

2.7.3. Перегруппировки углеродного скелета и некоторые возможности их использования в полном синтезе
Конструктивные и деструктивные реакции, которые мы до сих пор рассматривали, отличаются тем общим свойством, что в них затрагиваются (разрываются или образуются) лишь связи тех атомов, которые непосредственно входят в состав реакционных центров (функциональных групп) субстратов и реагентов. Наряду с этим в органической химии существует множество реакций более сложных типов. Это многочисленные перегруппировки, в которых изменения затрагивают не только функциональные группы, но и связи, удаленные от реагирующего центра. Некоторые классические примеры таких превращений приведены на схеме 2.153.
Схема 2.153 |
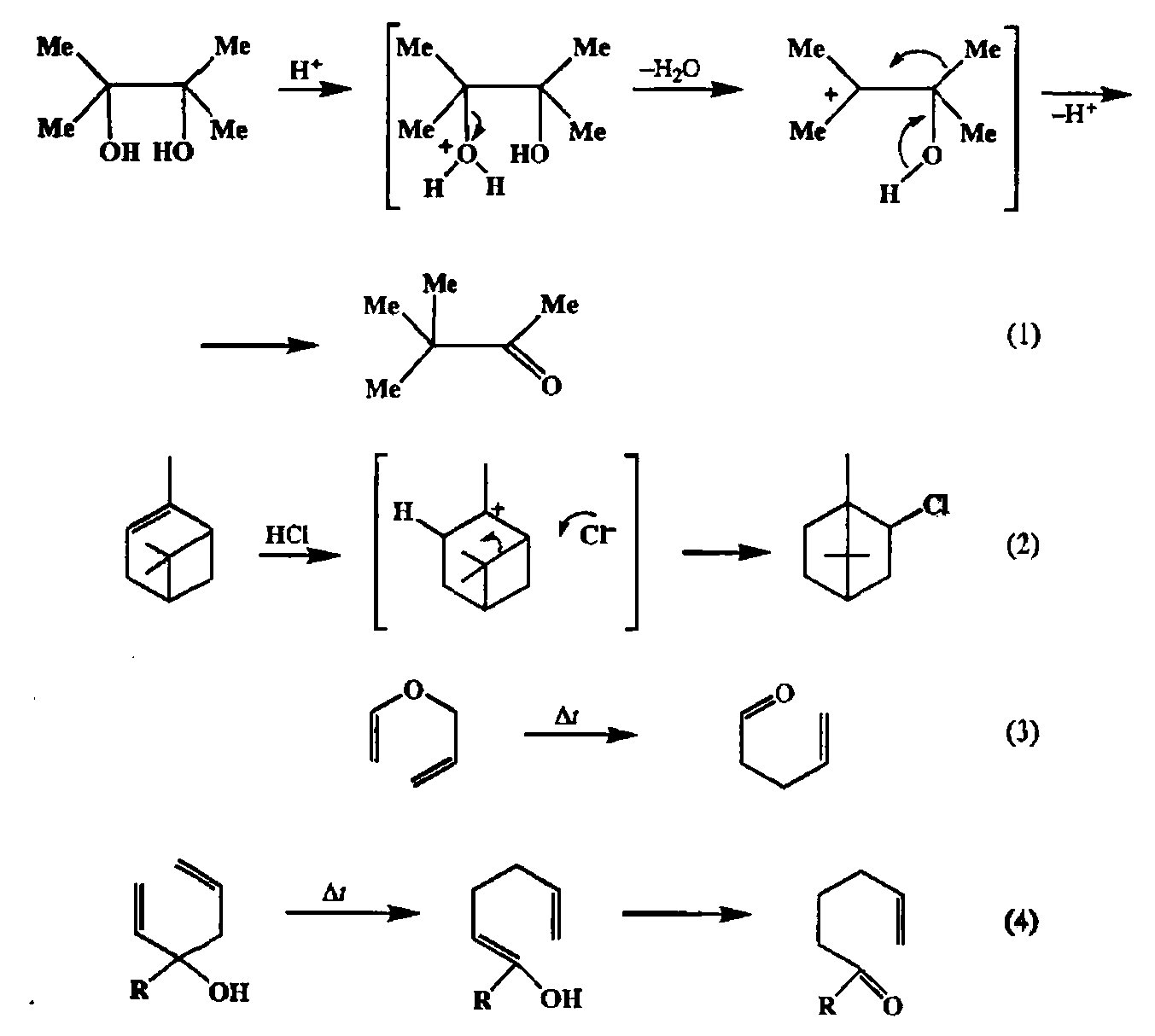
В числе этих примеров мы видим пинаколиновую перегруппировку (1) и близкородственную ей по химизму перегруппировку Вагнера—Меервейна (2), перегруппировку винилаллиловых эфиров (3) (перегруппировка Ююйзе-на) и превращение аллилвинилкарбинолов в 6,е-ненасыщенные карбонильные соединения (4) (гидрокси-перегруппировка Коупа). По существу перегруппировкой является также реакция Байера—Виллигера (см. выше), хотя традиционно ее называют «реакцией», а не «перегруппировкой».
