

Клинические проявления
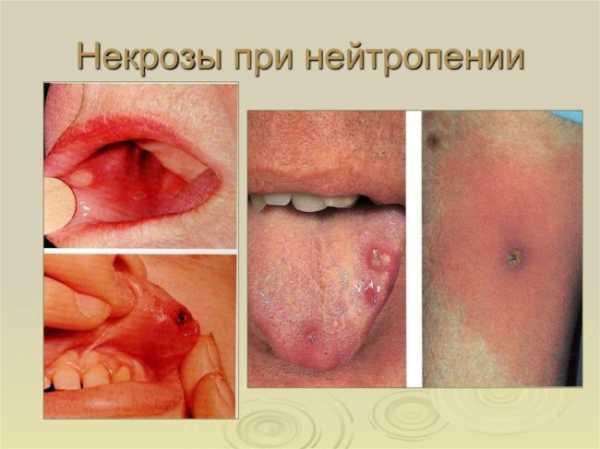
Рецидивирующие бактериальные инфекции, манифестация около 1 года. Частые проявления – неглубокие абсцессы, язвы в полости рта, инфекционные поражения кожи, омфалит, пневмония, средние отиты, неврологические нарушения.
Диагностика
Нейтропения, связанная с рецидивирующими инфекционными заболеваниями, диагностический критерий – содержание нейтрофилов меньше 500/мм3 + одновременное повышение чувствительности к рецидивирующим бактериальным инфекциям, начиная с раннего возраста. При исследовании костного мозга выявляют нарушенное созревание нейтрофилов из предшественников в ранней стадии (дифференцировка промиелоцитов и миелоцитов), клеточность обычно нормальная или слегка снижена, тогда как кол-во эозинофилов и моноцитов увеличено.
Лечение
Трансплантация костного мозга пациентам до 2-летнего возраста. Пациентам, которым не требуется пересадка костного мозга рекомендован рекомбинантный Г-КСФ (гранулоцитарный колониестимулирующий фактор), улучшает прогноз и качество жизни пациентов, 90% положительно отвечают на лечение увеличением числа нейтрофилов и снижением числа инфекционных заболеваний. Но! У пациентов с мутацией гена, кодирующего рецептор Г-КСФ, которые не отвечают на лечение Г-КСФ, продолжаются тяжелые бактериальные инфекции, развивается миелодисплазия. В этом случае требуется пересадка стволовых клеток.
Дефициты адгезии лимфоцитов
Недостаточность адгезии лейкоцитов (НАЛ) является следствием недостаточности адгезионных гликопротеинов на поверхности лейкоцитов, что приводит к нарушению межклеточных взаимодействий, прилипания клеток к стенкам кровеносных сосудов, миграции клеток и взаимодействия с компонентами системы комплемента. Такая недостаточность нарушает способность гранулоцитов (и лимфоцитов) мигрировать через стенки сосудов в ткани, участвовать в цитотоксических реакциях и фагоцитозе бактерий. Тяжесть заболевания коррелирует со степенью недостаточности.
Были идентифицированы три различных типа синдромов:
-
НАЛ 1 (недостаток или дефект β2-интегринов)
-
НАЛ 2 (отсутствие фукозилированных углеводных лигандов для селектинов)
-
НАЛ 3 (недостаточная активации всех β-интегринов [1, 2 и 3]).
Механизм развития
В норме при воспалении происходит накопление лейкоцитов в зоне поражения (трансмиграция этих клеток из крови через эндотелиальный барьер). Основная проблема лейкоцитарной адгезии – лейкоциты не могут покинуть кровеносные сосуды и мигрировать в зону воспаления.
При отсутствии адекватного лечения более 75% пациентов умирают до 2х летнего возраста.
Клинические проявления
Характеризуется кожными язвами, плохо заживающими ранами и рецидивирующими бактериальными инфекциями.
У пациентов выявляют аномалии рецепторов необратимой адгезии CD11/CD18 (интегрины). Если на клетке экспрессируется меньше 1% адгезивных молекул, развиваются угрожающие жизни инфекционные процессы. Если до 10%, то возможно развитие септицемии, гингивита, периодонтита, некротических поражений кожи, кишечных или перианальных свищей.
