

Взаимодействие лептина с другими звеньями нейроэндокринной системы
О наличии связи между лептином и гипоталамо-гипофизарной осью, контролирующей секреции гормона роста (СТГ) свидетельствуют (Акмаев И.Г., Сергеев В.Г., 2002): а) данные о низких уровнях СТГ в плазме крови у людей и грызунов с мутациями лептинового гена, или гена соответствующего рецептора; б) выраженное снижение СТГ в плазме крови крыс при нейтрализации действия лептина на структуры мозга с помощью антилептиновых антител или при их внутрижелудочковом введении; в) падение уровней лептина и СТГ при пищевой депривации и восстановление нормальной секреции СТГ при введении лептина в желудочки мозга.
Участие лептина в регуляции роста и в увеличении массы тела, по-видимому опосредуется его влиянием на нейроны, синтезирующие рилизинг-гормон гормона роста (РГГР), приводящему к увеличению продукции этого рилизин-гормона. Последующее воздействие РГГР на переднюю долю гипофиза обеспечивает усиление секреции гормона роста и, соответственно, стимуляцию роста тела. Нарушение этого процесса, напротив, вызывает задержку роста (рис.4).
Увеличение секреции гормона роста и
стимуляция роста тела; нарушение этого
процесса вызывает задержку роста


РГГР
ЛР
РГГР
Р

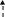



ЛЕПТИН
Нейроны, синтезирующие рилизинг-гормон
гормона роста
Передняя доля
гипофиза
Рис. 4 Центральный эффект лептина, обусловливающий его влияние на рост и массу тела
(Ю.А. Панков, 1999).
Правомерность такого механизма подтверждается данными о наличии рецепторов к лептину в паравентрикулярных и аркуатных ядрах гипоталамуса, где локализованы нейроны, синтезирующие РГГР и соматостатин, а также сведениями об отмене стимулирующего влияния лептина на секрецию СТГ в условиях блокады РГГР специфическими к РГГР антителами.
Вопрос о возможном непосредственном влиянии лептина на активность соматотропоцитов гипофиза остается открытым и требует дальнейшего изучения.
Аналогично представляется и центральный механизм влияния лептина на поддержание нормальной продукции тиреотропного и тиреоидных гормонов (рис. 5).
Увеличение секреции гипофизом
тиреотропного гормона и щитовидной
железой тиреоидных
гормонов
ТРГ
ЛР
ТРГ
Р



