

5.11. Особенности технологического процесса получения дисперсноупрочненных композиционных материалов «хрупкая матрица – хрупкий наполнитель».
Рассмотрим технологию композита, в составе которого матрица и наполнитель представлены диоксидом циркония, имеющим различные полиморфные модификации (матрица cZrO2 – наполнитель tZrO2), а также композиционного материала, у которого матрица и наполнитель отличаются по химическому составу (матрица Al2O3 – наполнитель tZrO2). Для начала укажем, что основное отличие этих технологий заключается в том, что в первом случае дисперсные включения tZrO2 выделяются в матрице композита в процессе термообработки благодаря специальному ее режиму, а во втором случае – их вводят специальным приемом в исходную порошковую смесь.
Технология композита cZrO2 (матрица) - tZrO2 (наполнитель). Для изготовления такого материала в начале синтезируют порошок ZrO2 частично стабилизированный каким-либо оксидом, например CaO (операция 1) (рис.5.28). Частичная стабилизация диоксида циркония достигается при содержании CaO в количестве 5-9% мол. При нагревании такого состава выше 1000С в нем наблюдается сосуществование кубической и тетрагональной модификаций диоксида циркония (рис.5.29). В порошок ZrO2 – CaO(5-9% мол) вводят органическую связку, после чего из полученной шихты формуют изделие (операция 2). После удаления связки путем ее выжига на воздухе (операция 3) проводят спекание изделия (операция 4) при температуре, соответствующей однофазной области существования
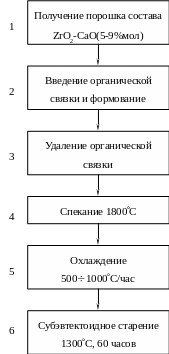
Рис. 5.28. Последовательность технологических операций для получения композита «хрупкая матрица (сZrO2) – хрупкий наполнитель (tZrO2)».
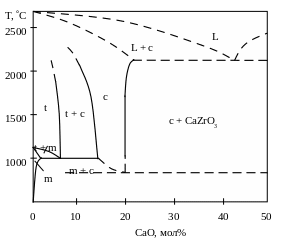
Рис. 5.29. Диаграмма состояния ZrO2-CaO (по Рою).
кубической модификации твердого раствора ( 1800С). Затем следует обеспечить быстрое охлаждение (операция 5) до субэвтектоидных температур. Высокая скорость охлаждения необходима для равномерного выделения в матрице зерен tZrO2. Тогда структура композита представлена крупными зернами cZrO2 (30-60 мкм), составляющими матрицу, и выделенными в ее объеме (при скоростном охлаждении) субмикронными (0,2 мкм) зернами tZrO2. Если скорость охлаждения мала, то происходит укрупнение зерен tZrO2 вследствие рекристаллизации до размеров выше критического, при которых происходит образование моноклинной фазы (в результате tm перехода). На заключительной стадии (операция 6) материал подвергают “субэвтектоидному старению”, то есть длительной изотермической выдержке при температуре ниже эвтектоидной для данного состава. При этой температуре происходит изменение размеров выделений фазы tZrO2, образование дополнительных продуктов эвтектоидного распада, а также потеря когерентности связи выделений tZrO2 с кубической матрицей. Таким образом, в результате “субэвтектоидного старения” можно изменять размер выделений tZrO2 и влиять на эффективность упрочнения. Режим такого старения подбирается индивидуально для каждого материала. Чрезмерное увеличение его продолжительности или температуры может привести к перестариванию композита и снижению его механических свойств.
Технология композита Al2O3 (матрица) - tZrO2 (наполнитель). Cогласно данной технологии (рис. 5.30) в качестве матричного порошка можно использовать производимые промышленностью алюмооксидные порошки марок (ГЛМК, ЦМ-332). В основе их получения (операция 1) лежит химическая переработка боксита по методу Байера. Бокситы – это природные гидраты глинозема преи-
2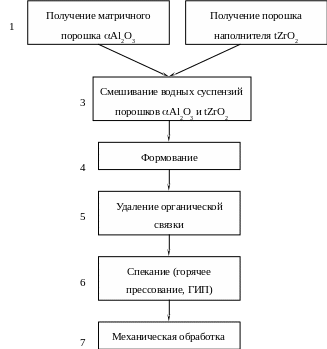
Рис. 5.30. Последовательность технологических операций для получения композита «хрупкая матрица (Al2O3) – хрупкий наполнитель (tZrO2)».
мущественного состава Al2O3 3H2O и Al2O3 H2O, их разлагают раствором едкой щелочи с образованием раствора алюмината натрия:
Al2O3 3H2O + NaOH NaAlO2 (раствор).
При этом, сопутствующие примеси (SiO2, Fe2O3 и др.) остаются вне раствора. После очистки раствора алюмината натрия от примесей его подвергают гидролизу:
NaAlO2 + H2O Al(OH)3 + NaOH.
