

2.6.3. Циклоприсоединение - методы, специально созданные для получения циклических структур
Нетрудно заметить, что все ранее рассмотренные методы циклообразования имеют одну общую особенность: циклизация осуществляется как внутримолекулярная реакция замыкания единственной связи, недостающей до циклической структуры.
Существует, однако, обширный класс методов, основанных на ином топологическом принципе. В этих методах образование цикла может происходить как в результате внутримолекулярной реакции, так и межмолекулярным путем, но во всех случаях оно происходит за счет образования двух или более связей в пределах одного реакционного акта. Это — реакции циклоприсоеди-нения. Примечательной особенностью всех этих реакций является высокая селективность циклообразования, что предопределено самим химизмом взаимодействия, по своей природе исключающим возможность альтернативных путей, таких, как олигомеризация или образование циклов иного размера.
2.6.3.1. [4 + 2]-Циклоприсоединение
Среди множества реакций, относящихся к этому классу, особое место занимает [4 + 2]-циклоприсоединение. Это — реакция Дильса—Альдера [2а], как правило, не требующая катализа или иницирования облучением. В этой реакции происходит образование шестичленного цикла в результате взаимодействия сопряженного диена ^-компоненты) и диенофила ((^-компоненты) с образованием циклического переходного состояния, в котором шесть л-электро-нов исходных соединений образуют единое электронное облако квазиароматического типа (324, схема 2.117) [31а,B].
Именно благодаря такому эффекту стабилизации переходного состояния последнее оказывается достаточно энергетически выгодным, а потому энергия активации реакции относительно низкой. Для несимметрично замещенных диенов и диенофилов возможно образование более чем одного переходного состояния типа 324. Однако эти изомерные переходные состояния достаточно различны по энергии, вследствие чего наиболее обычным результатом реакции Дильса—Альдера является исключительное, или по крайней мере преимущественное, образование одного из возможных изомерных продуктов (по положению или взаимной ориентации заместителей). Ход реакции адекватно объясняется в рамках концепции сохранения орбитальной симметрии Вудворда—Хоффмана [31с], и, как правило, конечный результат реакции хорошо предсказуем даже для очень непростых случаев [31d].
EWG – электроноакцепторная группа Схема 2.117 |

В классическом варианте реакции Дильса—Альдера в качестве 4л-компо-ненты используются различные соединения, содержащие 1,3-диеновый фрагмент, а в качестве 2я-компоненты, диенофила, — алкены или алкины, содержащие электроноакцепторные группы (EWG), такие, как сопряженные альдегиды, кетоны, кислоты и их производные, нитроалкены и т. д. Типичный набор карбо- и гетероциклических структур, получаемых с помощью этой реакции, показан на схеме 2.118.
Схема 2.118 |

Для того чтобы более полно охарактеризовать достоинства реакции Дильса—Альдера как препаративного метода, полезно хотя бы вкратце остановиться на особенностях регио- и стереонаправленности этой реакции. Прежде всего, следует отметить, что сам механизм реакции подразумевает, что образование циклических продуктов протекает с полным сохранением'конфигурации заместителей в обоих фрагментах (см. превращения 325 ->• 326 или 327 -> 328 на схеме 2.119). Для несимметричных диенов и диенофилов наблюдается преимущественное образование одного из возможных региоизо-меров (см., например, образование 329 или 330). Для 1,4-дизамещенных диенов возможно образование двух переходных состояний, отвечающих экзо-или энйо-ориентации рееагентов. Типичным результатом циклоприсоедине-ния в таких случаях является преимущественное или даже исключительное образование эядо-аддуктов (см., например, 331а).
В рамках общей схемы [4 + 2]-циклоприсоединения можно реализовать синтезы с введением в состав собираемого шестичленного фрагмента самых неожиданных (на первый взгляд!) заместителей, что может быть обеспечено подбором соответствующих структур 4л- или 2я-компоненты с тем, чтобы они содержали легко трансформируемые или удаляемые группы.
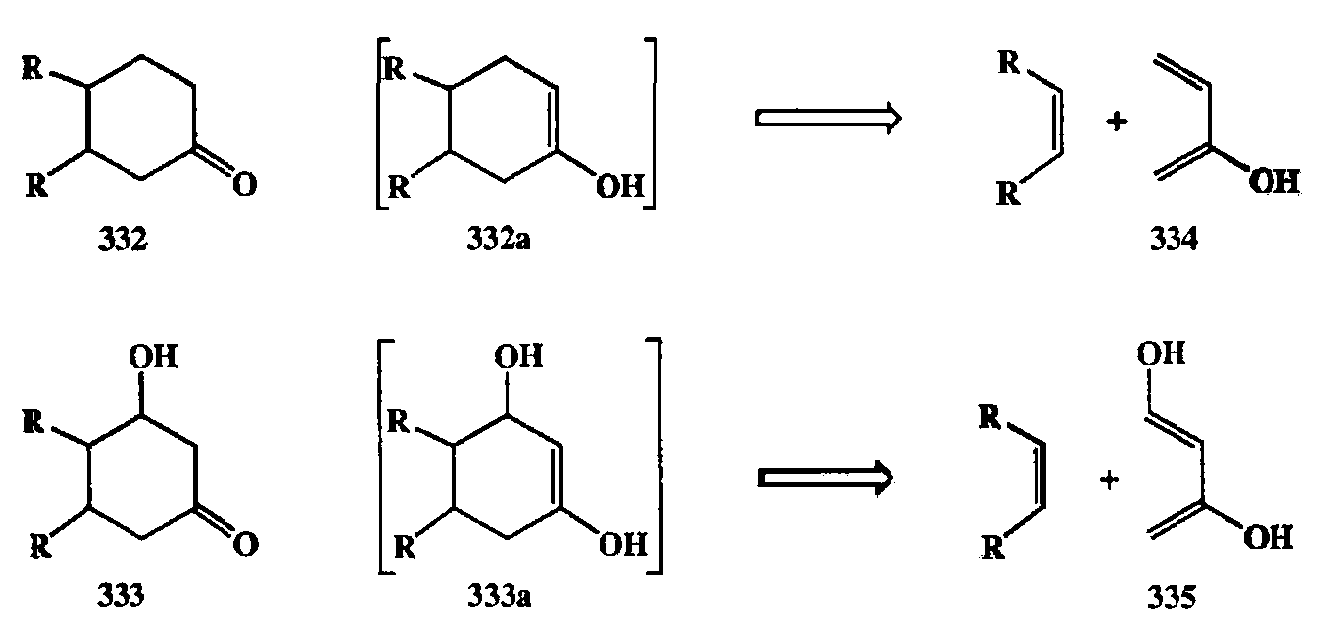
Рассмотрим, например, ретросинтетический анализ структур 332 или 333 на схеме 2.120. Для того чтобы представить себе возможность сборки таких продуктов по схеме диенового синтеза, полезно прежде всего превратить их (ретросинтетически) в енолы 332а и 333а соответственно. Если далее применить разборку по схеме ретродиенового синтеза, то мы автоматически придем к структурам 334 и 335 для диеновых компонент.