ЛЕПТИН
Нейроны, синтезирующие тиреотропин-рилизинг- гормон
Передняя доля
гипофиза
Рис. 5 Центральный эффект лептина, обусловливающий его влияние на продукцию тиреотропного и тиреоидных гормонов (Ю.А. Панков, 1999).
Известно, что пищевая депривация, сопровождаемая падением секреции лептина, характеризуется низкими уровнями Т3 и Т4 в крови, равно как и снижением синтеза тиреотропин-рилизинг-гормона (ТРГ) и тиреотропного гормона. Введение лептина голодающим крысам восстанавливает до нормальных значений сниженные показатели синтеза мРНК про-ТРГ в нейронах паравентрикулярных ядер. Такие параллели позволяют предполагать, что падение уровня лептина во время голодания служит сигналом для снижения синтеза тиреоидных гормонов щитовидной железой.
Остается невыясненным вопрос о способности лептина непосредственно регулировать активность щитовидной железы.
Приводятся данные о наличии механизма взаимодействия лептина и пролактина. Возможно, что пролактин стимулирует секрецию лептина, поскольку овариоэктомированные крысы после введения пролактина обнаруживают повышение мРНК лептина и уровня этого гормона в циркуляции. Однако такой стимулирующий эффект зависит от метаболического статуса организма и не наблюдается у животных в условиях пищевой депривации.
Влияние лептина на активность гипоталамо-гипофизарно-адренокортикальной системы (ГГАКС) носит репрессивный характер. Одной из примечательных особенностей генетически ожиривающих мышей и крыс является гиперкортикостеронемия, которая корректируется введением лептина. Лептин нивелирует повышения АКТГ и (или) кортикостерона, вызываемые голодом или стрессом. Поскольку лептин не меняет секрецию АКТГ in vitro, складывается впечатление, что он оказывает свое действие путем торможения секреции кортикотропин-рилизинг-гормона (Акмаев И.Г., Сергеев В.Г., 2002). В пользу этого предположения приводятся данные о присутствии лептиновых рецепторов в кортикотропин-рилизинг-содержащих нейронах мелкоклеточной части паравентрикулярных ядер.
Наряду с центральным регулирующим действием лептина на активность ГГАКС не исключена вероятность его влияния и на периферические звенья системы. Об этом свидетельствует наличие в коре надпочечников «длинноцепочечной» формы рецептора к лептину, что может обусловить непосредственное действие гормона на эндокринные клетки этого органа. Возможно, что при участии данных рецепторов происходит торможение лептином in vitro АКТГ-стимулируемой секреции кортизола в культуре клеток надпочечников человека и крыс. Однако такой ингибирующий эффект на секрецию кортизола со стороны лептина наблюдается лишь в случае его продолжительных инфузий. Это обстоятельство служит основанием в пользу представления о том, что быстрые изменения активности ГГАКС в ответ на изменения уровня лептина реализуются через центральные механизмы (Акмаев И.Г., Сергеев В.Г., 2002).
Таким образом, в организме реализуется сложная система взаимодействия лептина с различными нейроэндокринными механизмами, функционирование которой обеспечивается при участии как положительных, так и отрицательных обратных связей (рис.6). Он ак -
Рис. 6 Лептин и механизмы нейроэндокринной регуляции: прямые и обратные связи (Ch.S., Мantzoros 1999).
тивирует специфические лептиновые рецепторы в гипоталамусе и изменяет экспрессию нейропептидов, что приводит к снижению аппетита, повышению расхода энергии за счет изменения тонуса симпатического и парасимпатического отделов вегетативной нервной системы, а также нейроэндокринной функции. При повышении уровня лептина стимулируется выработка гормонов щитовидной железы, соматотропного и половых гормонов, подавляется активность гипоталамо-гипофизарно-адреналовой системы. За счет прямого или непрямого (опосредованного через другие гормоны и нейропептиды) действия лептин влияет на гемопоэз, иммунитет, оптимизирует метаболизм глюкозы и жиров. В свою очередь, изменение продукции различных гормонов и цитокинов и их содержания в крови стимулирует или подавляет выработку лептина жировыми клетками.
Роль лептина в развитии ожирения у человека
Оценка роли лептина в регуляции энергетического обмена позволила сформулировать принципиально новую концепцию патогенеза ожирения у человека (Ю.А. Панков, 1998-2000). Ее центральное положение заключается в том, что «ожирение развивается всегда, когда нарушается эндокринная функция жировой ткани» (рис. 7).
Традиционная концепция Новая концепция
Жировая ткань - инертное депо, в котором
сохраняются запасы биологической
энергии, необходимой для поддержания
жизни. 1г жира содержит значительно
больше энергии, чем белки и углеводы.
Поэтому жиры - наиболее удобные продукты
для накопления энергии в организме.
Жировая ткань - эндокринный орган,
