

Z глава 17. Нервно-мышечные заболевания
Наследственные нервно-мышечные заболевания - большая гетерогенная группа болезней, в основе которых лежит генетически детерминированное поражение нервно-мышечного аппарата (рис. 17.1, 17.2). Заболевание проявляется прогресирующими мышечными атрофиями, мышечной слабостью, парезами мускулатуры.
При диагностике учитывают возраст дебюта клинических проявлений, локализацию и прогрессирование миодистрофического процесса (наличие или отсутствие псевдогипертрофий, фасцикулярных подергиваний, эпизодов мышечной слабости, нарушений чувствительности), отягощенность семейного анамнеза, тип наследования заболевания.
К наследственным нервно-мышечным заболеваниям относят прогрессирующие мышечные дистрофии (первично-мышечные заболевания), наследственные мотосенсорные полинейропатии или невральные амиотрофии (заболевания с преимущественным первичным поражением двигательных волокон периферических нервов), а также спинальные амиотрофии (заболевания с первичным поражением мотонейронов спинного мозга).
17.1. Прогрессирующие мышечные дистрофии
В основе большинства миодистрофий лежат дефекты генов, кодирующих различные структурные белки мышечных волокон.
Прогрессирующая мышечная дистрофия Дюшенна
Заболевание связано с патологией гена, локализующегося на коротком плече Х-хромосомы в локусе Хр21 и ответственного за выработку дистрофина. До 60% всех случаев заболевания связано с делециями, в остальных случаях причинами заболевания являются дупликации или точечные мутации. Около 50% всех мутаций приходится на экзоны 5-20 или 45-53, что предположительно связано со структурой хроматина в этих областях. Продуктом гена является белок дистрофин, синтезирующийся в скелетных мышцах, миокарде и головном мозге.
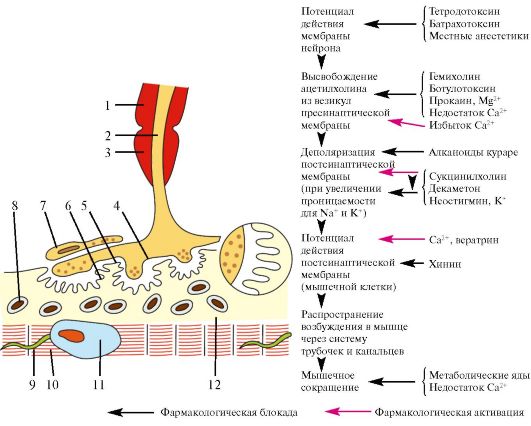 Рис.
17.1. Мионевральный
синапс. 1 - миелиновая оболочка; 2 - аксон;
3 - перехват Ранвье; 4 - пресинаптическая
мембрана; 5 - синаптическая щель; 6 -
постсинаптическая мембрана; 7- леммоцит
(шванновская клетка); 8 - митохондрия; 9
- поперечная система трубочек и канальцев;
10 - миофибриллы; 11 - ядро; 12 - саркоплазма
Рис.
17.1. Мионевральный
синапс. 1 - миелиновая оболочка; 2 - аксон;
3 - перехват Ранвье; 4 - пресинаптическая
мембрана; 5 - синаптическая щель; 6 -
постсинаптическая мембрана; 7- леммоцит
(шванновская клетка); 8 - митохондрия; 9
- поперечная система трубочек и канальцев;
10 - миофибриллы; 11 - ядро; 12 - саркоплазма
Дистрофин выполняет структурную функцию, а также различные модулирующие и сигнальные функции, связываясь с белками внеклеточного матрикса, плазматической мембраны, цитоскелета и других внутриклеточных структур. Отсутствие дистрофина в миофибриллах приводит к дезинтеграции дистрофингликанопротеинового комплекса, обеспечивающего структурно-функциональную организацию цитоскелета миофибрилл, утрате их устойчивости к циклическим актам сокращения и расслабления, разрывам. Его отсутствие повышает проницаемость мембраны для ионов кальция, что приводит к активации кальциевых протеаз, нарушению функционирования клетки и в конечном итоге к некрозу мышечных волокон.
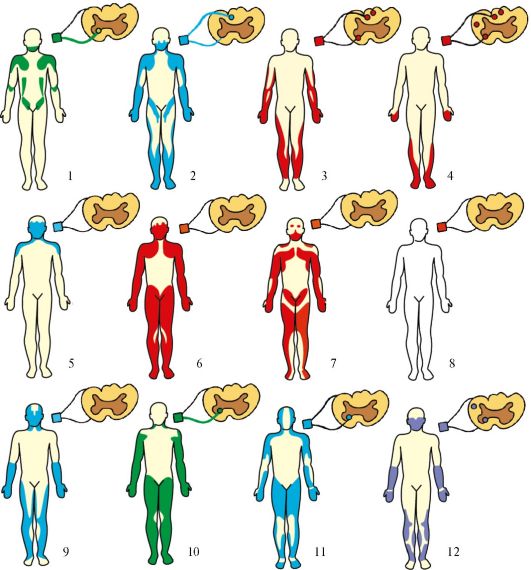 Рис.