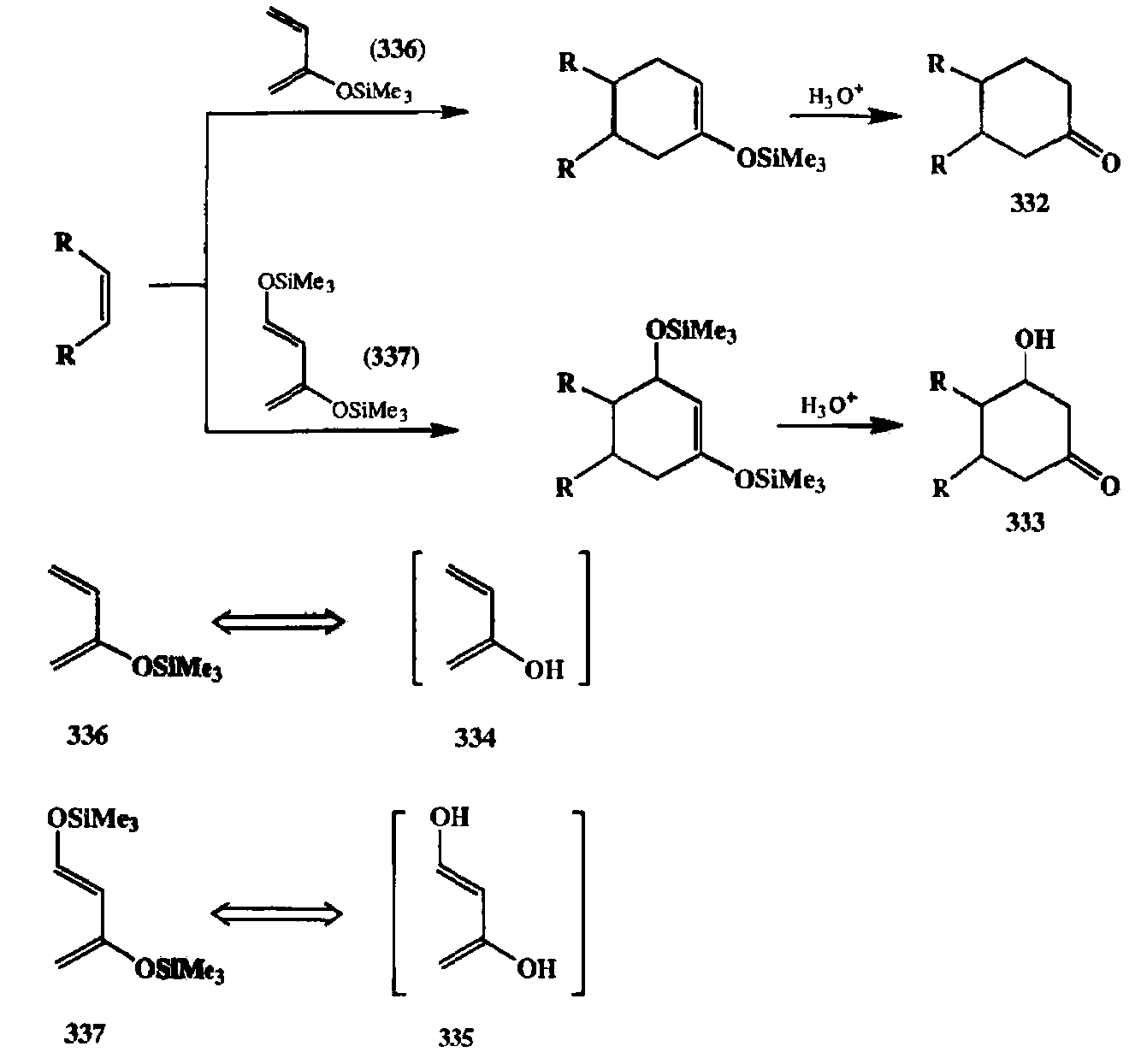
Может показаться, что такой путь разборки неконструктивен, так как подобного рода диены неспособны существовать как реагенты. Однако все становится на свои места, если вспомнить, что разнообразные силиловые эфиры енолов относятся к категории вполне доступных и стабильных производных. Понятно, что с помощью диенов типа силиловых эфиров 336 и 337, используемых в качестве стабильных эквивалентов диенов 334 и 335, требуемые превращения легко могут быть осуществлены [31е] (см. схему 2.121).
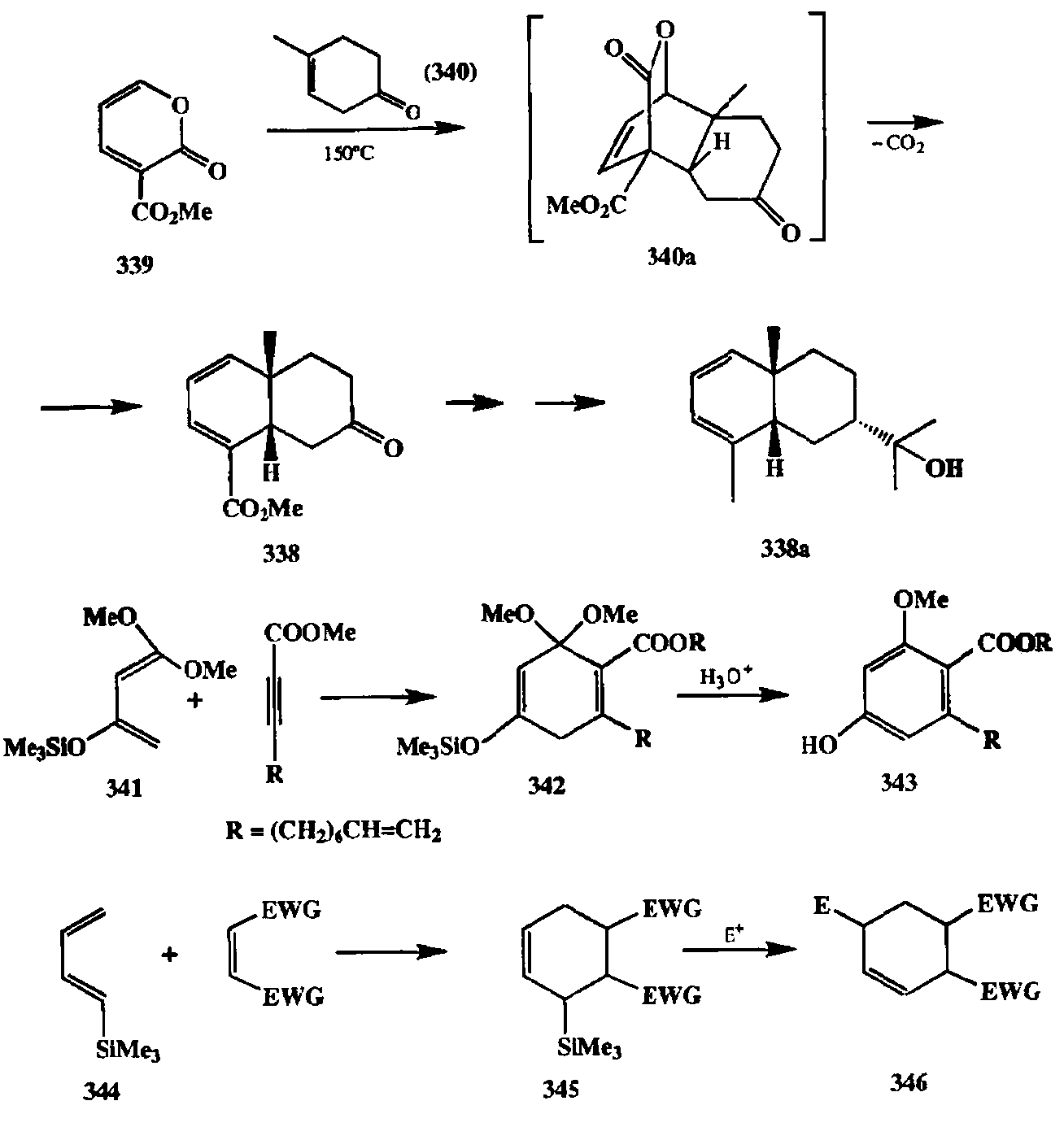
Другие примеры, иллюстрирующие некоторые неочевидные возможности Применения диенового синтеза с участием модифицированных диенов и диенофилов, приведены на схеме 2.122. В синтезе природного сесквитерпена гидроксиденталола (338а) ключевая стадии построения бициклического скелета, содержащего 1,3-диеновый фрагмент (продукт 338), выполняется по схеме диенового синтеза с использованием пирона-2 (339) как диеновой компоненты и диенофила 340. Первоначально получаемый при этом аддукт 340а легко подвергается декарбоксилированию в условиях реакции, что и приводит к получению требуемого промежуточного продукта 338 [31П. Использование как диена 338, так и производных, содержащих в своем составе подобного рода фрагмент, достаточно распространено в синтезе самых различных полициклических соединений, содержащих циклогексадиеновый остаток [31g]. Силоксидиен 341 был сконструирован для того, чтобы обеспечить возможность получения циклогексадиеновых и ароматических аддуктов с необычным типом замещения. Как показано на схеме 2.122, диеновый синтез с участием 341 и ацетиленового диенофила дает аддукт 342. Кислотный гидролиз последнего приводит в конечном счете к тетразамещенному ароматическому
Схема 2.119
|
Схема 2.120 |

продукту 343, получение которого другим путем было бы довольно затруднительно. Продукт 343 далее использовался в синтезе одного из природных ингибиторов роста растений, лазиодиплодина [31h].