Полученную гидроокись алюминия выделяют из раствора, высушивают и прокаливают на воздухе во вращающихся печах при температуре 1150 - 1200С:
Al(OH)3 Al2O3 + H2O (пар).
В результате получают белый сыпучий порошок – технический глинозем (Al2O3 с кубическим типом кристаллической решетки). Частицы Al2O3 имеют форму пористых (до 50%) сферолитов – это округлые агрегаты размером 40 – 70 мкм, состоящие из мельчайших кристаллов размером менее 0,1 мкм. Далее Al2O3 подвергают обжигу на воздухе при температуре 1450 - 1500С, в процессе которого Al2O3 переходит в высокотемпературную форму Al2O3 с гексагональной кристаллической решеткой. Сферолиты становятся непрочными и легко разрушаются при последующем помоле до размера частиц 1-5 мкм. В состав порошка Al2O3 можно вводить до 0,5 % масс MgO через водный раствор хлорида магния. Тогда после термообработки (1500С, воздух) на поверхности частиц порошка образуется тонкий субмикронный слой алюмомагнезиальной шпинели – MgO Al2O3, обеспечивающий торможение роста зерен вследствие собирательной (вторичной) рекристаллизации при спекании изделий. Порошок Al2O3, легированный MgO, является промышленным товарным продуктом.
Ультрадисперсный порошок наполнителя - t-ZrO2 чаще всего получают методом химического осаждения (операция 2), при этом, в состав порошка ZrO2 вводят добавку оксида иттрия - Y2O3 (3% мол.), способствующую сохранению тетрагональной модификации ZrO2 при охлаждении до комнатной температуры за счет частичной стабилизации ( в таком порошке содержание t-ZrO2 составляет не менее 60 % об, остальное – с-ZrO2) . Для этого используют следующие технологические приемы:
а) приготовление водных растворов солей и их смешивание:
![]()
в) осаждение (или соосаждение, поскольку при приливании к водному раствору смеси солей осадителя – NH4OH, одновременно выделяются два нерастворимых осадка):
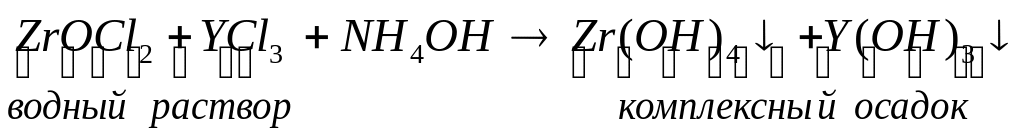
с) фильтрация, промывка, сушка комплексного осадка и его термообработка на воздухе при температуре 1200С:
![]()
В результате термообработки получается тетрагональная модификация ZrO2, легированная добавкой оксида – стабилизатора, в виде слабосвязанных агрегатов.
d) дезагрегация путем протирки через сито с размером ячеек 0,05 мм и получение сыпучего продукта – ультрадисперсного порошка с размерами частиц менее 1 мкм.
Особенностью данной технологии является необходимость смешивания матричного порошка с порошком наполнителя через их водные суспензии (операция 3). Такой способ смешивания необходим, поскольку он исключает tm переход в частицах наполнителя от ударного воздействия на них мелющих тел. Для его реализации приготавливают водные суспензии из матричного порошка и порошка наполнителя, затем их сливают в общую емкость и осуществляют перемешивание при помощи пропеллерной мешалки. После завершения смешивания порошковую композитную смесь выделяют из суспензии путем фильтрации и высушивают.
Рассмотрим физико-химические аспекты смешивания порошков через их водные суспензии. Водную суспензию получают путем введения частиц тонкодисперсного порошка в воду с добавлением электролита (например, HCl или NH4OH). В данной системе тонкодисперсный порошок является диспергируемой фазой, а вода с добавкой электролита рассматривается в качестве дисперсионной среды. Это коллоидная система, она должна быть кинетически и агрегативно устойчивой, тогда ее называют стабильной. Это означает, что частицы порошка должны как бы «зависнуть» в дисперсионной среде и не оседать на дно емкости под действием силы тяжести после окончания перемешивания и далее в течение достаточно длительного времени при хранении суспензии (это кинетический фактор устойчивости суспензии). Кроме того, они не должны соединяться с образованием крупных агрегатов, выделяющихся из суспензии, то есть не должны коагулировать (это агрегативный фактор устойчивости суспензии). Следует отметить, что процесс коагуляции термодинамически выгоден диспергированной системе, поскольку при этом понижается ее свободная энергия за счет снижения общей поверхности. Чтобы получить стабильную суспензию надо знать законы, по которым она «живет».