Диагностика
Сочетание ранних бактериальных инфекций с резко выраженной нейтрофилией (до 50000-100000/мкл) при инфицировании патогеном и в пределах 15000/мкл в отсутствии инфекции. Так же подтверждается диагноз экспрессией молекул CD18, CD15a на нейтрофилах. В крови выявляют массивный нейтрофильный лейкоцитоз до 15000-75000, и даже 100000 клеток в 1 мкл, что объясняется неспособностью этих клеток выйти на периферию из сосудистого русла.
Лечение
Антибиотикотерапия, рекомбинантный Г-КСФ (гранулоцитарный колониестимулирующий фактор), а также стволовые клетки. В будущем возможна генная терапия, методы которой разрабатываются.
Хроническая гранулематозная болезнь
Первичный иммунодефицит, при котором фагоциты не способны генерировать активные формы кислорода и нарушается “кислородный взрыв”, необходимый для уничтожения бактерий и грибов.
Механизм развития
Известно, что цитохром b558 в мембране фагоцитов представлен в форме гетеродимера двух цепей (массой 91 и 22 кДа – атомная единица массы). При активации фагоцитов компоненты NADPH-оксидазы в цитоплазме подвергаются фосфорилированию, мигрируют на поверхность мембраны и связывают цитохром b558. Развитие ХГБ обусловлено дефектами этих молекул.
К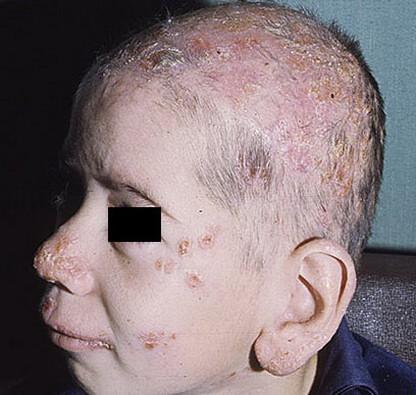 линические
проявления
линические
проявления
Могут возникать у детей в раннем возрасте и у подростков – гнойные поражения кожи (фурункулы, карбункулы, абсцессы с медленным прогрессированием). Заболевание проявляется в виде рецидивирующих инфекционных заболеваний, поражающих органы дыхания, кожу, лимфатические узлы, печень, почки. Характерно возникновение лимфаденита. Особенность ХГБ – формирование в любых органах гранулём, содержащих гигантские многоядерные клетки. У 70-80% детей может развиваться пневмония, осложняемая абсцедированием или эмпиемой плевры. У 15-30% развивается остеомиелит мелких костей, вызванный стафилококками и аспергиллами.
Диагностика
Тесты, помогающие выявить нарушения фагоцитоза – с нитросиним тетразолием, в иммунограмме – снижение высвобождения супероксида из стимулированных фагоцитов до 3-30% от нормы, повышенный уровень IgA, IgM, IgG.
Лечение
Пожизненная антибактериальная терапия, антимикотические препараты (флуконазол, тербинафин, микогептин и др.), генная терапия – введение в стволовые клетки костного мозга гена gp91phox, поврежденного мутацией при ХГБ.
Дефицит глюкозо-6-фосфатдегидрогеназы нейтрофилов
Наиболее частая форма энзимопатии, обычно служащая причиной несфероцитарной анемии.
Отсутствие глюкозо-6-фосфатдегидрогеназы в лейкоцитах приводит к неспособности лейкоцитов разрушать поглощенные бактерии.
Механизм развития
Заболевание возникает в результате мутации гена, кодирующего глюкозо-6-фосфатдегидрогеназу на хромосоме Xq28.
Клинические проявления
При тяжелых формах развиваются абсцессы в разных органах, тяжелые пневмонии и признаки гемолитической анемии.
Диагностика: симптомы гемолитической анемии в комбинации с “патологическим” тестом с нитросиним тетразолием, активность глюкозо-6-фосфатдегидрогеназы в эритроцитах и лейкоцитах определяют флюоресцентным методом.