Интересен синтетический потенциал, открываемый при проведении диенового синтеза с диеном 344. Непосредственно образуемые при этом аддук-ты типа 345 содержат группировку аллилсилана, что позволяет вводить их в реакции с различными электрофилами. Результатом последовательности превращений — диеновый синтез плюс электрофильное присоединение — является образование продуктов типа 346 (3 li] (схема 2.122), структура которых сама по себе никакие предполагает возможности их синтеза по схеме реакции Дильса—Альдера.
Сказанное выше в полной мере относится и к примерам вариаций на тему природы диенофила, показанным на схеме 2.122а. Так, диенофил 347 позволяет синтезировать (по реакции с циклопентадиеном) аддукт 348, из которого легко получаются аддукты 349 или 350 [31j]. В принципе, как 349, так к 350 могут быть получены непосредственным взаимодействием циклопентадиена с этиленом или соответствующим терминальным алкеном, однако условия подобных превращений с участием неактивированных диенофилов настолько жестки, что в препаративном плане такие возможности особого интереса не представляют. Напротив, благодаря использованию в качестве диенофила сульфона 347 как синтетического эквивалента алкеновых синтонов 347а и 347Ь, стало возможным применять реакцию Дильса—Альдера как общий метод синтеза циклических аддуктов, не содержащих электроноакцепторных групп.
Схема 2.121
Схема 2.122 |
Наличие трифенилфосфониевого фрагмента делает винильное производное 351 активным диенофилом. Легко видеть, что получаемая в результате реакции 351 с бутадиеном циклическая фосфониевая соль 352 представляет собой готовый реагент для генерации соответствующего фосфорана и синтеза (по Виттигу) диенов с экзоциклической двойной связью типа 353 [31k]. Получение последних непосредственно по схеме диенового синтеза потребовало бы использования в качестве диенофила аллена RCH=C=CH2, что мало перспективно из-за термической лабильности алленов.
Схема 2.122а |

Наконец, последний пример, показанный на схеме 2.122а, иллюстрирует возможность использования 1,1-дизамещенных диенофилов, например 354, как эквивалентов кетена. Действительно, аддукт 355, получаемый в результа-те взаимодействия замещенного циклопентадиена с диенофилом 354, может далее подвергаться щелочному гидролизу, что и приводит к кетону 356 [311], ключевому полупродукту в одном из первых стереоселективных синтезов простагландинов. Отметим, что 356 является продуктом формального диенового синтеза с участием кетена в качестве диенофила, однако из-за низкой активности (и малой термической стабилькости) кетена непосредственно реализовать такой синтез не удается.
Совершенно особое место занимает реакция Дильса—Альдера в синтезах полицикических структур каркасного типа, таких, как, например, баскетен (357) (схема 2.123) [31m]. В этом случае реакция между диеном 358 (в этой форме реагирует циклоктатетраен) и малеиновым ангидридом позволяет сразу получить с количественным выходом трициклическую структуру 359. Последующая стадия [2+2]-циклоприсоединения (об этой реакции см. следующий раздел) приводит к образовнию каркасного соединения 360, из которого в результате последовательности несложных стадий (омыление и окислительное декарбоксилирование) был получен целевой продукт 357.
Столь же эффективно используется диеновый синтез на ключевых стадиях получения кубана (361), пентапризмана (362) [31п] и многих других представителей этого экзотического класса органических соединений. Можно смело утверждать, что вообще синтетики вряд ли бы предпринимали синтез структур такой степени сложности, если бы не располагали столь мощным методом, как реакция Дильса— Альдера.
Начиная с 1960-х годов все более важную роль в полном синтезе начинает играть внутримолекулярная реакция Дильса—Альдера [31о]. На схеме 2.124 приведена выборка некоторых представительных примеров, позволяющая судить об особенностях протекания внутримолекулярного варианта диенового синтеза и специфике его использования в полном синтезе.
Как мы уже отмечали, алкены, не содержащие электроноакцелторных групп, являются очень «вялыми» диенофилами, и случаи их препаративного использования в межмолекулярном диеновом синтезе довольно редки. Показанный на схеме эффективный синтез крайне напряженной структуры брексена (363) в одну стадию из доступного предшественника 364 (через стадию равновесной изомеризации последнего в 364а) по схеме внутримолекулярного диенового синтеза наглядно показывает уникальные препаративные возможности этого метода [31р].
Схема 2.123 |

Превращения 365а -* 366а и 365Ь -> 366Ъ [3 lo,r] приведены как типичные примеры, иллюстрирующие особенности структурной и пространственной направленности реакции. Отметим, в частности, что региоселективность образования этих аддуктов обратна той, которую можно было бы ожидать на основании сравнения с аналогичным превращениям в межмолекулярном варинте 367 + 368 -> 369. Интересно также что, если образование 366а легко описывается в терминах стандартной схемы эидо-присоедине ния, то для интерпретации стереохимического результата превращения 365Ь -* 366Ь необходимо предположить исключительную экзо-ориентацию реагирующих фрагментов. Возможность реализации схем как эндо-, так и экзо-присоединения может показаться серьезным недостатком внутримолекулярной реакции Дильса—Альдера, однако на самом деле обычно бывает нетрудно предсказать результат для того или иного конкретного примера на основе рассмотрения особенностей несвязанных взаимодействий в альтернативных структурах соответствующих переходных состояний. Более того, удается также направлять реакцию по пути экзо-или эидо-присоединения подбором размера цепи, связывающей реагирующие центры и/или введением каких-либо вспомогательных групп [31 о].
Внутримолекулярное [4 + 2] -циклоприсоединение было использовано на ключевой стадии синтеза форскиолина (370) [31s] (схема 2.124), природного биологически активного вещества. Для получения требуемого предшествен ника легко доступный спирт 371 был превращен в эфир 372. Последний содержит в своем составе и диенофильную группу, и диеновый фрагмент. Хотя диены подобной степени замещенности обычно бывают крайне инертными компонентами в межмолекулярном варианте диенового синтеза, внутримолекулярная циклизация 372 -> 373 протекала достаточно легко и дала требуемый продукт с почти количественным выходом. Характер функциональности аддукта 373 позволил далее относительно легко завершить построение структуры 370.
