частота пульсу стає меншою за частоту скорочень серця (дефіцит пульсу). Найчастіше причиною розвитку миготливої аритмії є (правило 03):
а) стеноз лівого атріовентрикулярного отвору,
б) тиреотоксикоз;
в) атеросклеротичний кардіосклероз.
Нині найбільш визнаною теорією, що пояснює механізми розвитку миготливої аритмії (і екстрасистолії!), є теорія повторного входження імпульсів (re-entry) (рис. 127). Відповідно до неї, тріпотіння і миготіння виникають як наслідок порушень провідності, при яких поширення імпульсів припиняється тільки в одному (антеградному) напрямку і зберігається у зворотному (ретроградному). При цьому створюються умови для постійного руху імпульсів по колу в міокарді. У нормальних умовах хвиля збудження, виникнувши в одному місці, поширюється в обидва боки серцевої камери. Досягнувши протилежної стінки, вона згасає, зустрівшись з іншою хвилею, що залишила за собою зону рефрактерності. Якщо ж унаслідок виникнення тимчасового блоку або запізнення приходу збудження по деяких волокнах міокарда збудження приходить до місця, що вже вийшло зі стану рефрактерності, то створюються умови для тривалеї циркуляції імпульсу, що виник.
Рис. 127. Механізм повторного входження імпульсів
27.28. Що таке фібриляція шлуночків? Коли вона виникає й чим виявляє себе?
При деяких патогенних впливах (проходження електричного струму через серце, наркоз хлороформом або циклопропаном, закупорка вінцевих артерій або інші випадки різкої гіпоксії, травма серця, дія токсичних доз наперстянки і кальцію) виникає фібриляція шлуночків. При цьому внаслідок хаотичного скорочення окремих м'язових волокон пропульсивна сила скорочень практично зникає, кровообіг припиняється, швидко настає втрата свідомості і смерть. Фібриляції сприяють зменшення концентрації внутрішньоклітинного калію, що веде до зниження мембранного потенціалу кардіоміоцитів і легшого виникнення в них деполяризації та збудження, а також зміна вмісту нервових медіаторів, особливо катехоламінів.
При лікуванні фібриляції шлуночків найефективнішим є пропускання через серце короткого сильного одиничного електричного розряду (дефібриляція). При цьому відбувається одночасна деполяризація всіх волокон міокарда і припиняються асинхронні збудження м'язових волокон. Як захід, що попереджає розвиток фібриляції, застосовують корекцію електролітного складу крові.
27.29. Чим може бути обумовлений розвиток порушень скорочувальної функції міокарда ?
У клітинах робочого міокарда відбувається п'ять процесів, які визначають скорочувальну функцію серця. Усі вони можуть порушуватися і становити основу розвитку недостатності серця.
Розлади скорочувальної функції міокарда можуть бути обумовлені порушеннями:
1) збудження кардіоміоцитів;
2) електромеханічного спряження;
3) процесів власне скорочення;
4) процесів розслаблення;
5) енергозабезпечення міокарда.
27.30. Які фактори впливають на характер потенціалу дії і збудливість кардіоміоцитів? Якими електрофізіологічними ознаками виявляють себе зміни їхньої збудливості?
Основні характеристики потенціалу дії (тривалість, амплітуда, крутість наростання деполяризації) і збудливість кардіоміоцитів залежать від таких чинників (рис. 128):
Рис. 128. Потенціал дії скорочувальних клітин міокарда: І—фаза швидкої деполяризації; II- фаза повільноїреполяризації (фаза "плато "); III — фаза швидкої реполяризації
а) характеру електричної імпульсації, що надходить до кардіоміоцитів по провідниковій системі серця. При аритміях можливе порушення основних параметрів потенціалу дії волокон робочого міокарда і його збудливості;
б) стану (властивостей) сарколеми кардіоміоцитів, і насамперед іонних каналів. Нині відомі хімічні агенти, здатні вибірково порушувати провідність цих каналів. Це блокатори Na-каналів, Са-каналів, К-каналів;
в) концентрації позаклітинних іонів, що беруть участь у формуванні електричних потенціалів на мембрані кардіоміоцитів. В умовах in vivo найбільше значення має позаклітинна концентрація іонів калію.
Зміни збудливості волокон міокарда можуть виявляти себе:
1) змінами тривалості потенціалів дії, а отже, і сили серцевих скорочень. Зменшення тривалості потенціалів дії виникає при збільшенні частоти стимуляції, збільшенні позаклітинної концентрації іонів калію, при дії ацетилхоліну. Збільшення тривалості потенціалу дії характерно для охолодження;
2) змінами тривалості періодів абсолютної і відносної рефрактерності, а отже, здатності сприймати імпульсацію (засвоювати ритм). Тривалість цих періодів залежить від тривалості потенціалів дії.
27.31. Як впливають зміни позаклітинної концентрації іонів калію на збудливість міокарда?
Позаклітинна концентрація іонів калію впливає на збудливість міокарда через рівень потенціалу спокою кардіоміоцитів.