Электролит в воде диссоциирует на противоположно заряженные ионы: HCl H+ + Cl- (в случае использования в качестве электролита раствора соляной кислоты), NH4OH NH4+ + OH- (в случае использования в качестве электролита водного раствора аммиака). Концентрация ионов H+ (в первом случае), либо OH- (во втором случае) определяет величину pH суспензии: pH 7 – кислая среда, pH 7 – основная среда. При введении тонкодисперсного порошка в дисперсионную среду эти ионы (H+ либо OH-) покрывают монослоем поверхность каждой его частицы, притягивая противоположно заряженные ионы (Cl- либо NH4+), концентрация которых постепенно снижается по мере удаления от частицы. Таким образом, вокруг каждой частицы порошка образуется ионная оболочка, которую называют двойным электрическим слоем, либо ионогенным комплексом (7). Его структура, в случае использования в качестве электролита раствора соляной кислоты, показана на рис. 5.31. На поверхности порошковой частицы, которая является неким твердым ядром (1), образуется ионный слой H+ (2). Противоположно заряженные ионы формируют противоионный слой (3). В его составе есть ионы, близко расположенные по отношению к ионному слою (2), которые прочно удерживаются силами электростатического взаимодействия. Поэтому эти ионы, совокупно с ионным слоем, образуют устойчивый адсорбционный слой (4). В составе противоионного слоя имеются периферийные ионы, слабосвязанные со слоем (2) в силу их значительной удаленности от него. Они образуют диффузный слой (5), пределы которого они могут покидать вследствие диффузии, при этом их количество может восполняться за счет диффундирующих ионов диффузных слоев соседних частиц. Твердое ядро совместно с двойным электрическим слоем образует мицеллу (6), которая в целом - электронейтральна.
Если в суспензию погрузить электроды (4) и создать электрическое поле (рис. 5.32), то будет наблюдаться разрыв двойного электрического слоя в структуре каждой мицеллы по границе адсорбционного и диффузного слоев. Тогда ядро, а также ионный слой и часть ионов противоионного слоя, будут перемещаться к катоду (так как при этом знак заряда ядра определяется зарядом ионного слоя), а все ионы диффузного слоя – к аноду. Такое явление передвижения коллоидных частиц в электрическом поле носит название электрофореза. Ясно, что в этом случае электронейтральность мицеллы нарушена и по границе адсорбционного и диффузного слоев, вследствие отделения части компенсирующих ионов, возникает потенциал, называемый электро-
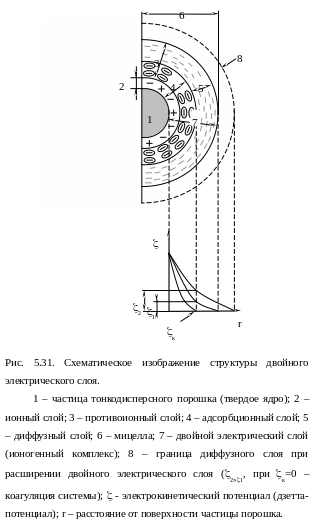

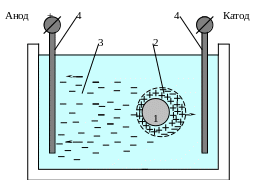
Рис. 5.32. Схематическое изображение разрыва двойного электрического слоя по границе адсорбционного и диффузного слоев в электрическом поле.
1 – твердое ядро; 2 – ионный слой и часть ионов противоионного слоя; 3 – ионы диффузного слоя; 4 – электроды.
кинетическим потенциалом или «дзета» - потенциалом, он обозначается буквой . Его величина соответствует напряжению, которое необходимо приложить для разрыва двойного электрического слоя (например, = 44 мв для суспензии, в которой диспергируемой фазой являются частицы SiO2). Видно (рис. 5.31), что размер диффузного слоя, в составе двойного электрического слоя, определяет величину - потенциала: чем он больше, тем выше - потенциал (2 1; диффузный слой, соответствующий 2, обозначен пунктирной линией). В то же время, ясно, что расширение двойного электрического слоя будет приводить к увеличению расстояния между частицами порошка в суспензии, способствуя увеличению ее текучести. Это можно объяснить тем, что в этом случае возрастает толщина жидкой прослойки между твердыми ядрами, что снижает внутреннее трение в жидкости. Таким образом, величина потенциала может использоваться в качестве характеристики суспензии, определяющей ее текучесть: чем больше - потенциал, тем выше текучесть суспензии.