17.2. Основные
типы мышечных атрофий. 1 - спинальная
прогрессирующая детская амиотрофия
Верднига-Гоффманна; 2 - интерстициальная
гипертрофическая невропатия Дежерина-Сотта;
3 - невральная амиотрофия (прогрессирующая
невральная перонеальная мышечная
атрофия Шарко-Мари-Тута); 4 - болезнь
Фридрейха (семейная спинно-мозжечковая
атаксия); 5 - миастения; 6 - пароксизмальная
миоплегия (семейный периодический
паралич); 7 - прогрессирующая мышечная
дистрофия; 8 - врожденная миотония Томсена
(неатрофическая); 9 - атрофическая
миотония; 10 - врожденная амиотония
Оппенгейма; 11 - хроническая прогрессирующая
семейная спинальная амиотрофия взрослых
Арана-Дюшенна; 12 - боковой амиотрофический
склероз Шарко. Зеленым цветом обозначен
дебют заболевания в детстве, красным -
в подростковом возрасте, синим - в зрелом
возрасте, фиолетовым - в пожилом
Рис.
17.2. Основные
типы мышечных атрофий. 1 - спинальная
прогрессирующая детская амиотрофия
Верднига-Гоффманна; 2 - интерстициальная
гипертрофическая невропатия Дежерина-Сотта;
3 - невральная амиотрофия (прогрессирующая
невральная перонеальная мышечная
атрофия Шарко-Мари-Тута); 4 - болезнь
Фридрейха (семейная спинно-мозжечковая
атаксия); 5 - миастения; 6 - пароксизмальная
миоплегия (семейный периодический
паралич); 7 - прогрессирующая мышечная
дистрофия; 8 - врожденная миотония Томсена
(неатрофическая); 9 - атрофическая
миотония; 10 - врожденная амиотония
Оппенгейма; 11 - хроническая прогрессирующая
семейная спинальная амиотрофия взрослых
Арана-Дюшенна; 12 - боковой амиотрофический
склероз Шарко. Зеленым цветом обозначен
дебют заболевания в детстве, красным -
в подростковом возрасте, синим - в зрелом
возрасте, фиолетовым - в пожилом
Мозговая изоформа экспрессируется в коре больших полушарий, гиппокампе и клетках Пуркинье. В головном мозге дистрофин участвует в процессах нейрональной пластичности, синаптической стабильности и интеграции сигнала на клеточном уровне. Кроме того, дистрофин принимает участие в нормальном функционировании глии. Снижение интеллекта, наблюдаемое примерно у 10-20% больных с формой Дюшенна, связано с нарушением синтеза этой изоформы дистрофина.
|
Частота заболевания составляет 3,3 на 100 000 населения, 14 на 100 000 родившихся. В большинстве случаев болеют мальчики. Случаи заболевания у девочек редки и возможны при кариотипе Х0, мозаицизме Х0/ХХ, Х0/ХХХ, Х0/ХХХ/ХХХ и при структурных аномалиях хромосом.
Патоморфология. Разрушение мышечных волокон, их замещение соединительной и жировой тканью.
Клинические проявления. Признаки заболевания проявляются на 1-3 годах жизни. Уже на 1-м году жизни обращает на себя внимание отставание детей в моторном развитии. Они с задержкой начинают садиться, вставать, ходить. Движения неловкие, при ходьбе дети неустойчивы, часто спотыкаются, падают. В 2-3 года появляются мышечная слабость, изменения походки по типу «утиной», что связано с поражением ягодичных мышц. Наблюдается своеобразная «стереотипная» динамика движений детей во время вставания из горизонтального положения, из положения на корточках или со стула. Вставание происходит поэтапно, с активным использованием рук - «взбирание лесенкой» или «взбирание по самому себе».
Атрофии мышц всегда симметричны. Сначала они локализуются в проксимальных группах мышц нижних конечностей и в мышцах тазового пояса, в 1-3 года распространяются на проксимальные группы мышц верхних конечностей - плечевой пояс, мышцы спины. Вследствие атрофий появляются лордоз, «крыловидные» лопатки, «осиная» талия (рис. 17.3). Типичным симптомом заболевания является псевдогипертрофия икроножных мышц. При пальпации мышцы плотны, безболезненны. Мышечный тонус снижен преимущественно в проксимальных группах мышц. Сухожильные рефлексы изменяются с различной последовательностью: в ранних стадиях заболевания исчезают коленные рефлексы, затем рефлексы с двуглавой и трехглавой мышц. Ахилловы рефлексы могут долго оставаться сохранными.