Збільшення концентрації іонів калію — гіперкаліємія - викликає деполяризацію мембрани м'язових волокон. При цьому характер змін збудливості міокарда залежить від рівня гіперкаліємії. Виділяють такі діапазони концентрацій іонів калію, для яких характерні певні картини порушень:
1) від 4 до 8 ммоль/л. Відбувається незначна деполяризація, мембранний потенціал падає від -90 мВ до -80 мВ. При цьому стан Na-каналів сарколеми істотно не змінюється. Унаслідок того, що величина мембранного потенціалу наближається до критичного рівня деполяризації, збудливість м'язових волокон і швидкість проведення імпульсів зростають;
2) від 8 до 35 ммоль/л. Мембранний потенціал падає від -80 мВ до -40 мВ. При такому рівні деполяризації значно зменшується провідність швидких потенціал-залежних Na-каналів (період відносної рефрактерності). Наслідком є зменшення збудливості і провідності, а також зміни характеру потенціалів дії (зменшення тривалості, амплітуди, крутості);
3) понад 35 ммоль/л. Мембранний потенціал стає менше -40 мВ. При цьому всі швидкі потенціалзалежні Na-канали перебувають у стані інактивації (абсолютна рефрактерність) — відбувається зупинка серця.
Зменшення позаклітинної концентрації іонів калію — гіпокаліємія — викликає гі-перполяризацію мембрани. Якщо гіпокаліємія досягає рівня 2—3 ммоль/л, дещо збільшується поріг деполяризації, зменшується збудливість, збільшуються тривалість потенціалів дії і сила скорочень серця.
27.32. Що таке кардіоплегія? Які використовують підходи до її здійснення?
Кардіоплегія — це штучна зупинка серця, яку застосовують під час операцій на "сухому" серці.
Розроблено такі підходи до здійснення кардіоплегії: а) ішемічна кардіоплегія. Перетискають аорту до відходження вінцевих артерій. Оскільки через 30 хв. починаються необоротні зміни в міокарді, цей підхід можна використати при короткочасних операціях (до 10—15 хв.);
б) хімічна кардіоплегія. У вінцеві артерії вводять кардіоплегічні розчини, до складу яких входять високі концентрації іонів калію, ацетилхолін і деякі інші препарати. Цей метод дозволяє проводити операції на "сухому" серці протягом 40-60 хв.;
в) холодова кардіоплегія. її досягають зрошенням серця охолодженим до +4 °С... — 5 °С фізіологічним розчином. Тривалість операцій на серці при цьому може становити 60 хв.
27.33. Які фактори впливають на характер електромеханічного спряження в міокарді?
1. Тривалість потенціалу дії.
2. Частота потенціалів дії.
3. Концентрація іонів кальцію в позаклітинній рідині.
4. Стан Са-каналів сарколеми кардіоміоцитів.
5. Стан систем видалення іонів кальцію із цитоплазми м'язових волокон.
27.34. Як зміни тривалості і частоти потенціалів дії впливають на силу серцевих скорочень?
Зміни тривалості потенціалів дії в кардіоміоцитах відбуваються за рахунок укорочення або подовження фази "плато ", під час якої іони Са2+ через потенціалзалеж-ні Са-канали сарколеми надходять у саркоплазму.
З урахуванням того, що концентрація іонів Са2+ у цитоплазмі м'язових волокон визначає кількість актоміозинових комплексів, що утворюються, а отже, і силу скорочень, можна зробити такий висновок. При зменшенні тривалості потенціалу дії зменшується надходження іонів Са2+ у саркоплазму і, як наслідок, зменшується сила скорочення м'язового волокна. При збільшенні тривалості потенціалу дії — все навпаки.
Тривалість потенціалу дії кардіоміоцитів зменшується при гіперкаліємії, при дії на м'язові волокна ацетилхоліну. Збільшення тривалості потенціалу дії характерне для гіпокаліємії, охолодження серця.
При збільшенні частоти потенціалів дії, незважаючи на те, що тривалість кожного окремого потенціалу, а отже, і фази "плато" зменшується, сумарна тривалість зазначеної фази за одиницю часу збільшується. Це означає, що в кардіоміоцитах створюються більш високі концентрації Са2+ і, як наслідок, збільшується сила скорочення м'язових волокон. Цим, зокрема, пояснюється хроноінотропний механізм негайної компенсації серця (див. запит. 27.10).
При зменшенні частоти потенціалів дії (наприклад, при брадикардії) сумарна тривалість фази "плато" за одиницю часу зменшується, що веде до зменшення концентрації Са2+ у саркоплазмі і зменшення сили скорочень серця.
27.35. Як впливає позаклітинна концентрація іонів кальцію на силу скорочень серця? Що таке "кальцієвий парадокс"?
На початку XX ст. Рінгером було показано, що в розчині, позбавленому іонів кальцію, ізольоване серце швидко зупиняється. Нині відомо, що причиною цього є повне роз'єднання збудження і скорочення. В умовах норми іони кальцію, що надходять під час фази "плато" потенціалу дії з позаклітинного середовища в саркоплазму
кардіоміоцитів, "запускають" вивільнення Са2+ із саркоплазматичного ретикулуму й поповнюють його запаси в цих структурах м'язових волокон. При зменшенні позаклітинної концентрації Са2+ його запаси в саркоплазматичному ретикулумі швидко виснажуються, концентрація Са2+ у саркоплазмі падає й, отже, зменшується сила серцевих скорочень.