Помимо этого, расширение двойного
электрического слоя за счет увеличения
диффузного слоя, значительно способствует
повышению агрегативной устойчивости
суспензии, поскольку диффузные слои
препятствуют сближению частиц на
расстояние, при котором энергия их
взаимного притяжения (Еп) становится
больше энергии Броуновского движения
(ЕБр), отдаляющего частицы друг
от друга. Этим предупреждается их
объединение в агрегаты и последующая
коагуляция. При этом, силы электростатического
отталкивания ионных оболочек мицелл в
суспензии возрастают, что препятствует
их оседанию на дно емкости. Здесь важно
отметить, что максимально возможная
ширина диффузного слоя достигается при
оптимальном количестве добавки
электролита. Превышение этого количества
приводит к коагуляции (Еп
ЕБр), поскольку ширина диффузного
слоя резко уменьшается до размера
адсорбционного слоя. Вводится понятие
порога коагуляции – это минимальное
количество данного электролита (мол/на1л
суспензии), которое необходимо добавить
к стабильной суспензии, чтобы вызвать
начало коагуляции. Описанный эффект
коагуляции при добавлении избытка
электролита объясняется с позиции
теории сильных электролитов, согласно
которой каждый ион в суспензии окружен
ионной атмосферой, радиус (R) которой
зависит от ионной силы раствора – Fион.
Она равна сумме концентраций всех ионов,
умноженной на квадрат их валентности
(Fион =
![]() сi
zi2,
где сi zi
– концентрация и валентность i – го
иона, R1/ Fион).
Ясно, что при наличии избытка электролита
Fион возрастает, а ионная атмосфера
каждого иона сжимается, что, в общем,
приводит к сжатию диффузного слоя. Если,
при этом, размер диффузного слоя
становится равным адсорбционному, тогда
дзета потенциал колоидной системы
становится равным нулю (к
= 0 – изоэлектрическое состояние)
(рис. 5.31). В этом случае диффузные слои
больше не выполняют роль экранирующих
жидких прослоек между частицами порошка,
наступает коагуляция.
сi
zi2,
где сi zi
– концентрация и валентность i – го
иона, R1/ Fион).
Ясно, что при наличии избытка электролита
Fион возрастает, а ионная атмосфера
каждого иона сжимается, что, в общем,
приводит к сжатию диффузного слоя. Если,
при этом, размер диффузного слоя
становится равным адсорбционному, тогда
дзета потенциал колоидной системы
становится равным нулю (к
= 0 – изоэлектрическое состояние)
(рис. 5.31). В этом случае диффузные слои
больше не выполняют роль экранирующих
жидких прослоек между частицами порошка,
наступает коагуляция.
Отметим также, что поскольку концентрация ионов электролита связана с величиной рН суспензии, то оптимальное количество добавки электролита и порог коагуляции можно контролировать при помощи указанной характеристики. Например, при смешивании водных суспензий матричного порошка - Al2O3 и порошка наполнителя - t-ZrO2 в качестве электролита выбирают раствор HCl либо NH4OH. Тогда, используя для приготовления суспензий какой-либо из указанных электролитов, определяют оптимальное значение рН – среды, при котором коллоидные системы являются агрегативно устойчивыми. Следует стремиться, чтобы устойчивость предварительно приготовленных суспензий перед их смешиванием достигалась при использовании одного и того же электролита и при одинаковых или близких значениях рН. В противном случае, после их сливания в общую емкость и перемешивания пропеллерной мешалкой, избыточное количество электролита, присущее одной из смешиваемых суспензий (с существенно отличным значением рН), будет вызывать коагуляцию другой смешиваемой суспензии. Из опытных данных известно, что оптимальная величина рН – среды, при использовании кислоты, составляет 3-5, а при использовании основания 10-12.
Для формования изделий (операция 4) порошковую композитную смесь пластифицируют за счет добавления органической связки, а затем подвергают, как правило, прессованию или экструзии. Часто используют метод горячего шликерного литья, либо литье водных суспензий в гипсовые формы. В последнем случае нет необходимости во введении в смесь органической связки.
Органическую связку удаляют (операция 5) путем ее выжига в результате термообработки сырых изделий на воздухе при температуре 600 - 800С.
Спекание материала (операция 6) можно проводить на воздухе, либо в защитной газовой среде. Возможно использование горячего прессования (ГП) или горячего изостатического прессования (ГИП), в этих случаях можно не вводить в порошковую композитную смесь органическую связку. Очень важным является подбор режима спекания. Температура и время спекания, а также приложенное давление (в случае ГП и ГИП) выбираются таким образом, чтобы исключить рекристаллизационный рост частиц наполнителя до размеров более 1 мкм. Необходимость этого обсуждалась в разделе 5.10. Обычно используемый режим ГП для получения композита: температура спекания ~ 1500С, время изотермической выдержки - 30-60 мин., давление - 10-50 МПа.
Механическая обработка спеченного изделия производится алмазным инструментом (операция 7). Ее особенностью является использование «мягких» режимов обработки, при которых исключается значительное нагружение поверхностного слоя обрабатываемого материала путем прижима инструмента. Это необходимо соблюдать во избежание преждевременного инициирования tm перехода определенной доли дисперсных включений ZrO2 в матрице.