Очевидные преимущества внутримолекулярной реакции Дильса—Альде-ра стимулировали поиски с целью разработки способов создания временных мостиков между диеном и диенофилом. Особенно плодотворными оказались исследования в этой области, выполненные в группе Сторка [31t], целью которых было выяснение возможностей использования в качестве мо-стиковых групп фрагментов, содержащих атомы кремния, магния или алюминия. Эффективность такого подхода показана на примере разных вариантов модельного превращения 374 -> 375 через промежуточные стадии 374а и 374Ь (схема 2.124). Примечательно, что диеновый синтез протекал особенно легко в том случае, когда в качестве мостиковых гетероатомов использовалась пара O-Mg (374a, Z=Mg). Подкупающе проста процедура получения подобного рода производных взаимодействием литиевого алкоголята спирта 374 с винилмагнийбромидом. Хотя диенофилом в структуре 374а является по сути дела карбанионный фрагмент, тем не менее превращение 374а (Z=Mg) -> 374b протекало в довольно мягких условиях (нагревание при 80°С в течении нескольких часов). Та же реакция другого, явно ковалентного производного — кремнийсодержащего аналога 374а (Z=SiMe2) — требовала нагревания при 160°С. Отметим, что для всех производных типа 374Ь завершающая стадия — превращение в продукт 375 — легко достигалось путем гидролиза. Примечательная эффективность внутримолекулярного диенового синтеза побудила более внимательно изучить вопросы биогенеза некоторых типов полициклических соединений, синтез которых по этой схеме казался вполне реальным с точки зрения синтетиков. Оказалось, что Природа не менее нашего «осведомлена» о подобных возможностях, и действительно для ряда примеров было экспериментально доказано использование внутримолекулярной циклизции по Дильсу—Альдеру как одной из стадий биосинтеза. Так, из патогенных грибов Alternaria solan's были выделены фитотоксины, со-ланопироны А и D (376а и 376Ь). Сам факт выделения пары диастереомеров из одного и того же природного источника был достаточно необычным, ибо в подавляющем большинстве случаев биосинтетические превращения протекают с абсолютной стереоселективностью. Изучение биосинтеза этих соединений с использованием изотопно меченых соединений показало, что они образуются в результате циклизации общего предшественника 377 [31u,v], как это показано на схеме 2.125.
Схема 2.124 |

Схема 2.125 |
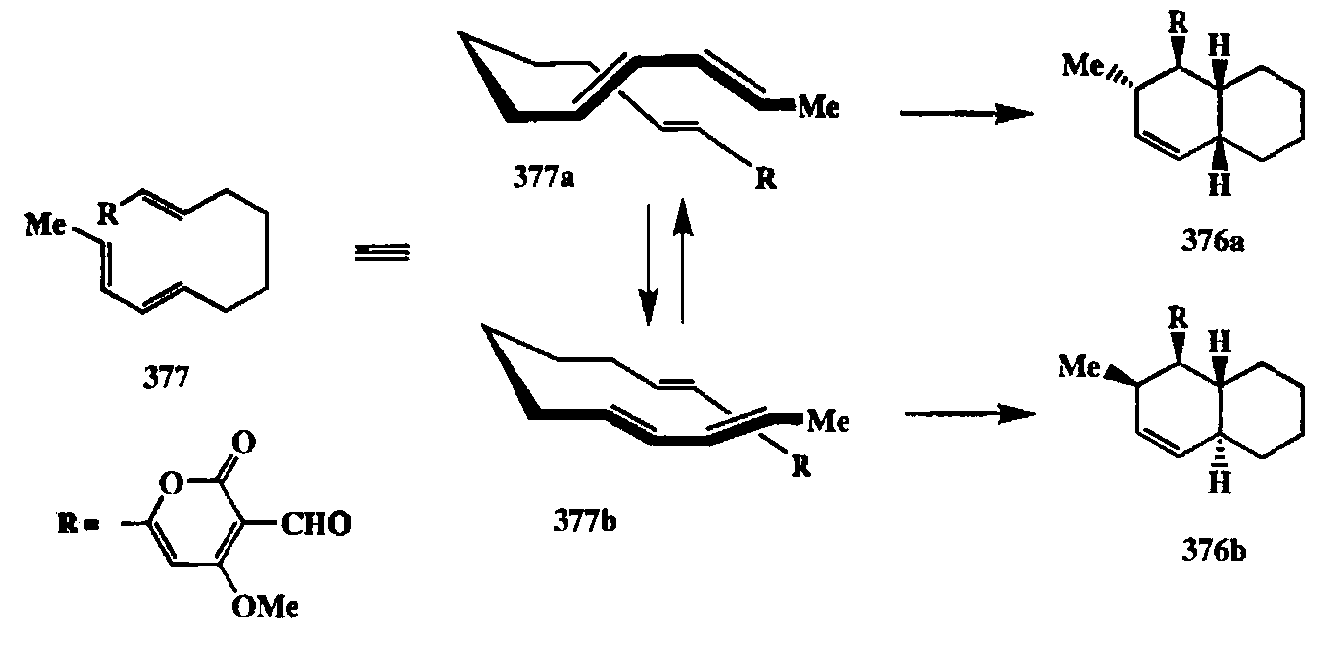
Та же циклизация триена 377 при проведении ее в колбе (in vitro) протекала при нагревании и привела к образованию тех же диастереомеров 376а и 376Ь примерно в том же соотношении, в каком они образуются в клетке (in vivo) [31w]. Рассмотрение моделей показало, что циклизация 377 может проходить как по схеме эндо-присоединения (для конформера 377а), так и по схеме экзо-присоединения (для конформера 377Ь). Расчеты также показали, что эти конформеры, как и образующиеся из них переходные состояния циклизации, очень мало различаются по своей энергии. По-видимому, именно благодаря этим факторам циклизация 377 как in vivo, так и in vitro протекает с низкой стереоселективностью.
В последующих разделах мы еще неоднократно будем рассматривать примеры применения самых различных вариантов реакций Дильса—Альдера в синтезе.
2.6.3.2. [2 + 2]-Циклоприсоединение в синтезе производных циклобутана
[2 + 2)-Циклоприсоединенис относится к категории важнейших синтетических методов, поскольку эта реакция позволяет получать различные производные циклобутана по схеме сборки из двух алкеновых фрагментов. Этот процесс может протекать как по согласованому механизму через образование циклического переходного состояния (а), так и по стадиям, включающим промежуточное образование бирадикального (B) или биполярного (с) интермедиата (схема 2.126). Реализация того или иного из этих механизмов зависит как от строения реагентов, так и от условий проведения реакции.
Схема 2.126 |
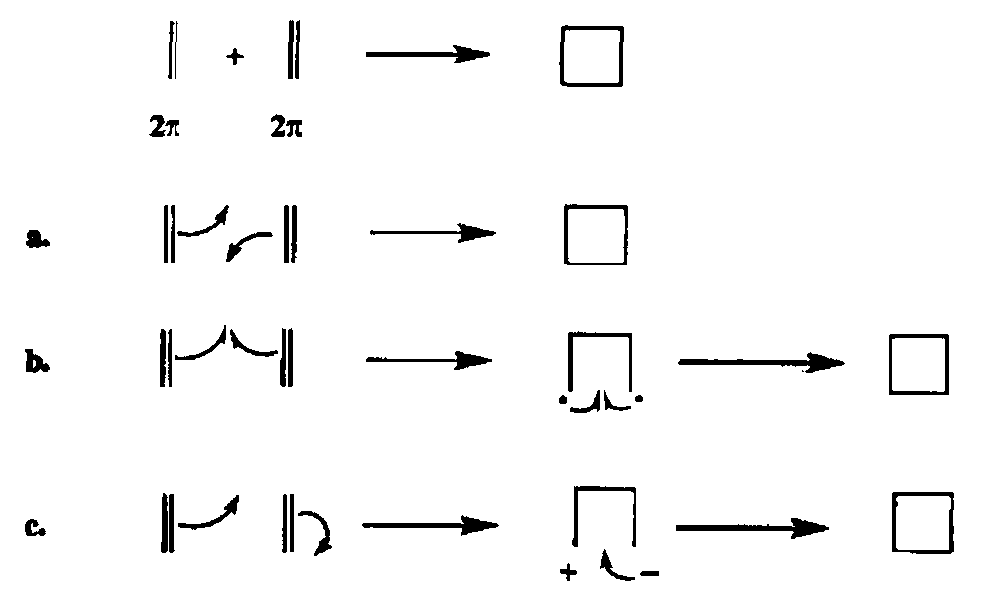
В случае термически индуцированного процесса согласованный механизм реализуется лишь для ограниченной категории реакций, таких, как взаимодействие кетенов с алкенами или алкинами. Результатом реакции всегда является образование продуктов чис-присоединения [32а]. Сам кетен в силу своей неустойчивости относительно редко используется в этой реакции, и чаще всего для синтеза циклобутанонов применяют более стабильные а-хлоркетены, которые генерируются in situ (действием триэтиламина на хло-рангидриды а-хлорзамещенных кислот) в присутствии второго компонента циклоприсоединения. На схеме 2.127 приведены типичные примгры этой реакции — образование циклоаддуктов 378 [32Ь] и 379 [32с]. Отметим, что, хотя в последнем случае реакция протекала с довольно скромным выходом (около 20%), однако она оказалась самым удобным методом синтеза аддукта 379, из которого в одну стадию был получен микотоксин монилиформин (380), природное соединение уникальной структуры, которую довольно затруднительно получить другим способом.