"Кальцієвий парадокс" — це експериментальний феномен, що розвивається після того, як у безкальцієвий розчин, яким здійснювали перфузію серця протягом декількох хвилин, вносять іони кальцію. При цьому розвивається необоротне ушкодження міокарда: зменшується вміст АТФ і креатинфосфату, з кардіоміоцитів виходять білки, у тому числі ферменти (міоглобін, креатинкіназа), руйнується саркоплаз-матичний ретикулум.
"Кальцієвий парадокс" пояснюють тим, що в безкальцієвому розчині відбувається розшарування глікокалікса кардіоміоцитів (зовнішній шар глікокалікса відокремлюється від внутрішнього), у результаті чого значно зростає проникність сарколеми до іонів кальцію. При наступному внесенні в середовище іонів Са2+ відбувається їх масивне надходження у клітини, різко зростає його концентрація в саркоплазмі, що "запускає" кальцієві механізми ушкодження (див. розд. 11).
27.36. Які фактори впливають на стан кальцієвих каналів сарколеми кардіоміоцитів? Чим можуть бути обумовлені їх активація і блокада?
У сарколемі кардіоміоцитів є потенціолзалежні повільні Са-канали. Це білкові молекули, вмонтовані в плазматичну мембрану і здатні пропускати через себе іони кальцію в тому випадку, коли вони відкриті. Відкриття Са-каналів відбувається при деполяризації мембрани. Відкриватися здатні не всі наявні канали, а тільки фосфо-ровані (активовані).
Під активацією Са-каиалів розуміють їх фосфорування, у результаті чого збільшується кількість Са-каналів, здатних відкриватися при виникненні потенціалу дії. В основі фосфорування Са-каналів лежить збільшення концентрації цАМФ у саркоплазмі м'язових волокон. Усі фактори, що викликають утворення цАМФ, ведуть до активації Са-каналів. До таких, зокрема, належать:
а) катехоламіни і фармакологічні агенти, що активують fi-адренорецептори;
б) інгібітори фосфодіестерази (метилкеантини: теофілін, кофеїн та ін.). Зазначені агенти збільшують вміст цАМФ або через активацію аденілатциклази (через 0-адренорецептори), або через пригнічення руйнування цАМФ (інгібітори фосфодіестерази).
З активацією Са-каналів пов'язаний позитивний інотропний ефект катехола-мінів/Q основі його така послідовність подій: катехоламіни → активація р-адрено-рецепторів → активація аденілатциклази → утворення цАМФ → активація проте-їнкіназ → фосфорування Са-каналів сарколеми → збільшення надходження Са2+ у саркоплазму під час потенціалів дії → збільшення концентрації Са2+ у саркоплазмі → збільшення сили скорочень серця.
В умовах блокади Са-капалів зменшується надходження іонів Са в саркоплазму кардіоміоцитів і, отже, зменшується сила серцевих скорочень.
Блокаду Са-каналів викликають:
а) ендогенні фактори — дефіцит АТФ, дефіцит цАМФ, іони водню (ацидоз);
б) екзогенні фактори — двовалентні іони (Ni2+, Со2+, Мп2+), деякі тривалентні іони (La3+), органічні сполуки, що їх застосовують у практичній медицині (верапаміл, ніфедипін та ін.).
27.37. Які фактори впливають на стан систем видалення іонів кальцію з кардіоміоцитів і, як наслідок, на силу серцевих скорочень?
До таких чинників відносять:
а) серцеві глікозиди — фармакологічні препарати рослинного походження, що підвищують силу скорочень серця (препарати дигіталісу, строфантин та ін.);
б) ендогенні дигіталісоподібні і строфантиноподібні фактори. Вважають, що серцеві глікозиди, яких здавна використовують в медицині, імітують дію цих, відносно недавно відкритих, ендогенних природних факторів. Фізіологічне значення останніх сьогодні ще не встановлено.
Механізм дії серцевих глікозидів і подібних до них ендогенних факторів пов'язаний з пригніченням активності Na-K-АТФ-ази кардіоміоцитів. Наслідком цього є порушення роботи Na-K-насосів сарколеми, зменшення градієнта концентрацій іонів Na+ по обидва боки плазматичної мембрани м'язових волокон. Це призводить до порушення Na-Ca-обмінного механізму в кардіоміоцитах, у результаті чого зменшується видалення Са2+ із саркоплазми в позаклітинне середовище і збільшується внутрішньоклітинна концентрація Са2+. Останнє і обумовлює збільшення сили серцевих скорочень.
27.38. Від чого залежить сила скорочень окремих кардіоміоцитів?