Открытие скелетных перегруппировок в начале XX в. породило немало проблем, поскольку превращения подобного типа казались совершенно необъяснимыми в рамках существовавших в то время представлений о реакционной способности органических соединении. Однако именно возникновение такого рода проблем и явилось одним из стимулов для развития современной структурной теории органической химии. Достаточно напомнить, что создание концепции карбокатионных интермедиатов было самым непосредственным образом связано с изучением закономерностей хода перегруппировок типа перегруппировки Вагнера—Меервейна. Благодаря успехам, достигнутым в ходе углубленного исследования механизма скелетных перегруппировок различных типов, удалось создать цельную систему концепций, позволяющую не только грамотно объяснять особенности протекания такого рода превращений для модельных простых примеров, но и надежно прогнозировать результаты их применения к новым более сложным системам. Тем самым были созданы предпосылки для использования реакций, протекающих с изменением углеродного скелета в качестве надежного инс-тумента направленного органического синтеза [40а].
Известно несколько десятков реакций самых различных типов, результатом которых является скелетная перегруппировка. Ниже мы рассмотрим лишь те из них, которые наиболее часто применяются в полном синтезе.
2.7.3.1. Перегруппировка Кляйзена-Джонсона—Айрленда и гидрокси-перегруппировка Коупа
Как показано в общем виде на схеме 2.154, синтетический результат перегруппировки Кляйзена сводится к введению аллильного фрагмента по а-ато-му исходного карбонильного соединения через промежуточную стадию превращения кетона или альдегида в аллильный эфир енола 480 [40Ь]. Формально тот же результат (образование у,5-непредельного карбонильного производного 481) может быть получен по уже известной нам реакции адкилиро-вания ионных енолятов с помощью аллильных электрофилов. Однако как требования к природе субстратов, используемых в этих методах, так и механизм и условия проведения показанных реакций, резко различны, что и предопределяет синтетическое использование обоих показанных вариантов в качестве дополняющих друг друга альтернатив.
Схема 2.154 |

Одним из методов получения аллилвиниловых эфиров служит переэтери-фикация алкилвиниловых эфиров аллиловыми спиртами, катализируемая солями ртути. Мягкость условий проведения этой реакции делает возможным ее применение для получения субстратов перегруппировки Кляйзена содержащих самые разные структурные фрагменты в составе винильного или аллильного остатков. Это позволяет применять тандем реакций перс-этерификация — перегруппировка Кляйзена в качестве эффективного метода решения целого ряда синтетических задач, трудно решаемых другими способами. Некоторые типовые примеры даны на схеме 2.155.
Схема 2.155 |

Превращение аллилвинилового эфира 482 в альдегид 483 иллюстрирует уникальность синтетического потенциала перегруппировки Кляйзена как метода получения ангулярно замещенных производных с использованием легко доступных исходных веществ, таких, как спирт 484 [40с]. Получение даже такой относительно несложной структуры, как альдегид 483, другими путями было бы очень непростой синтетической задачей. Неудивительно, что реакции, подобные показанному превращению 482-»483, нашли широкое применение в синтезе различных полициклических природных соединений [40а].
Перегруппировка Кляйзена относится к типу перициклических [3.3]-сиг-матропных перегруппировок, протекающих через образование квазициклического переходного состояния. Стерическая направленность такого рода превращений зависит от наличия и природы заместителей, определяющих предпочтительную конформацию переходного состояния [40Ь]. Учет этих соображений и детальный анализ моделей переходного состояния перегруппировки Кляйзена позволили разработать оригинальный и простой метод сте-реоселективного синтеза олефиновых производных с Е-конфигурацией двойной связи с помощью этой перегруппировки. Особенно эффектный пример использования достоинств такого подхода был дан в работах Джонсона [40d] по стерсонаправленному итеративному синтезу регулярных изопренои-дов. Исходным соединением в таком синтезе служил бис-аллиловый спирт Сш (485), который по схеме переэтерификации с избытком 2-метоксиизопре-на (486) превращался в бис-эфир 487. Последний при нагревании легко претерпевает двойную перегруппировку Кляйзена, что приводит к стереоселек-тивному получению дикетона 488, в котором обе двойные связи внутри центрального фрагмента имеют требуемую Е-конгигурацию. Восстаношгение обеих кетогрупп этого продукта и повторение стадий переэтерификации под действием 486 и перегруппировки Кляйзена дает аддукт 489. Последний уже содержит в готовом виде С^-скелет природного тритерпена, сквалена, который таким образом был получен по беспрецедентно короткой схеме симметричного удлинения цепи, отвечающей сборке из блоков С10 + 2 С5 + 2 С5.