Схема 2.127 |

а-Хлорциклобутаноны, образующиеся в рассматриваемой реакции, достаточно просто могут быть превращены в соответствующие циклобутаноны (восстановительным дехлорированием) или их можно непосредственно использовать в дальнейших превращениях в качестве субстратов, содержащих хорошую уходящую группу (а-хлорзаместитель) (см. далее).
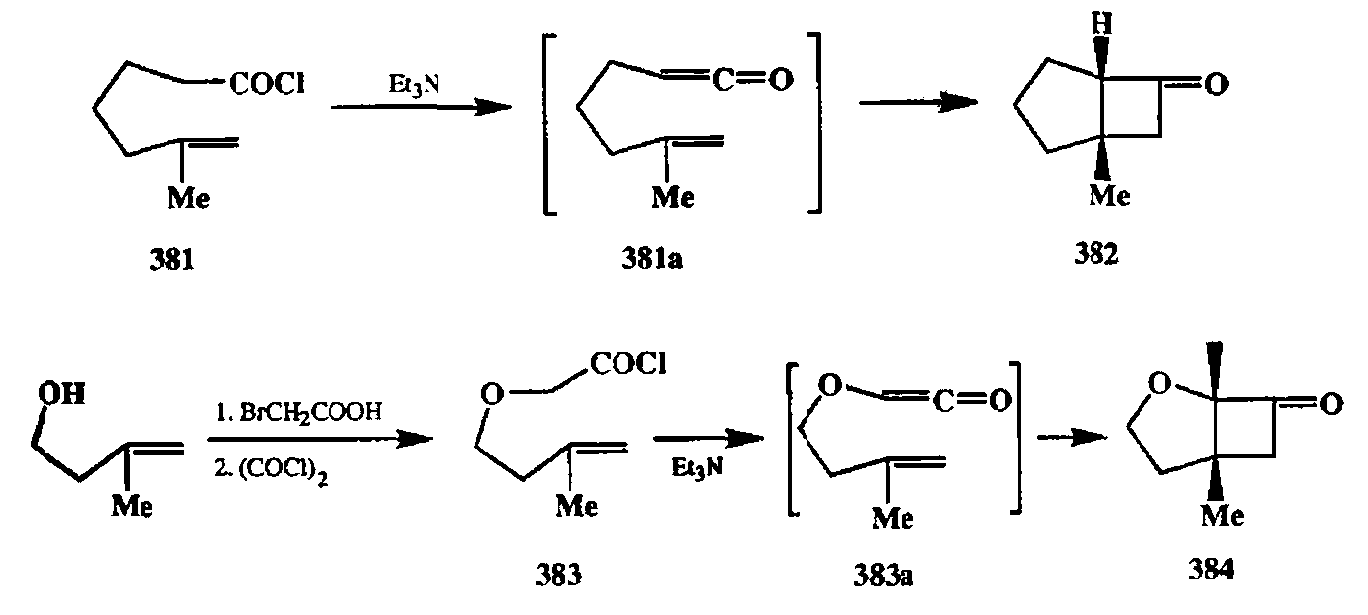
Важным для синтеза является внутримолекулярное [2 + 21-циклопри-соединение кетенов к алкенам. Для этой реакции оптимальное расстояние между двумя кратными связями соответствует мостику из трех звеньев [32d]. Поэтому этот путь особенно удобен для получения различных структур, содержащих 4,5-сочлененный бициклический фрагмент. Типичными примерами являются показанные на схеме 2.128 превращения 381 -> 382 [32е] и 383 -> 384 [32f]. В этих превращениях генерация кетеновой функции также проводится in situ, но в этом случае, благодаря легкости протекания внутримолекулярной циклизации, уже не требуется наличия а-хлорзаместителя в кетеновом фрагменте (см. структуры интермедиатов 381а и 383а). Очевидным достоинством метода является легкость получения требуемых предшественников, как это показано на примере синтеза эфира 383.
Схема 2.128 |
Фотохимически индуцированное [2 + 21-циклоприсоединение относится к категории важнейших реакций, особенно б синтезе стерически затрудненных соединений самых экзотических структурных типов [32g|. Эта реакция может протекать в соответствии с правилами сохранения орбитальной симметрии по согласованному механизму, но чаще реализуется бирадикальный механизм (пугьЬ, схема 2.126). В препаративной практике чаще всего применяется фотохимически индуцированное [2 + 2]-циклоприсоединение оле-фина к енону. Для этой реакции характерны высокая региоселективность, но подчас довольно низкая стереоселективность, что вполне согласуется с упомянутым бирадикальным механизмом.
Один из первых примеров, показавших полезность этой реакции для получения полициклических соединений, был описанный Кори [32h] синтез природного сесквитерпсна, а-кариофиллена (385) (схема 2.129). В качестве исходного вещества здесь был использован бициклический аддукт 386, полученный с высоким выходом по схеме фотоциклоприсоединения изобутиле-на к циклогексенону.