|
Одной из отличительных особенностей формы Дюшенна является патология костносуставной, сердечно-сосудистой и нейроэндокринной систем. Костно-суставные нарушения включают в себя деформации
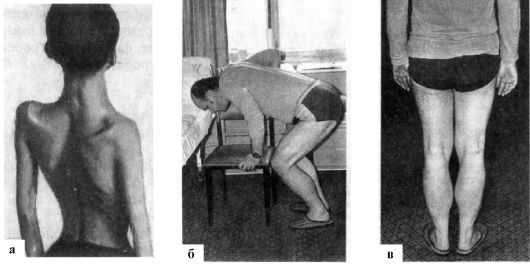 Рис.
17.3. Мышечная
дистрофия. а -
гипотрофия мышц плечевого и тазового
пояса, проксимальных отделов конечностей;
«крыловидные» лопатки, «осиная»
талия; б -
вставание с корточек «лесенкой» (прием
миопата); в -
псевдогипертрофия мышц икроножной
группы
Рис.
17.3. Мышечная
дистрофия. а -
гипотрофия мышц плечевого и тазового
пояса, проксимальных отделов конечностей;
«крыловидные» лопатки, «осиная»
талия; б -
вставание с корточек «лесенкой» (прием
миопата); в -
псевдогипертрофия мышц икроножной
группы
позвоночника, стоп, грудины. У многих больных в результате избирательного и неравномерного поражения различных групп мышц рано возникают мышечные контрактуры и ретракции сухожилий. Сердечнососудистые расстройства проявляются дилатационной кардиомиопатией, которая может быть одной из причин неблагоприятного исхода заболевания. На ЭКГ регистрируются изменения миокарда (блокада пучка Гиса и др.). Среди нейроэндокринных расстройств чаще встречаются синдромы Иценко-Кушинга, Бабинского-Фрелиха.
Снижение интеллекта разной выраженности отмечается у большинства больных и, как правило, не соотносится с тяжестью поражения скелетной мускулатуры и тяжестью самого заболевания. Наиболее часто у детей выявляются относительно неспецифические изменения в виде затруднения концентрации внимания, сложности в воспроизведении недавно полученной информации, нарушения слуховой памяти, произношения, усвоения материала.
При позитронной эмиссионной томографии и при 31Р МР-спектроскопии наиболее выраженные изменения, отражающие нарушение утилизации глюкозы (по данным ПЭТ) и изменение соотношения неорганического фосфора, АТФ, фосфомоноэстеров и фосфокреатинина (МР-спектроскопия), отмечаются в коре лобных долей больших полушарий и в мозжечке, в меньшей степени - в гиппокампе.
|
Форма Беккера является аллельным вариантом миодистрофии Дюшенна и также связана со структурным дефектом гена дистрофина. Наследуется по рецессивному типу, сцепленному с Х-хромосомой. Первые клинические проявления отмечаются позднее, чем при форме Дюшенна, чаще в возрасте 10-15 лет, а само заболевание протекает значительно мягче. Мышечная слабость, повышенная мышечная утомляемость при физической нагрузке, псевдогипертрофии икроножных мышц не достигают такой выраженности, как при форме Дюшенна. Мышечный тонус снижен незначительно. Сухожильные рефлексы долго остаются сохранными. В поздних стадиях болезни могут наблюдаться изменения походки по типу «утиной», компенсаторные миопатические приемы при вставании. Заболевание прогрессирует медленно на протяжении многих лет. Сердечно-сосудистые расстройства выражены умеренно. Иногда наблюдается блокада ножек пучка Гиса. Эндокринные нарушения проявляются гинекомастией, снижением либидо, импотенцией. Выраженных изменений интеллекта не отмечается. Больные долго сохраняют работоспособность.
Диагностика и дифференциальная диагностика. Диагноз устанавливают на основании клинических проявлений, данных биохимических исследований (повышение в крови активности КФК, ЛДГ), электромиографии (признаки первичного мышечного поражения) и молекулярно-генетического анализа. Для уточнения аллельной формы заболевания проводится биопсия мышц для определения дистрофина (при форме Дюшенна в скелетных мышцах дистрофин не выявляется; при форме Беккера дистрофин синтезируется, но в большинстве случаев его уровень снижен). При обследовании матерей - носителей патологического гена (биопсия ворсин хориона на 8-9 неделе) выявляют заболевание у мальчиков.
Дифференцировать форму Дюшенна следует от спинальной амиотрофии Верднига-Гофмана, а форму Беккера - от прогрессирующих мышечных дистрофий Дюшенна, конечностно-поясных форм прогрессирующих мышечных дистрофий, спинальной амиотрофии Кугельберга-Веландера, метаболических и эндокринных миопатических синдромов.
|