На силу скорочень м'язових волокон серця впливають:
1) концентрація іонів Са2+у саркоплазмі. Залежність тут така: що вищий вміст Са2+ у саркоплазмі, то більше утворюється комплексів Са2+ з тропоніном С, то більше вивільняється центрів зв'язування (активних центрів) на актинових міофіламентах, то більше утворюється "містків" між актином і головками міозину, то більшою буде сила скорочення м'язового волокна. При зменшенні концентрації Са2+ у саркоплазмі — навпаки;
2) ступінь спорідненості тропоніну С до іонів кальцію. Іони водню й неорганічного фосфату, зв'язуючись з тропоніном С, унеможливлюють взаємодію цього білка з Са2+, у результаті чого сила скорочень кардіоміоцитів зменшується;
3) стан скорочувальних білків — актину і міозину. Велике значення має взаємне розташування актинових і міозинових міофіламентів. Воно лежить в основі залежності, що її описує закон Франка-Старлінга. При дуже сильному розтягненні м'язових волокон кількість утворюваних актоміозинових "містків" зменшується - зазначений закон не "спрацьовує", сила скорочень серця падає;
4) концентрація АТФ, енергія гідролізу якого забезпечує ковзання м'язових філамен-тів один відносно одного.
27.39. Що лежить в основі розслаблення кардіоміоцитів? Які механізми забезпечують видалення іонів кальцію із саркоплазми волокон міокарда? Що може бути причиною порушень розслаблення серцевого м'яза?
Основний процес, що визначає розслаблення кардіоміоцитів, — це видалення іонів кальцію із саркоплазми, у результаті чого концентрація Са2+ у ній зменшується і стає нижче 10"7 моль/л. При цьому комплекси Са2+ з тропоніном С розпадаються, тро-поміозин зміщується по відношенню до актинових філаментів і закриває їхні активні центри — скорочення припиняється.
Існує три механізми видалення іонів Са2 + із саркоплазми кардіоміоцитів:
1) Са-насоси плазматичної мембрани і саркоплазматичного ретикулуму. Видаляють Са2+ у позаклітинне середовище і цистерни саркоплазматичного ретикулуму. Складовою їх частиною є Са-АТФ-аза, яка для здійснення активного транспорту іонів Са2+ використовує енергію АТФ;
2) Na-Ca-обмінний механізм. Видаляє іони Са2+ у позаклітинне середовище. Є різновидом вторинного активного транспорту (антипорту). Використовує енергію градієнта концентрацій іонів натрію по обидва боки плазматичної мембрани, тому залежить від роботи Na-K-насоса, що створює цей градієнт;
3) Са-акумулятивна функція мітохондрій. Активується тільки при значному підвищенні вмісту іонів Са2+ у саркоплазмі, що найчастіше буває в умовах патології. Видалення Са2+ із саркоплазми в матрикс мітохондрій відбувається за рахунок енергії, що вивільняється в процесі транспорту електронів по дихальному ланцюгу. Використання цієї енергії на активний транспорт іонів Са2+ у мітохондрії є альтернативою окисному фосфоруванню.
Основними причинами порушень розслаблення кардіоміоцитів є:
а) дефіцит АТФ. При цьому порушується енергозабезпечення Са- і Na-K-насосів, а також не відбувається розщеплення актоміозинових "містків", що утворилися в процесі скорочення;
б) порушення роботи Са-транспортних систем. Відомі спадково обумовлені дефекти білків Са-насосів, що призводять до розвитку кардіопатії.
Порушення розслаблення кардіоміоцитів виявляють себе розвитком м'язових контрактур.
27.40. Яке значення має АТФ у забезпеченні функцій клітин міокарда? Чим можуть бути обумовлені порушення енергетичного обміну в серцевому м 'язі?
Енергія гідролізу АТФ у функціонально активних кардіоміоцитах забезпечує:
1) механічну роботу скорочень міофібрил (ковзання міофіламентів один відносно одного);
2) осмотичну роботу — активний транспорт іонів Са2+, Na +, К+ проти градієнтів концентрацій (робота іонних насосів);
3) фосфорування білків Са-каналів, фосфоламбану (білка Са-насосів саркоплазматичного ретикулуму).
Розлади енергозабезпечення кардіоміоцитів можуть бути пов'язані з порушеннями:
а) ресинтезу АТФ (гіпоксія, голодування, дефіцит вітамінів, зменшення активності ферментів енергетичного обміну);
б) транспорту АТФ із мітохондрій до місць його використання (порушення креатин-кіназної транспортної системи);
в) утилізації АТФ (зменшення АТФ-азної активності структур кардіоміоцитів).
27.41. Дайте порівняльну характеристику гіпо- і гіперкальцієвого варіантів недостатності серця.
Сучасний рівень знань про молекулярні механізми скорочувальної функції серця дозволяє виділити два принципово різних патогенетичних варіанти недостатності серця: гіпокальцієвий і гіперкальцієвий, для яких характерне відповідно зменшення і збільшення концентрації іонів Са2+ у саркоплазмі кардіоміоцитів.
Гіпокальцієвий варіант розвивається в результаті порушень збудження і електромеханічного спряження у волокнах міокарда. Це буває при аритміях (брадикардії різного походження, блокади), короткочасній ішемії міокарда (порушується фосфорування Са-каналів у результаті дефіциту АТФ), ацидозі (блокада Са-каналів іонами водню), гіпокальціємії. Виявляє себе зменшенням сили серцевих скорочень. Основний принцип лікування — підвищення вмісту іонів Са2+ у саркоплазмі кардіоміоцитів під час систоли серця. Для цього використовують: а) серцеві глікозиди; б) катехола-міни і р-адреноміметики; в) парні електричні стимули.