Схема 2.129 |
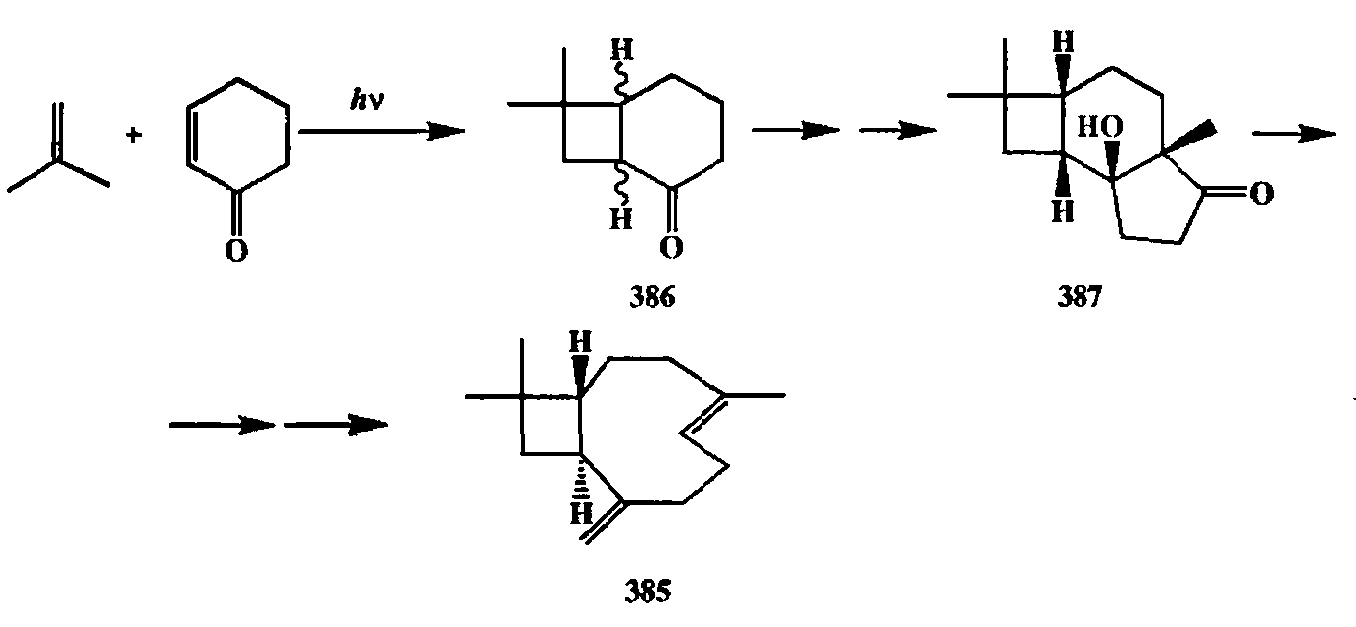
В структуре адцукта 386 уже содержался требуемый фрагмент 1,1-диме-тилциклобутана. Наличие в этом адцукте карбонильной функции позволило далее относительно легко достроить третий цикл и получить таким образом аддукт 387, уже содержавший 14 из 15 требуемых атомов углерода молекулы 385. Дальнейшая последовательность превращений, помимо трансформаций функциональных групп, включала также стадии разрыва центральной связи гидриндановой системы (по реакции фрагментации) с образованием 9-членного цикла и метиленирования по Виттигу,
Синтез а-кариофиллена наглядно продемонстрировал богатые синтетические возможности фотоциклоприсоединия олефина к енону, и вслед за ним последовало еще множество полных синтезов, в которых эта реакция использовалась на той или иной из ключевых стадий [32i].
Схема 2.130 |
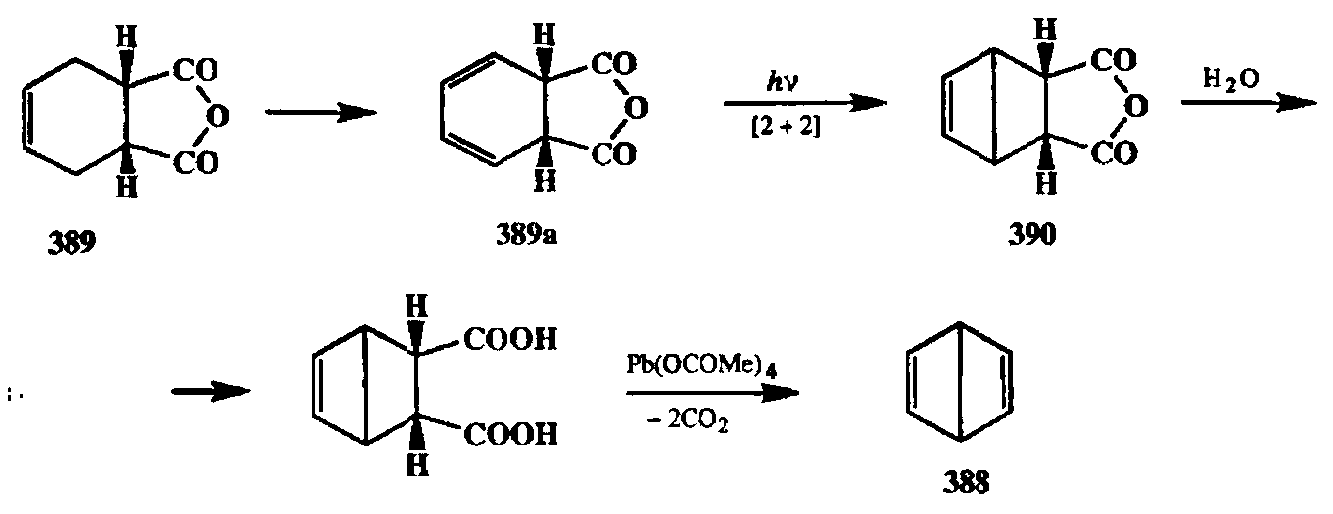
Фотохимически индуцированное [2 + 2]-циклоприсоединение во внутримолекулярном варианте проведения реакции оказалось незаменимым методом построения высоконапряженных молекул. Именно с помощью этой реакции за последние несколько десятилетий удалось получить «во плоти» множество структур, возникших «на кончике пера» как плод творческого воображения химиков-органиков. Одним из первых достижений такого рода был выполенный ван Тамеленом [laj удивительно короткий синтез бензола Дьюара (388), показанный на схеме 2.130.
На начальной стадии этого синтеза легко доступный продукт реакции Дильса—Альдера 389 с помощью последовательности бромирование/дегидробромирование превращался в диен 389а. При облучении последнего легко протекало [2 + 2]-циклоприсоединение с образованием трициклического продукта 390. Завершающие стадии синтеза 388 включали омыление ангидридного цикла и окислительное декарбоксилирование полученной дикислоты.
Отметим также, что в уже упоминавшемся синтезе баскетена (357) (схема 2.123) образование последней недостающей связи каркасной системы было также осуществлено с помощью [2 + 2]-фотоциклоприсоединения.
Внутримолекулярное [2 + 2]-фотоциклоприсоединение олефина к енону находит широкое применение в синтезе полициклических соединений как ключевая стадия, обеспечивющая быстрое усложнение скелета собираемой молекулярной конструкции [32j]. Хорошей иллюстрацией эффективности такого подхода могут служить превращения, показанные на схеме 2.131.
Схема 2.131 |
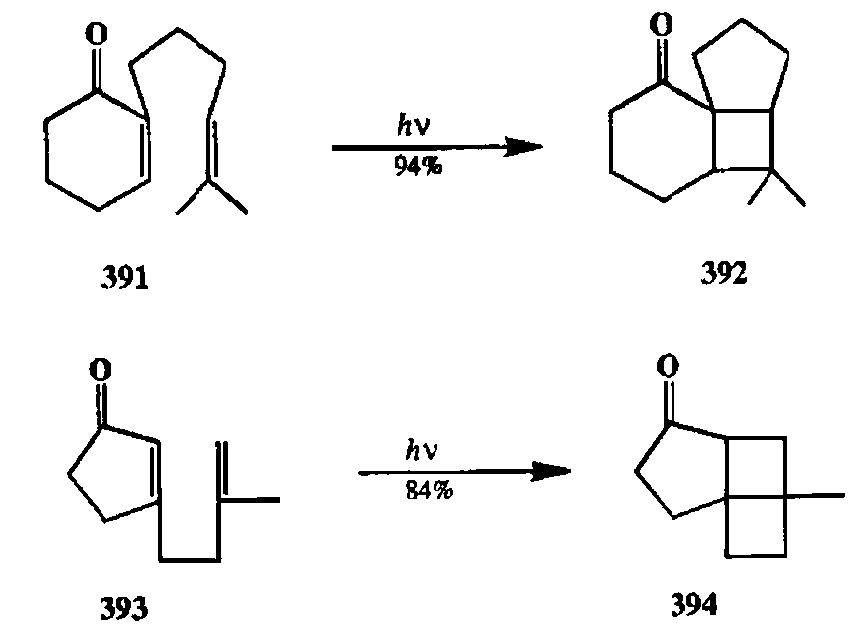
Действительно, превращения 391->392 [32k] и 393->394 [321] (равно как и многие другие, им подобные, см., например [32т]) протекают при комнатной температуре достаточно быстро с хорошим выходом, с высокой регио- и стереоселективностью и не требуют никаких реагентов или катализаторов, а только облучения. К этому следует добавить, что синтез требуемых диенонов типа 391 или 393 также несложен и легко может быть выполнен с помощью последовательности хорошо отработанных реакций, таких, как реакция Михаэля и ачкилирование енолятов. Таким образом, вырисовывается общая схема синтеза полициклических соединений, содержащих разные комбинации линейно и ангулярно сочлененных циклов, который включает две основные стадии, а именно: синтез полифункционального субстрата и его [2 + 2]-фотоциклоприсоединение. Дополнительная препаративная ценность такого протокола обусловлена тем, что получаемые напряженные системы могут легко претерпевать реакции с раскрытием циклобутанового фрагмента, а также скелетные перегруппировки [32т] (см. ниже, разд. 2.7.3.2.).
