

2.4.2 Селективность обеспечивается выбором подходящей реакции
Наиболее простой пример такого подхода мы рассматривали на примере бро-мирования толуола (см. разд. 2.1.3). Действительно, в толуоле имеются две функциональные группы, способные легко реагировать с бромом: метильная группа и ароматическое ядро. Тем не менее, как мы видели, нетрудно направить бромирование селективно в ядро или метальную группу путем правильного выбора типа реакции: при ионном бромировании — в ядро, при радикальном — в боковую цепь. Другим примером является селективное присоединение водорода по двойным связям ароматической системы толуола: при каталитическом гидрировании — насыщение всех трех двойных связей, при восстановлении по Берчу — селективное восстановление одной из них.
В этих случаях речь шла об обеспечении хсмоселективности превращения субстрата. Разберем несколько примеров иного типа, иллюстрирующих возможность управления региоселективностью процесса также за счет выбора надлежащей реакции.
Рассмотрим для начала модельную структуру, диен 151 (схема 2.71), содержащую два алкеновых фрагмента, которые различаются только тем, что в одном из них имеется ди-, а в другом тризамещенная двойная связь. Каким же образом можно провести региоселекгивное восстановление одной из связей, а или B? Хорошо известно, что скорость каталитического гидрирования зависит от стерической доступности двойной связи, и поэтому в условиях гидрирования такого диена, например, над палладием, восстановлению будет предпочтительно подвергаться менее затрудненная двойная связь а, что и приведет к желаемому продукту 152. Альтернативный результат — селективное восстановление связи B с образованием изомерного алкена 153 — достигается с помощью ионного гидрирования (системой трифторуксусная кислота + триэтилсилан) [19k]. Механизм этой реакции принципиально отличается от механизма каталитического гидрирования. Ключевой стадией этой реакции является образование карбокатионного интермедиага за счет протонирования двойной связи с последующим переносом гидрид-иона с кремния на электрофильный центр. Селективность ионного гидрирования определяется относительной устойчивостью промежуточных карбокатио-нов. В рассматриваемом случае протонирование диенового субстрата 151 происходит почти исключительно как атака по связи B, поскольку при этом образуется более стабильный третичный карбокатион 154.
Схема 2.71 |

В рассмотренных случаях субстраты содержали две независимые функциональные группы, и требовалось провесги селективно реакцию по одной или Другой из этих функций. Нередки и иные ситуации, когда в пределах одной и той же функции возможны два направления атаки реагента, ведущие к альтернативным продуктам. Типичный пример показан на схеме 2.72.
Схема 2.72 |
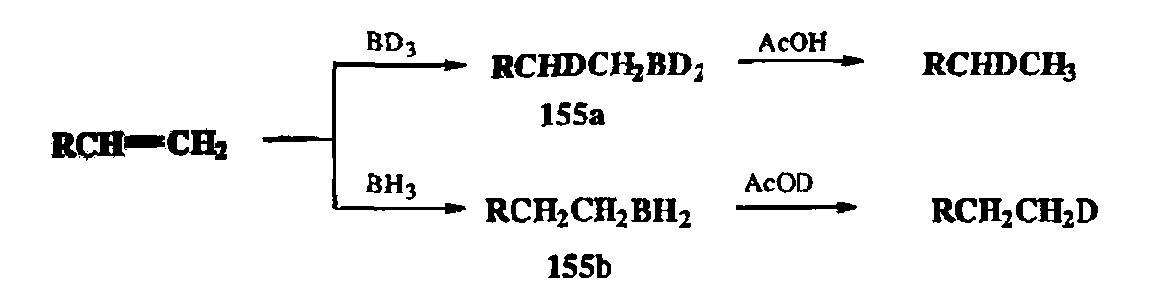
Как уже говорилось, само по себе превращение алкена в алкан не составляет какой-либо проблемы и может быть легко выполнено тем или другим из упомянутых выше методов. Однако эта задача перестает быть тривиальной, если в результате такого превращения требуется получить монодейтерированный алкан, содержащий метку в заданном положении. Очевидно, что для этой цели нельзя применить каталитическое гидрирование. Казалось бы этот результат может быть достигнут с помощью ионного гидрирования (скажем, с использованием дейтерированного триэтилсилана или дейтеротрифторуксусной кислоты). Однако чисто провести эту реакцию не удастся из-за легкости протонного обмена карбокатионного интермедиата типа 154 с сильной кислотой. Эффективным и практичным способом решения такой проблемы является использование совершенно иного метода превращения алкена в алкан, а именно последовательности стадий гидроборирования и протолиза получающихся алкилборанов. Если при этом используется дейтерированный боран, то на первой стадии образуется алкилборан 155а, протолиз которого чисто приводит к получению алкена, содержащего дейтерий у вторичного атома углерода. Если жг на Первой стадии использован обычный боран, то результатом реакции образующегося интермедиата 155Ь с дейтероуксусной кислотой будет столь же чистое образование алкана, содержащего дейтерий при первичном углеродном атоме.
В качестве дополнительной иллюстрации возможностей управления се-лективностью превращений, основанного на выборе подходящей реакции, сошлемся на уже рассмотренные выше примеры региоселективной гидрата-ции алкенов с получением продуктов присоединения по правилу Марковни-кова (М-аддукта, схема 2.10) или против правила Марковникова (аМ-аддук-та, схема 2.47). В этих превращениях формально одинаковый результат — присоединение воды — достигается за счет использования различных по химизму реакций, чем и обеспечивается требуемая селективность образования одного или другого из изомерных спиртов.
Однако далеко не во всех случаях целесообразно искать решение задачи селективности путем выбора природы подходящей реакции. Очень многого можно подчас добиться даже в пределах одной и той же по химизму реакции, если грамотно использовать возможности вариаций конкретной природы применимых реагентов.