Гіперкальцієвий варіант розвивається в результаті посиленого надходження іонів Са2+ у саркоплазму кардіоміоцитів з позаклітинного середовища (усі види ушкодження сарколеми, при яких підвищується її проникність) або внаслідок порушень видалення Са2+ із саркоплазми (дефіцит АТФ, порушення функції Са-транспортних систем). Виявляється розвитком контрактури (перескорочення) міофібрил, у результаті чого стає неможливим розслаблення м'язових волокон, а отже, і наступне їх скорочення. Основний принцип лікування — зменшення вмісту іонів Са2+ у саркоплазмі кардіоміоцитів. Для цього використовують: а) β-адреноблокатори; б) блокатори Са-каналів.
Позаміокардіальна недостатність серця. Ураження перикарда. Гостра тампонада серця, прояви та наслідки.
Позаміокардіальна недостатність розвивається в тих випадках, коли до серця притікає мало крові по венах або коли воно не в змозі прийняти всю кров, що надходить. Перше буває при гіповолемії (крововтрата) або різкому розширенні судин (колапс), друге - при накопиченні рідини в порожнині перикарда, що утруднює розширення порожнин під час діастоли.
Накопичення рідини в порожнині перикарда може відбуватися швидко і повільно. Швидке накопичення відбувається внаслідок крововиливу при пораненні або розриві серця, при перикардиті, що швидко розвивається. Через погану розтяжність перикарда в порожнині підвищується тиск, що перешкоджає діастолічному розширенню сер-
ця, виникає гостра тампонада серця. В експерименті цей процес моделюють введенням рідини в порожнину перикарда (О. Фохт). При цьому спостерігають, як зменшується кро-вонаповнення порожнин серця, знижується ударний об'єм і падає артеріальний тиск. Між внутрішньоперикардіальним і артеріальним тиском існує чітка обернена залежність: що більший внутрішньоперикардіальний тиск, то нижчий артеріальний. Венозний тиск при цьому підвищується.
Вмикання компенсаторних механізмів при перикардиті відбувається рефлекторно за участю сигналів, що надходять з трьох рецепторних полів:
1) отворів порожнистих і легеневих вен — підвищення тиску на шляхах припливу;
2) аорти і сонних синусів (синокаротидні зони) — зниження тиску на шляхах відтоку;
3) перикарда — підвищення внутрішньоперикардіального тиску. При перетинанні блукаючих і депресорних нервів, а також при вимиканні рецепторних полів за допомогою новокаїну пристосувальні реакції не здійснюються - порушення кровообігу мають набагато тяжчий перебіг. При тампонаді серця мобілізація потужних механізмів компенсації, які ведуть до посилення скорочень серця (гомео- і гетерометричні механізми, інотропний ефект катехоламінів), малоефективна або неможлива. Тому діють тільки відносно малопотужні і енергетично невигідні механізми компенсації і підтримки артеріального тиску - збільшення частоти серцевих скорочень і звуження периферичних судин. Цим і пояснюється важкий клінічний перебіг гострої тампонади серця.
При повільнішому накопиченні рідини в перикарді робота компенсаторних механізмів виявляється ефективнішою, підвищення внутрішньоперикардіального тиску протягом деякого часу може компенсуватися. Повільне накопичення рідини, що спостерігається при хронічному ексудативному перикардиті і гідроперикарді, супроводжується поступовим розтягуванням перикарда і збільшенням об'єму навколосерцевої сумки. Унаслідок цього внутрішньоперикардіальний тиск змінюється порівняно мало, а порушення кровообігу не виникають тривалий час
Артеріальна гіпертензія: визначення поняття, принципи класифікації. Первинна та вторинна артеріальна гіпертензія. Гемодинамічні варіанти. Особливості у дітей.
28.22. Що таке первинна і вторинна артеріальна гіпертензія?
Розрізняють первинну і вторинну артеріальну гіпертензію.
При первинній артеріальній гіпертензії підвищення артеріального тиску не може бути пов'язане з конкретним захворюванням або патологічним процесом у тих чи тих органах і системах: причина підвищення артеріального тиску залишається неясною. Для позначення цієї форми гіпертензії в різних країнах використовують два рівнозначних терміни: "есенціальна гіпертонія " і "гіпертонічна хвороба ".
Вторинна артеріальна гіпертензія виникає як наслідок патологічних процесів у різних органах і системах. Вона характерна для:
а) захворювань нирок (гломерулонефрит, пієлонефрит, полікістоз нирок та ін.);
б) пухлин надниркових залоз (феохромоцитома, альдостерома);
в) уражень серця й судин (деякі вади серця, коарктація аорти);
г) захворювань нервової системи (бульбарний поліомієліт, енцефаліти; травми, струси мозку та ін.).
У всіх цих випадках причина гіпертензії ясна. її усунення, як правило, веде до нормалізації артеріального тиску.
28.23. Які виділяють гемодинамічні варіанти артеріальної гіпертензії?