Область препаративной применимости перегруппировки Кляйзена значительно расширилась, когда было найдено, что с ее помощью можно получать не только у,6-непредельные альдегиды и кетсны, но и эфиры у,5-непредельных карбоновых кислот. В последовательности превращений, разработанной Джонсоном для этой цели (схема 2.156), все стадии — получение смешанного ортоэфира 490 переэтерификацией исходного аллилового эфира, образование промежуточного кетенацеталя 491 и его перегуппировка с образованием конечного продукта, эфира 492 — осуществляются в одной колбе, этот ор-тоэфирный вариант перегруппировки Кляйзена, известный под названием метода Джонсона—Кляйзена, является сейчас одним из самых эффективных и простых в исполнении путей синтеза прозводных типа 492 [40d,e].
Дальнейшее усовершенствование этого метода — вариант, предложенный Айрлендом [40f[, в котором исходными субстратами служат легко получаемые аллиловыс эфиры кислот типа 493 (схема 2.156). Обработка этих эфиров в обычных условиях генерации енолятов (действием диизопропил-амида лития, LDA) приводит к литиевым производных 494, из которых при действии триметилхлорсилилана образуются соответствующие силиловые производные 495. Перегруппировка 494 → 496 протекает уже при понижен-ноитемпературе, в то время как превращение 495 → 496 осуществляется при комнатнойтеипературе. Отметим, что как в классическом варианте проведения перегруппировки Кляйзена, так и в варианте Джонсона, реакция протекает, как правило, в интервале температур 140-160˚С.
Схема 2.156 |
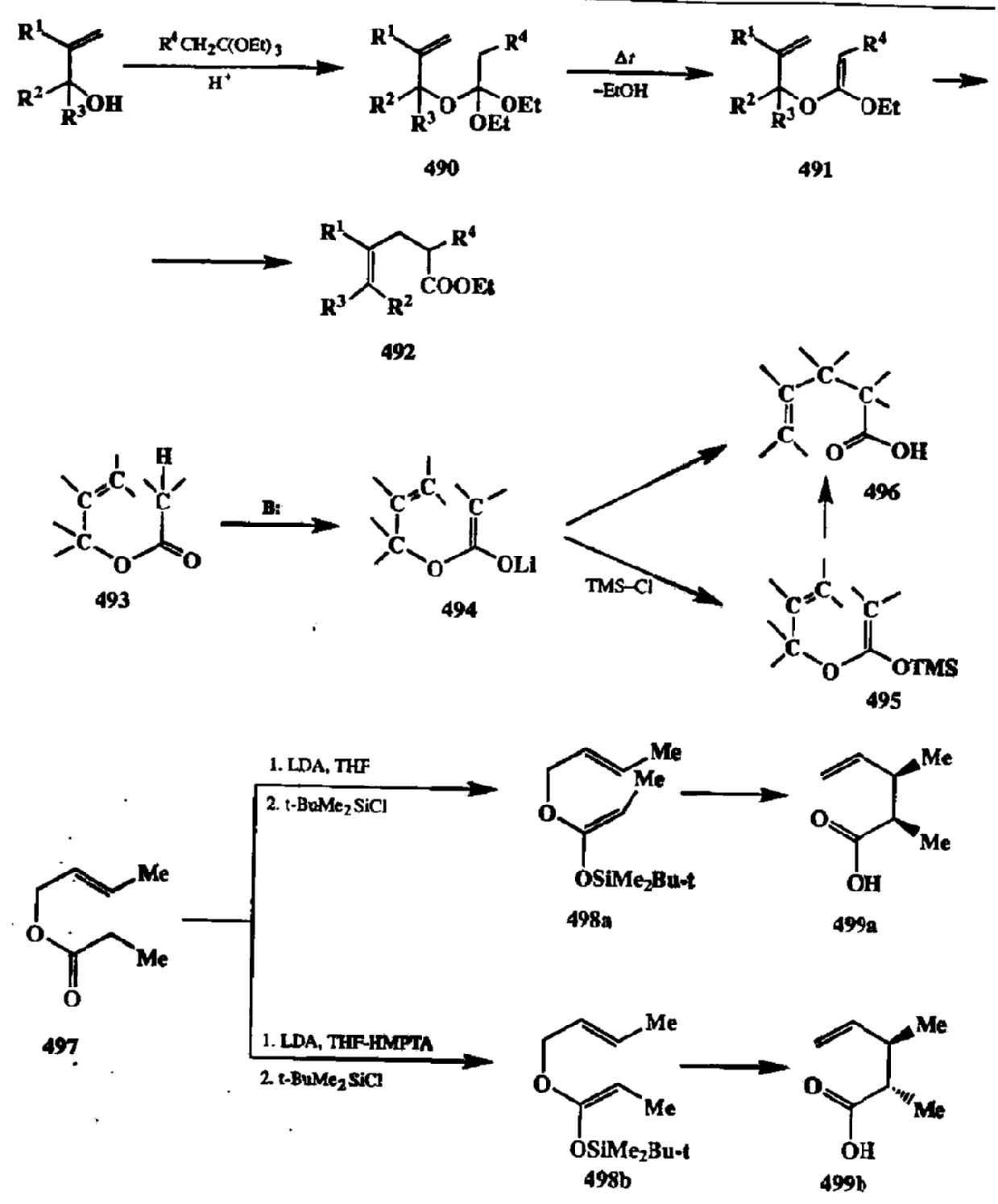
Вариант Айрленда, предусматривающий промежуточное образование триметилсилилкетенацеталей (495), оказался особенно удобным для стерео-контролируемого синтеза кислот типа 496. Дело в том, что конфигурацию двойной связи в промежуточно образующемся кетенацетальном фрагменте можно контролировать подбором условий образования енолятов, как это показано на схеме 2.156 на примере получения из пропионата 497 как Е-, так и Z-изомеров силилкетенацеталей 498а и 498Ь соответственно. Эти еноляты могут быть легко выделены и очищены от примесей изомеров. Последующая перегруппировка Кляйзена приводит к стереоспецифическому образованию диастереомеров 499а и 499b [40b,g].
Гидрокси-перегруппировка Коупа относится к тому же типу перицикли-ческих [3.3] сигматропных перегруппировок [40h], и се механизм также предполагает образование квазициклического шестичленного переходного состояния. Это превращение является общим методом трансформации 3-гидроксизамещенных гексадиенов-1,5 500 в 5,Б-ненасышенные карбонильные соединения типа 501 (схема 2.157). Одно из достоинств этого спссо ба — возможность достаточно легкого получения требуемых субстратов, например, с помощью методов, представленных на схеме.