В последующих разделах этой, а также других глав, будет приведено еще немало примеров использования [2 + 2]-фотоциклоприсоединення (как в варианте алкен+ачкен, так и в варианте алкен+енон) для решения задач построения самых различных структур. Здесь уместно сделать еще одно замечание более общего характера. Структуры типа баскетена (357) или бензола Дьюара (388) относятся к числу богатых энергией жестких структур с системой напряженных связей С—С. По сути дела в ходе образования подобного рода систем происходит преобразование лучистой энергии в энергию химической связи. Ясно также, что превращения таких соединений, протекающие с разрывом напряженных фрагментов (например, под действием катализаторов), должны сопровождаться выделением энергии, запасенной при их синтезе. Поэтому внутримолекулярное фотоциклоприсоединение рассматривается сейчас не только как один из полезнейших инструментов органического синтеза, но и как перспективный путь создания систем, способных аккумулировать лучистую (в том числе солнечную) энергию в форме химической энергии, удобной для практического использования.
2.6.3.3. Синтез циклопропанов путем [2 + 1]-циклоприсоединения
Синтез трехчленных циклов по схеме циклоприсоединения должен, очевидно, включать взаимодействие непредельного субстрата, например алкена, с каким-либо Срреагентом, выступающим в роли синтетического эквивалента карбена 395 или его замещенных производных (схема 2.132).
Схема 2.132 |

Карбены — крайне нестабильные, реакционноспособные частицы [33а], которые можно зафиксировать лишь в аргоновой матрице при низких температурах (ниже 77К) [ЗЗЬ]. Некоторые сведения о природе их реакционной способности удалось получить из данных по газофазным превращениям [33с]. В большинстве методов синтеза циклопропанов, формально описываемых как присоединение карбена, на самом деле в реакции с непредельным субстратом участвует не карбен, как таковой, а какой-либо реагент, который выступает в роли переносчика карбена. При этом далеко не во всех случаях можно считать, что в таких реакциях карбен может действительно образовываться, хотя бы в качестве интермедиата [33d]. Поэтому часто используют более нейтральный термин — карбеноид, подразумевающий, что эффективным итогом реакции с рассматриваемым интгрмедиатом является перенос карбена, т. е. в уже привычных читателю терминах такой интермедиат, равно как и его предшественник, являются синтетическими эквивалентами карбена. Типичные реакции, используемые для генерации такого рода ин-термедиатов, представлены на схеме 2.133.
Схема 2.133 |
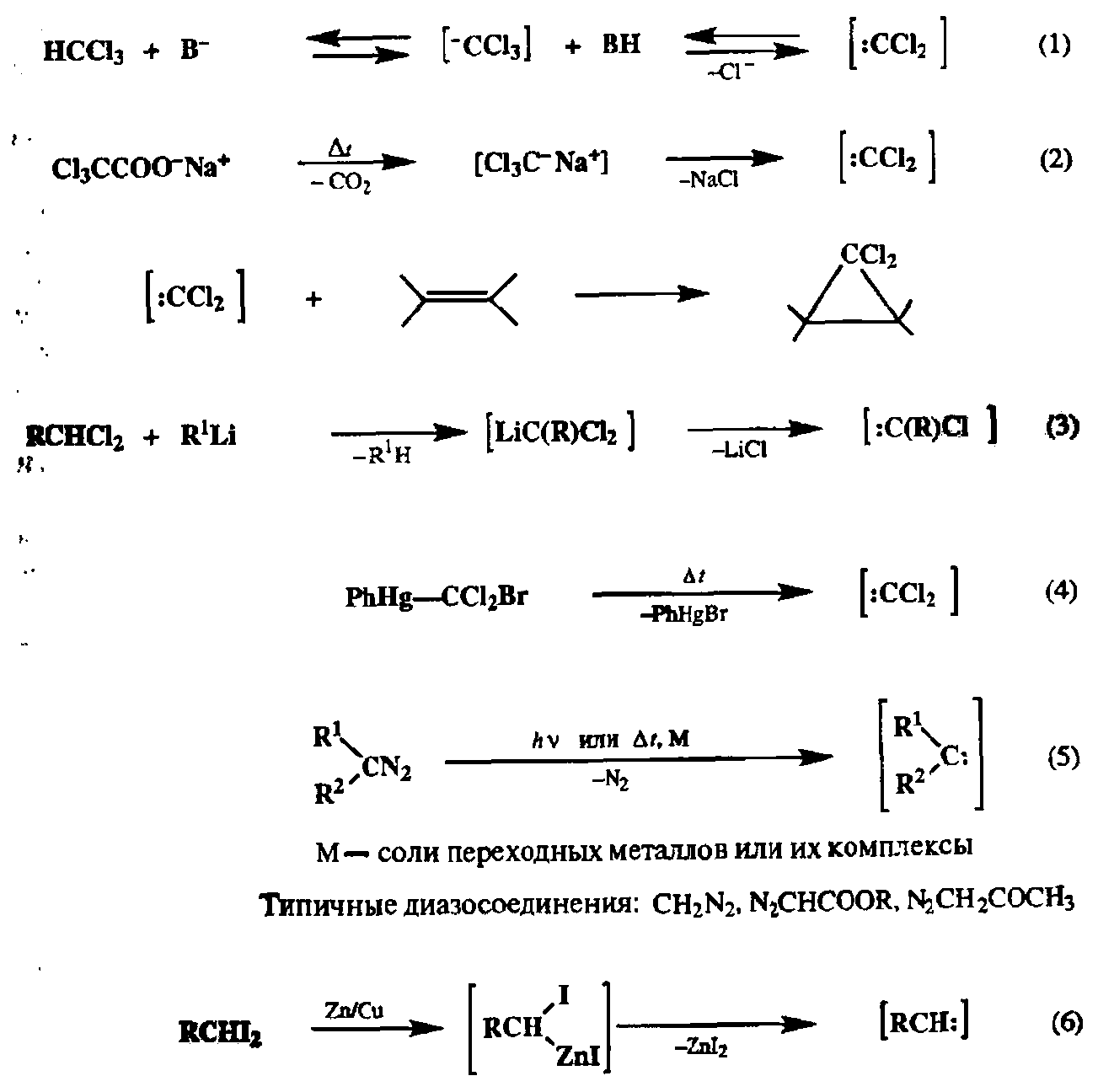
Среди показанных на схеме 2.133 реакций препаративно наиболее важна реакция (1), для которой разработана очень удобная методика проведения в условиях межфазного катализа. Согласно этой методике, раствор непредельного субстрата в хлороформе перемешивают с водным раствором щелочи в присутствии катализатора фазового переноса, например тетрабутиламмониевой соли. При этом на границе раздела фаз происходит генерация дихлоркарбена, который немедленно реагирует с субстратом, содержащимся в хлороформном слое. Альтернативный путь генерации дихлоркарбена, не требующий использования основания или каких-либо катализаторов, основан на декарбоксилировании трихлорацегата натрия при умеренном нагревании (2). Метод (3) является менее общим, и его используют обычно в случае внутримолекулярных вариантов [2 + 1]-циклоприсоединения. Метод (4) удобен для исследований, направленных на выяснение особенностей механизма реакций с участием дихлоркабе-нов. Как видно, методы (1), (2) и (4) ориентированы на генерацию дихлоркар-бенов, и, следовательно, в результате реакций получаемых реагентов с алкена-ми будут образовываться дихлорциклопропаны. Последние достаточно легко могут быть превращены в монохлорпроизводные или соответствующие углеводороды, так что по своему конечному результату использование этих реагентов и реакций эквивалентно циклопропанированию с помощью карбена.