Оскільки величина артеріального тиску визначається загальним законом гемоди-наміки, відповідно до якого
P = QR,
де Р — артеріальний тиск; Q — хвилинний об'єм серця; R — загальний периферичний опір,
то артеріальна гіпертензія може бути обумовлена збільшенням хвилинного об'єму серця (Q), збільшенням загального периферичного опору (R) або тим та іншим одночасно.
Відповідно до цього виділяють три гемодинамічних варіанти артеріальної гіпертензії.
1. Гіперкінетичнш тип. Обумовлений істотним збільшенням роботи серця, у результаті чого зростає його хвилинний об'єм (Q).
2. Еукінетичний тип. Виникає при помірному збільшенні хвилинного об'єму серця (Q) і загального периферичного опору (R).
3. Гіпокінетичний тип. Його розвиток пов'язаний з істотним збільшенням загального периферичного опору (R).
28.24. Які існують експериментальні моделі артеріальної гіпертензії?
Жодна хвороба людини не має такої великої кількості різних експериментальних моделей, як артеріальна гіпертензія. Нині артеріальну гіпертензію вивчають на мишах, щурах, кролях, кішках, собаках, свинях, мавпах.
За методами відтворення всі моделі артеріальної гіпертензії можна поділити на кілька великих груп.
I. Порушення функції центральної нервової системи:
а) зіткнення процесів умовного збудження і гальмування, що призводить до розвитку у тварин (собак, мавп) неврозу;
б) моделювання психоемоційної напруги шляхом створення зоосоціального конфлікту (у мавп), змін біоритмів, іммобілізації тварин;
в) електрична і хімічна стимуляція лімбічних структур головного мозку.
II. Порушення мозкового крово- і лімфообігу:
а) одно- і двостороннє перев'язування сонних і вертебральних артерій, що живлять мозок (центрально-ішемічна артеріальна гіпертензія)',
б) блокада лімфовідведення по периневральних і периваскулярних лімфатичних шляхах за допомогою каоліну, що його вводять у велику цистерну мозку.
III. Порушення функції депресорних регуляторних систем:
а) двостороннє перетинання у кролів і собак депресорних і синусних нервів, у результаті чого знімаються гальмівні впливи з барорецепторів рефлексогенних зон дуги аорти і каротидного синуса (рефлексогенна гіпертензія, або гіпертензія розгальмування);
б) центральна деаферентація барорецепторів, що її викликають ушкодженням ядра солітарного тракту;
в) пригнічення синтезу простагландинів за допомогою індометацину.
IV. Порушення функції нирок:
а) звужування обох ниркових артерій або звужування однієї ниркової артерії з видаленням другої контрлатеральної нирки (реноваскулярна гіпертензія). Виникнення артеріальної гіпертензії в цьому випадку пов'язане з активацією ренін-ангіотензинної системи;
б) видалення обох нирок і переведення тварин на гемодіаліз для запобігання уремії (ренопривна гіпертензія). її розвиток пояснюють припиненням депресорних функцій нирок;
в) обгортання нирок целофаном, шовком. При цьому виникає перинефрит:' здавлюється ниркова паренхіма, розвивається венозний застій і гіпоксія нирок, активується ренін-ангіотензинна система.
V. Порушення гормонального стану:
а) введення тваринам адреналіну;
в) введення вазопресину;
в) субтотальне видалення кори надниркових залоз. При цьому відбувається посилення регенерації залозистої тканини з посиленою продукцією кортикостероїдів, особливо альдостерону (надшрнико-регенераційна гіпертензія).
VI. Порушення водно-сольового обміну:
а) введення тваринам великої кількості кухонної солі (сольова гіпертензія)',
б) введення мінералокортикоїдів (дезоксикортикостерону, альдостерону) - мі-нералокортикоїдна гіпертензія;
в) поєднане введення кухонної солі й мінералокортикоїдів.
VII. Моделі генетично обумовленої артеріальної гіпертензії. У багатьох лабораторіях світу виведено чисті лінії щурів, характерною рисою яких є гіпертензія -ознака, що спадкується. Це, зокрема, щури зі спонтанною гіпертензією (лінія Окамото - Аокі); щури, схильні до інсультів; новозеландські щури, міланські щури; щури, чутливі до сольової дієти, та ін.
28.25. Яка етіологія первинної артеріальної гіпертензії?
Нині виділяють ряд факторів, що мають безпосередній стосунок до виникнення
гіпертонічної хвороби (есенціальної гіпертензії).
1. Психоемоційна перенапруга. Установлено більшу поширеність артеріальної гіпертензії серед людей, характер роботи яких пов'язаний з постійною психоемоційною напругою, наприклад серед телефоністок і телеграфістів, студентів у період екзаменаційної сесії, у дітей і підлітків, що навчаються у спеціалізованих математичних і деяких інших школах з напруженим режимом занять. Відомо, що під час другої світової війни в Ленінграді під час облоги виникла була ціла "гіпертонічна" епідемія.
Російський учений Ланг уперше висловив думку, що першопричиною гіпертонічної хвороби є психоемоційні перенапруги, які ведуть до розвитку невротичних
порушень вищої нервової діяльності і підвищення артеріального тиску як вегетативного компонента цих порушень. Основну роль Ланг відводив так званим "не-відреагованим" негативним емоціям, тобто таким, при яких сильні вегетативні реакції, зокрема з боку серцево-судинної системи, не супроводжуються адекватними руховими реакціями.