Схема 2.157 |

Однако классический вариант требовал довольно жестких условий проведения реакции (термолиз в интервале температур 150—200°С), что существенно ограничивало возможности его синтетического применения. Ситуации резко изменилась после того, как было обнаружено, что скорость этой перегруппировки сильно возрастает (в Ю10—1015 раз!), если в качестве субстрата реакции использовать не спирты типа 500, а их алкоголяты [40i]. Так, например, для превращения спирта 502 в кетон 503 требуется термолиз в течении нескольких часов при 170-200'С. То же превращение для калиевого алкоголята 502а протекает в кипящем тетрагидрофуране за несколько минут (интересно, что этот эффект гораздо слабее выражен для литиевого или на триевого алкоголята). Если же в среду добавить 18-краун-б, то перегруппировка калиевого алкоголята проходит уже при 0°С.
В результате разработки алкоголятного способа проведения гидрокси-перегруппировка Коупа приобрела значение действительно общего метода, исключительной синтетической важности [40J]. Ниже мы рассмотрим примеры, иллюстрирующие некоторые типичные аспекты препаративного использования этой реакции.
Основная проблема полного синтеза перипланона В (504), полового ат-трактанта таракана, — построение функционализированного 10-членного кольца. Как уже отмечалось ранее, синтез циклов подобного размера не является тривиальной задачей. Эта проблема была эффективно рещена с помощью гидрокси-перегруппировки Коупа. В разработанном группой Стилла синтезе 504 [40k] непосредственным предшественником для перехода к деся-тичленной системе служил карбинол 505, получение которого из циклогек-сенона 506 было осуществлено с помощью короткой последовательности несложных превращений (схема 2.158).
Перегруппировка 505 протекала без осложнений и с хорошим выходом привела к продукту 507. Наличие в последнем заместителей и двух двойных связей в требуемых положениях позволило далее получить не только 504, но и использовать тот же предшественник 507 для синтеза еще двух диастереомеров 504, что было необходимо для подтверждения стереохимии природного продукта {см. разд. 1.4). Уместно отметить, что во втором синтезе 504, осуществленном несколько позднее по совершенно другой схеме [401], построение 10-членного цикла также проводилось с помощью гидрокси-перегруппировки Коупа.
Схема 2.158 |

Превращение 505 -> 507 представляет собой лишь один из примеров общей реакции расширения циклов 1,2-дивинилциклоалканолов в циклоалке-ноны с увеличением размера цикла на четыре звена. Этот путь часто применяется на практике, как это проиллюстрировано на примере синтеза цикло-ноненона (508) и циклогексадеценона (509) из 1,2-дивинилциклопентанола (510) и циклододеканола (511) соответственно [40j] (схема 2.158).