Диазомеган, равно как и многие замещенные диазоалканы, широко применяются для циклопропанирования, поскольку под действием света или при нагревании в присутствии солей тяжелых металлов (Си, Rh) из них легко генерируются карбены {реакция (5)] [ЗЗе]. Однако простейшие диазоалканы не могут долго храниться даже в растворах, и каждый раз их приходиться получать из таких предшественников, как N-нитрозометилмочевина или ее аналоги. Напротив, производные диазоалканов, содержащие электроноакцепторные заместители, такие, как диазоуксусный эфир или диазоацетон, достаточно стабильны и удобны в применении для синтеза замешенных циклопропанов.
Наконец, к числу препаративно удобных методов циклопропанирования относится также реакция Симмонса—Смита, в которой в качестве синтетического эквивалента карбена используется карбеноидная частица ICH(R)ZnI, генерируемая in situ при действии цинк-медной пары на 1,1-ди-иодалканы [реакция (6)] [33f,g].
Некоторые примеры циклопропанирования с помощью рассмотренных методов показаны на схеме 2.134.
Схема 2.134 |
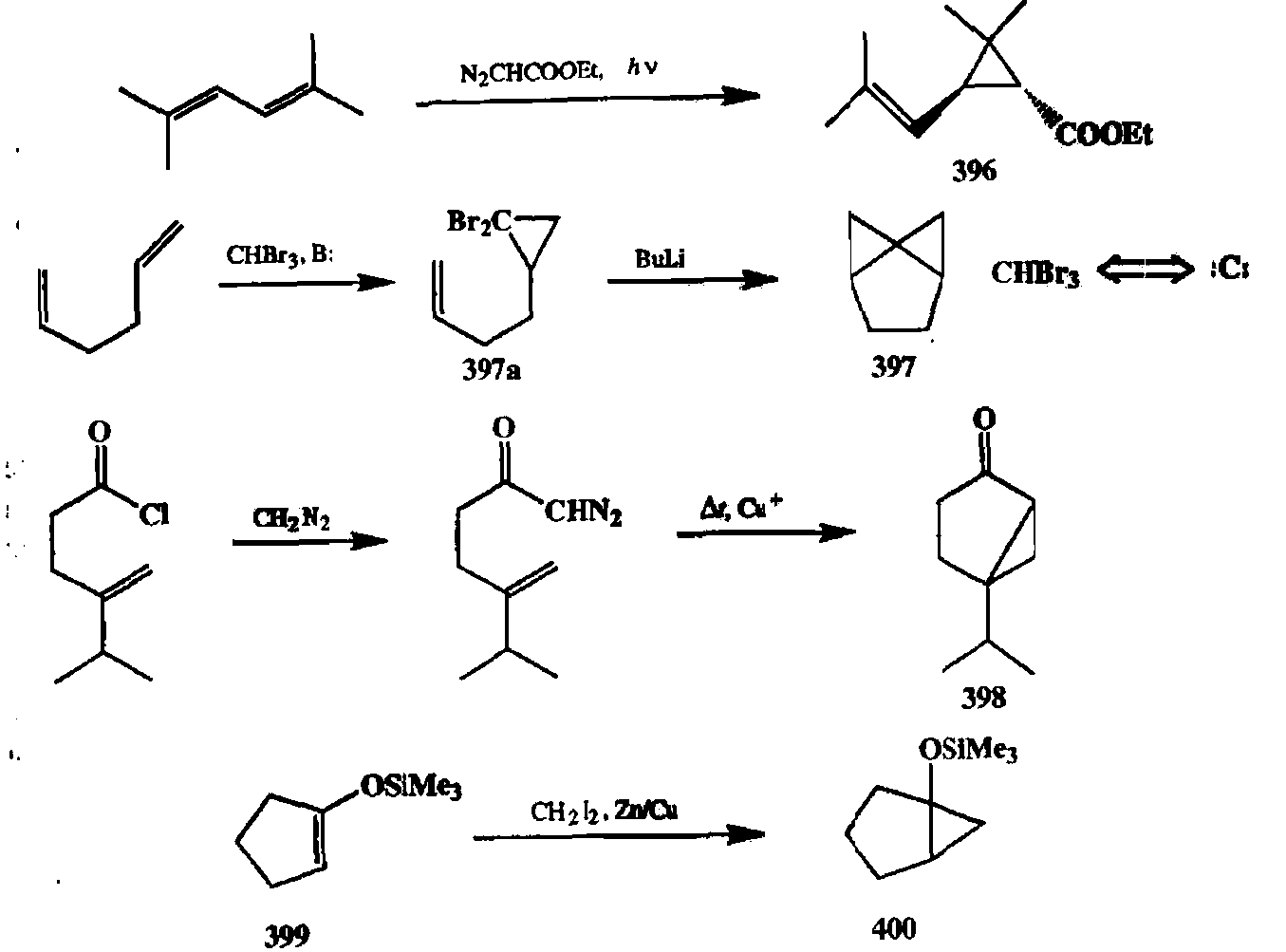
транс-Хризантемовая кислота (396), природный инсектицид, как и многочисленные синтетические аналоги этого соединения, чаще всего получают по схеме циклопропанирования с использованием почти всего спектра реагентов, упоминавшихся выше. Некоторые из полученных веществ нашли широкое применение как эффективные и экологически чистые инсектициды. На схеме 2.134 представлен один из первых синтезов 396, основанный на моноциклопропанировании 2,5-диметилгексадиена-2,4 [33h].
Показанный на этой же схеме синтез трициклического углеводорода 397 из гексадиена-1,5 включает последовательность двух реакций циклопропанирования, межмолекулярной и внутримолекулярной [33i]. Исходный реагент, бромоформ СНВгз, используется в первой из этих реакций как предшествен-ник дибромкарбена. Из получаемого на этой стадии дибромциклопропана 397а далее под действием BuLi снова генерируется карбеноидная частица, реагирующая со второй двойной связью. Суммарный итог этих двух реакций соответствует использованию СНВгз в качестве эквивалента тетрадентатного синтона — бис-карбена :С:.
Примером эффективности внутримолекулярного [2 + 1]-циклоприсоеди-нения для построения полициклических систем, содержащих циклопропа-новый фрагмент, может служить синтез [3,1 ]-бициклогексанового производного 398 [33j]. Требуемые для подобной циклизации диазокетоны могут быть легко получены по достаточно тривиальной реакции хлорангидридов с ди-азометаном.
Условия циклопропанирования по Симмонсу—Смиту |33f] особенно удобны для применения к енолъным производным, таким, как силиловые эфйры 399. Производные типа показанного на схеме 2.134 силилового эфира 400 находят разнообразное применение в синтезе как замаскированные эквиваленты нестабильного циклопропанола. Помимо этого, такие эфиры являются промежуточными продуктами при секо-алкилировании — методе селективного введения одного алхильного заместителя в ct-положение кетонов, основанного на кислотном расщеплении трехчленного цикла силило-вых эфиров типа 400, что непосредственно приводит к ос-алкилкетояам.
Отметим в заключение этого раздела, что методы циклопропанирования самых различных алкеновых производных в целом очень хорошо отработаны. Поэтому, если целевая структура содержит циклопропанозое кольцо, то можно с уверенностью включать в число рассматриваемых вариантов ретро-синтеза те из них, которые предусмативают разборку по этому фрагменту.