Лечение
-
Воздействие на причину дефицита глюкозо-6-фосфат-дегидрогеназы – в настоящее время невозможно. Проводятся генетические исследования (то есть исследования генов – носителей наследственной информации) с целью внедрения в организма пациента генов, обеспечивающих нормальный уровень глюкозо-6-фосфат-дегидрогеназы.
-
Трансплантация костного мозга проводится в случаях тяжелого дефицита активности глюкозо-6-фосфат-дегидрогеназы. Трансплантация костного мозга позволяет частично заменить костный мозг пациента с нарушенной структурой клеток на здоровый донорский костный мозг. Появление в кровотоке полноценных эритроцитов значительно снижает риск возникновения гемолитических кризов.
-
Быстрое восполнение количества эритроцитов (красных клеток крови) – переливание эритроцитарной массы (эритроцитов, выделенных из донорской крови) или (предпочтительнее) отмытых эритроцитов (донорских эритроцитов, лишенных потенциально опасных белков донора на их поверхности) по жизненным показаниям (то есть при угрозе для жизни пациента). Угрозой для жизни пациента с анемией являются два состояния:
- анемическая кома (утрата сознания с отсутствием реакции на внешние раздражители вследствие недостаточного поступления кислорода к головному мозгу в результате значительного или быстро развившегося снижения количества эритроцитов);
- тяжелая степень анемии (то есть уровень гемоглобина крови ниже 70 г/л).
Дефицит миелопероксидазы
Дефицит миелопероксидазы — наиболее частый дефект фагоцитов, выявляемый с частотой 1:4000. Вследствие этого дефицита нарушается образование фагоцитами гипохлорной кислоты. Впервые описан у больных с диссеминированным кандидозом, у которых в нейтрофилах и моноцитах не была выявлена активность лизосомальных ферментов, при этом активность других гранулосвязанных ферментов оставалась в норме.
Механизм развития
Миелопероксидаза находится в азурофильных гранулах и катализирует превращение H2O2 в гипохлорную кислоту. Ген расположен на хромосоме 17q23. Дефицит миелопероксидазы – врожденное аутосомно-рецессивное заболевание. Описаны вторичные формы дефицита миелопероксидазы при тяжелых инфекционных заболеваниях, СД, миелоидной лейкемии, ходжкинской лимфоме.
Клинические проявления
Больше чем у 95% клинические симптомы не проявляются, не смотря на дефекты киллинга нейтрофилов. При наличии симптоматики выявляют кандидозную инфекцию и иногда СД. Тяжелые инфекционные поражения костей и сепсис развиваются редко.
Диагностика
Определяют дефицит фермента в клетках. Антитела к миелопероксидазе нейтрофилов в крови - в норме антитела к миелопероксидазе нейтрофилов в сыворотке крови отсутствуют.
Лечение
Противомикробное лечение, базисная терапия с применением препаратов гамма-интерферона и так называемых модуляторов фагоцитоза (например, производных етиленпиперазину), которые можно сочетать с иммуноглобулинами, учитывая опсониз-ю активность антител.
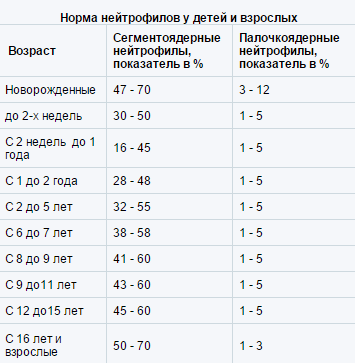
Циклическая нейтропения
Характеризуется нейтропенией, возникающей каждые 3 недели и сохраняющейся в течение 3-6 сут.
Впервые заболевание описано в 1910 г доктором Чарльзом Лилем у младенцев с рецидивирующими эпизодами лихорадки, инфекционным поражением кожи, стоматитами, нейтропенией.
У пациентов уровень нейтрофилов колеблется от нормального содержания до полного отсутствия с периодичностью 21 сут.
При полном исчезновении нейтрофилов повышается риск развития инфекционных заболеваний.
 В
целом благоприятное течение, для лечения
применяют – Г-КСФ
(гранулоцитарный колониестимулирующий
фактор).
В
целом благоприятное течение, для лечения
применяют – Г-КСФ
(гранулоцитарный колониестимулирующий
фактор).