

Последствия энзиматического блока
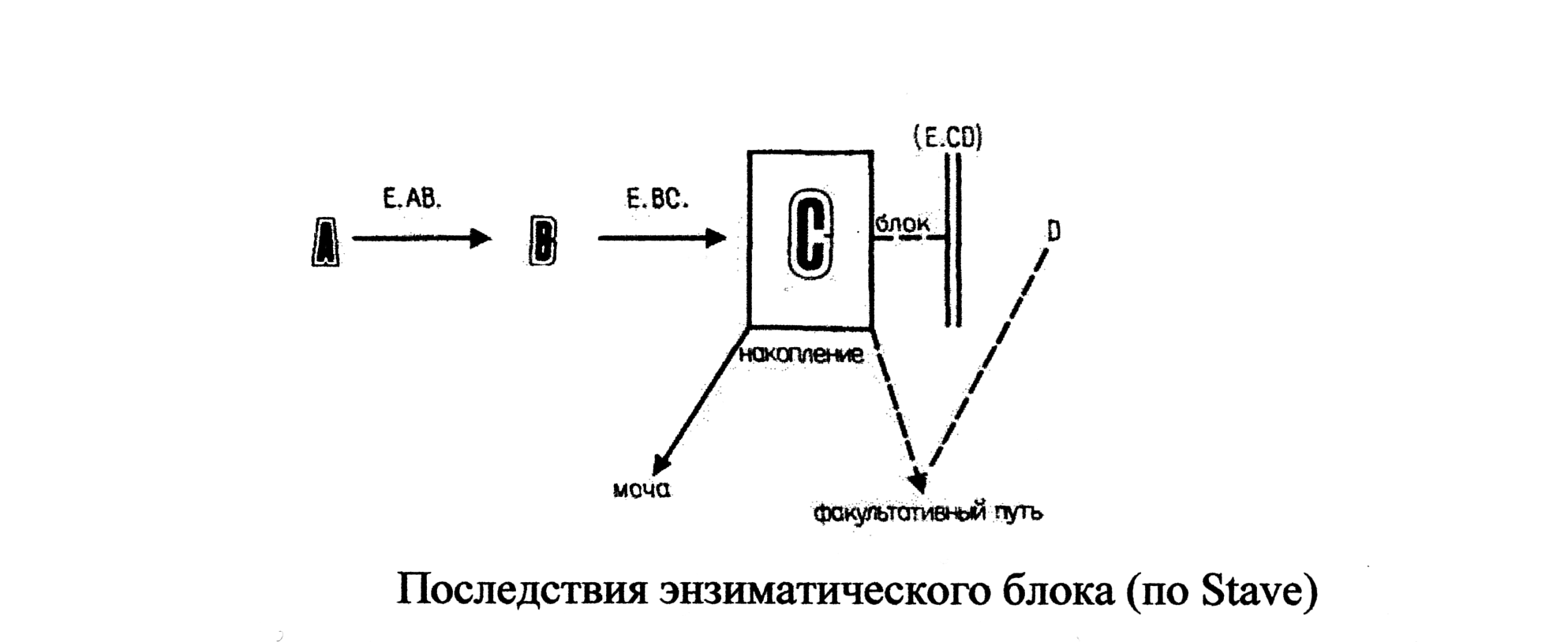
Исходя из вышесказанного, можно сделать вывод о том, что при диагностике энзимопатий возможно использование не только данных о наличии или отсутствии активности энзима, но и изменении концентрации веществ, участвующих в данной реакции.
Наследственные классические энзимопатии могут нарушать обмен белков, жиров, углеводов, нуклеиновых кислот.
Энзимопатии обмена аминокислот
Нарушения обмена фенилаланина и тирозина (рисунок 3)
Примером врождённого дефекта обмена фенилаланина может служить фенилкетонурия (фенилпировиноградная олигофрения). При данном заболевании снижен синтез фермента фенилаланинмонооксигеназы (фенилаланингидроксилазы), которая в печени, почках и поджелудочной железе катализирует окисление аминокислоты фенилаланина в тирозин (блок1). В результате указанного ферментативного блока повышается содержание фенилаланина, который затем трансформируется в фенилпировиноградную и фенилуксусную кислоты с последующим выведением их в составе мочи. Эти необычные метаболиты обмена фенилаланина оказывают токсическое действие на ткань головного мозга. У детей при фенилкетонурии развивается слабоумие (олигофрения). Возникающий при заболевании дефицит тирозина приводит к снижению синтеза из него нейроактивных биогенных аминов. Повышение концентрации фенилаланина нарушает обмен многих аминокислот в головном мозге и сопровождается нарушением регуляции синтеза нейромедиаторов.
Вариантом гиперфениланемии является недостаточность фермента дигидроптеридинредуктазы. Этот фермент катализирует образование восстановленного тетрагидробиоптерина, являющегося кофактором фенилаланинмонооксигеназы. В отличие от классической фенилкетонурии эта патология не поддаётся лечению ранним ограничением содержания фенилаланина в пище.
К аферментозам обмена аминокислоты тирозина относятся алкаптонурия, альбинизм и тирозинемии (тирозинозы).
При алкаптонурии снижен синтез фермента оксидазы гомогентизиновой кислоты, катализирущей одну из промежуточных стадий обмена тирозина – разрыва фенольного кольца в гомогентизиновой кислоте (блок 2). Недостаток данного фермента приводит к поступлению гомогентизиновой кислоты в кровь и выведению её с мочой. Признаки заболевания могут наблюдаться вскоре после рождения. Моча ребёнка окрашивается в чёрный цвет, т.к. гомогентизиновая кислота на воздухе переходит в меланиноподобное соединение. В детском возрасте алкаптонурия (чёрная моча) – единственное проявление дефекта, другими симптомами он клинически не проявляется. У взрослых может наблюдаться окрашивание хрящей носа, ушных раковин (охроноз).
При наследственной тирозинемии отсутствует фермент гидролаза фумарилацетоуксусной кислоты – промежуточного метаболита распада тирозина (блок 3). При данной патологии из фумариацетоуксусной кислоты образуется сукцинилацетон, вызывающий поражение печени и почек. Клинические симптомы заболевания возникают в первые месяцы жизни и проявляются гипотрофией, печёночной недостаточностью, кровоточивостью. Причиной тирозинозов других типов могут служить дефекты фермнтов тирозиновой трансаминазы и гидроксилазы парагидроксифенилпировиноградной
кислоты.
Транзиторная тирозинемия новорожденных встречается чаще других нарушений обмена аминокислот. При ней снижена активность оксидазы парагидроксифенилпировиноградной кислоты – фермента, катализирующего образование гомогентизиновой кислоты. Симптомы транзиторной тирозинемии уменьшаются ограничением количества белка в пище или введением витамина «С».
Альбинизм развивается при нарушении активности ферментов синтеза меланинов (блок 4). Дефицит меланинов снижает их фотопротекторное, антиоксидантное действие.
Описаны различные аферментозы в цикле биосинтеза мочевины
(рисунок 4).
Встречается недостаточность карбамоилфосфатсинтетазы – фермента, катализирующего включение аммония в цикл синтеза мочевины. Дефект данного фермента проявляется выраженной гипераммониемией I типа. При молниеносном течении заболевания развивается тяжёлое коматозное состояние.
В основе врождённой гипераммониемии II типа лежит недостаточность орнитинкарбамоилтрансферазы (орнитинтранскарбамилазы), катализирующей синтез цитруллина.
Рисунок 3