2. Спадковий фактор. Як доказ значення спадковості в етіології гіпертонічної хвороби наводять той факт, що артеріальну гіпертензію виявляють у парах однояйце-вих близнюків набагато частіше, ніж у парах близнюків двох'яйцевих. Крім того, поширеність артеріальної гіпертензії серед родичів хворих на гіпертонічну хворобу є значно більшою, ніж в популяції в цілому. Нарешті, експериментальні моделі генетично обумовленої гіпертензії у тварин за основними своїми характеристиками є найближчими до первинної артеріальної гіпертензії людини.
3. Надмірне споживання кухонної солі. В епідеміологічних дослідженнях відзначено низьку захворюваність на гіпертонічну хворобу в представників деяких етнічних груп, що живуть у різних регіонах нашої планети: гренландських ескімосів, аборигенів гір Китаю та Австралії, деяких племен індіанців, що живуть у Панамі. Загальним для всіх цих народностей є споживання малих кількостей кухонної солі (1—2 г на добу), якщо порівнювати з іншими людьми, що споживають її 10-1'5 г на добу.
Поряд з цим, там, де солі споживається більше, первинна артеріальна гіпертензія виявляється частіше. Високу захворюваність на гіпертонічну хворобу виявлено серед негритянського населення Багамських островів, у деяких регіонах Японії, де споживання солі досягає 20-50 г на добу.
В експерименті артеріальну гіпертензію можна одержати шляхом згодовування тваринам кухонної солі.
28.26. Які існують концепції патогенезу первинної артеріальної гіпертензії?
Нині уявлення про патогенез гіпертонічної хвороби розвиваються в основному в рамках двох концепцій: дисрегуляторної і мембранної.
Дисрегуляшорна концепція пояснює виникнення первинної артеріальної гіпертензії порушеннями механізмів регуляції артеріального тиску.
В основі мембранної концепції лежить положення про те, що первинна артеріальна гіпертензія виникає як наслідок первинних порушень у гладком'язових клітинах артеріол.
28.27. Який патогенез первинної артеріальної гіпертензії з погляду дисрегуляторної концепції її розвитку?
У патогенезі гіпертонічної хвороби розрізняють дві фази: гіперкінетичну і гіпо-кінетичену.
Гіперкінетична фаза характеризується переважно збільшенням хвилинного об'єму серця, наслідком чого і є підвищення артеріального тиску (рис. 132).
У її розвитку виділяють ряд послідовних етапів.
I етап — активація симпатоадреналової системи. Відбувається в результаті частих стресів, психоемоційних перенапруг, що зумовлюють появу осередків постійного тривалого збудження в центральній нервовій системі (патологічна домінанта). Катехоламіни, які виділяються при цьому, викликають щонайменше три важливих для подальшого розвитку гіпертензії ефекти:
а) збільшують хвилинний об'єм серця;
б) збільшують загальний периферичний опір;
в) викликаючи спазм приносних артеріол нирок і безпосередньо діючи на клітини юкстагломерулярного апарату, сприяють виділенню в кров реніну.
II етап — активація ренін-ангіотензинної системи. Надходження реніну в кров зумовлює ряд послідовних біохімічних реакцій, у результаті яких утворюється ангіо-тензин Пі ангіотензин III. З цими пептидами пов'язані такі зміни:
а) скорочення гладких м'язів артеріол (ангіоспазм);
б) збудження структур центральної нервової системи, що беруть участь у регуляції артеріального тиску;
в) вивільнення в кров альдостерону клітинами клубочкової зони кори надниркових залоз.
III етап — активація альдостерон-вазопресинової системи. Надходження в кров альдостерону, а також надмірне надходження в організм хлориду натрію викликають розвиток гіпернатрієміїІ підвищення у зв'язку з цим осмотичного тиску плазми крові. Збудження центральних і периферичних осморецепторів, що настає в цих умовах, активує секрецію вазопресину (антидіуретичного гормону) у ядрах гіпоталамуса. Вазопресин, впливаючи на нирки, викликає збільшення факультативної реабсорбції води.Це, у свою чергу, веде до збільшення об'єму циркулюючої крові (гіперволемія), хвилинного об'єму серця, а отже, і артеріального тиску. Зазначений механізм доповнюється безпосередньою судинозвужувальною дією вазопресину.
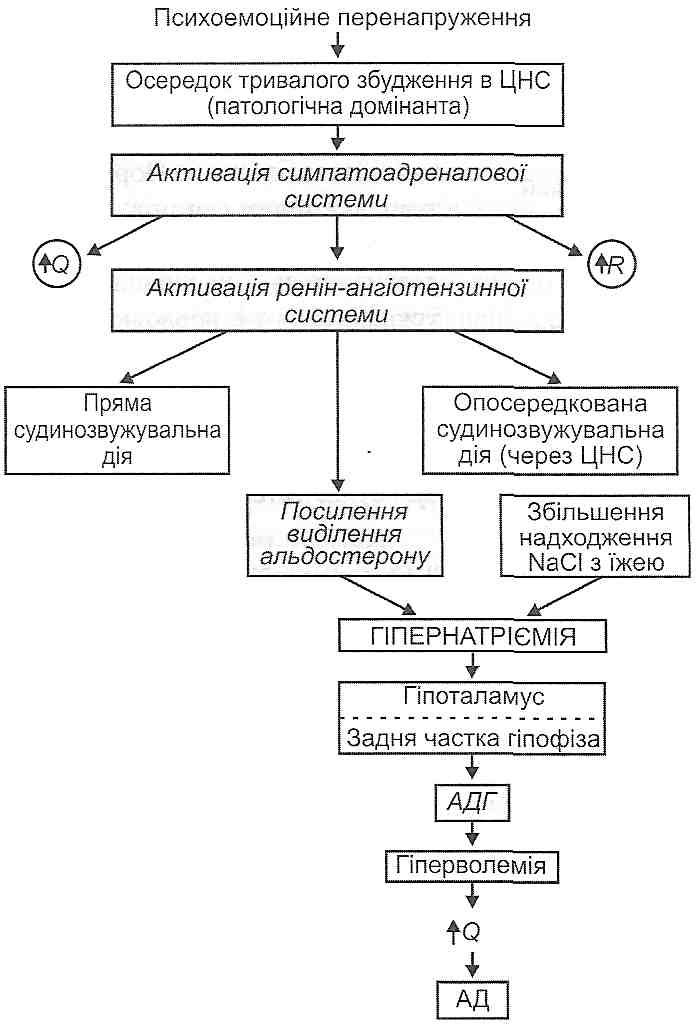
Рис. 132. Механізми гіпєркінетичноїфази розвитку первинної артеріальної гіпертензії: Q - хвилинний об'єм серця; R - загальний периферичний опір; AT— артеріальний тиск; АДГ
- антидіуретичшй гормон
Гіпокінетична фаза характеризується необоротними структурними змінами ре-зистивних судин, у результаті чого загальний периферичний опір і артеріальний тиск постійно збільшені (рис. 133).
У розвитку цієї фази можна виділити ряд послідовних стадій:
1) ауторегуляторний спазм артеріол. Виникає як наслідок збільшення хвилинного об'єму серця. Є реакцією, спрямованою на підтримку сталості кровообігу в тканинах (попереджає надходження надлишкової кількості крові);
2) гіпертрофія гладких м 'язів артеріол. Є структурним проявом гіперфункції гладких м'язів, що виникає при часто повторюваних спазмах;
3) артеріолосклероз. Гіпертрофовані гладком'язові клітини поступово зазнають дистрофічних змін і гинуть, відбувається їх заміщення сполучною тканиною - розви-
вається артеріолосклероз. Артеріоли перетворюються у ригідні сполучнотканинні трубки, не здатні ні до скорочення, ні до розслаблення. Загальний периферичний опір, а отже, і артеріальний тиск постійно збільшені. Порушується живлення життєво важливих органів: головного мозку, серця, нирок. Можливий розрив змінених артеріол — тоді розвивається крововилив. Найнебезпечнішим ускладненням є крововилив у мозок — геморагічний інсульт.
Рис. 133. Механізми
гіпокінетичної фази розвитку
первинної артеріальної гіпертензії
28.28. У чому сутність мембранної концепції патогенезу первинної артеріальної гіпертензії?
Автори мембранної концепції (Ю. Постнов, Р. Орлов) головну роль у розвитку гіпертонічної хвороби відводять спадково обумовленим порушенням іонних насосів мембран гладком'язових клітин.
У рамках цієї концепції нині розвиваються два напрями, що вивчають роль відповідно Са- і Na-K-насосів у порушенні функції гладком'язових клітин артеріол.
1. Дефекти Са-насосів клітинних мембран спричиняються до порушень видалення іонів кальцію із цитоплазми клітин і збільшення їхньої внутрішньоклітинної концентрації. Це викликає постійну контрактуру (перескорочення) гладких м'язів артеріол, що виявляється збільшенням загального периферичного опору і артеріального тиску. Крім того, надлишок іонів Са2+ у цитоплазмі клітин викликає їх ушкодження (див. розд. 11) і є, таким чином, передумовою розвитку артеріоло-склерозу.
2. Пригнічення роботи Na-K-насосів плазматичної мембрани гладком'язових клітин є однією з ознак первинної артеріальної гіпертензії. Про це, зокрема, свідчить той факт, що у багатьох хворих на гіпертонічну хворобу виявляють так званий ендогенний строфантиноподібний фактор, що, як і відомі серцеві глікозиди (строфантин, оуабаїн), пригнічує роботу Na-K-насосів.
У результаті порушень діяльності Na-K-насосів у цитоплазмі поступово збільшується концентрація іонів Na+ і виникає набряк гладком'язових клітин артеріол. Це має кілька наслідків:
а) потовщення стінки і зменшення просвіту артеріол;
б) збільшення чутливості гладком'язових клітин до дії ендогенних катехоламінів;
в) ушкодження і загибель клітин з наступним розвитком артеріосклерозу.
Усі наведені зміни викликають стійке збільшення загального периферичного опору і підвищення артеріального тиску.
Причини і механізми розвитку вторинних артеріальних гіпертензій, експериментальне моделювання.