продуцирующий лептин. Снижение
продукции лептина или проявления его
биологического действия приводит к
ряду негативных последствий в виде
подавления репродуктивной функции,
бесплодия, задержки роста и полового
развития, развития инсулинрезистентности,
повышенной секреции инсулина и др.
Ожирение -
результат избыточного накопления
энергетического субстрата
вследствие нарушения действия на
жировую ткань гормонов, секретируемых
эндокринными органами (снижение действия
жиромобилизующих гормонов и усиление
действия анаболических гормонов)
Ожирение - результат нарушенной
эндокринной функции самой жировой
ткани и представляет собой ее
гипертрофию, направленную на увеличение
секреции лептина, призванную компенсировать
дефицит гормона либо его пониженную
биологическую активность
Рис. 7. Традиционная и современная концепции патогенеза ожирения
(по Ю.А. Панкову, 1998; 2000; 2003)
Нарушение этой функции может быть обусловлено либо неспособностью жировой ткани к продукции лептина – абсолютная недостаточность (мутация гена ob), либо при сохраняющейся способности к секреции гормона – недостаточностью его биологического действия - относительная недостаточность. Оценивая возможность формирования абсолютной и относительной недостаточности лептина, следует отметить, что согласно современным данным у людей, страдающих ожирением, снижение синтеза и секреции лептина – довольно редкое явление. Ситуация характеризуется как раз отсутствием рецепрокности между этими показателями, а содержание лептина в циркуляции четко коррелирует с массой жировой ткани. Это означает, что в подавляющем большинстве случаев центральным звеном патогенеза ожирения выступает несостоятельность биологического действия лептина, т.е. его относительная недостаточность. Последняя может определяться разными механизмами, включая: а) мутации гена рецептора лептина; б) мутации гена рецептора а-МСГ (МК 4-Р); в) снижение транспорта лептина через ГЭБ (например, вследствие нарушения сплайсинга про-мРНК-рецепторов к лептину с ограничением к синтезу растворимой формы рецептора; г) дефектностью пострецепторной трансдукции лептинового сигнала.
В качестве механизма, определяющего резистентность к лептину на уровне ЦНС, детально рассматривается нарушение его транспорта через гематоэнцефалический барьер (ГЭБ). Имея ограниченную пропускную способность, эта транспортная система может служить препятствием для реализации эффектов лептина. Зачастую одни и те же дозы гормона, не оказывающие никакого воздействия при периферическом введении, эффективно снижают аппетит при центральном введении (Мantzoros Ch.S., 1999). Еще одним доказательством в пользу возможного участия транспортного механизма в формировании резистентности к лептину служат сведения о том, что при ожирении наблюдается падение соотношения концентраций лептина в спинномозговой жидкости и в сыворотке крови. Причем, не только за счет возрастания уровня гормона в циркуляции, но и снижение его в ликворе. В этом аспекте представляется весьма важным то обстоятельство, что при достижении определенной концентрации лептина в сыворотке крови (25-30 нг/мл) дальнейшее ее повышение в циркуляции не сопровождается параллельным увеличением концентрации гормона в ткани мозга и спинномозговой жидкости (Мantzoros Ch.S., 1999).
В последнее время получены данные (Benks W.A. et al., 2004), свидетельствующие о том, что блокировка транспорта лептина через ГЭБ опосредована триацилглицеридами (ТАГ). Индукция их повышенного уровня (при голодании, ожирении или при использовании молока, содержащего 98% ТАГ) немедленно ограничивало этот транспорт, также как и в случае прямого внутривенного введения ТАГ. Применение инфиброзила – препарата, специфически редуцирующего уровень ТАГ, отменяло данный эффект. И хотя конкретный механизм такого влияния ТАГ остается невыясненным, представленные сведения показывают, что триацилглицериды являются важным фактором в формировании лептиновой резистентности. С другой стороны, они демонстрируют возможность усиления аноректического эффекта лептина путем повышения его транспорта через ГЭБ в условиях снижения уровня ТАГ.
Еще один вероятный механизм, определяющий резистентность к лептину – нарушение его взаимодействия с рецептором в ЦНС, может иметь в своей основе не только мутацию гена рецептора db, распространенность которой в общей популяции неизвестна, хотя должна быть очень низкой, но и действие некоторых периферических сигнальных гормонов, в частности, глюкокортикоидов (Мantzoros Ch.S., 1999).
Что касается участия в развитии резистентности к лептину таких факторов как снижение его транспорта в крови в результате нарушения связывания со специфическими белками или усиление катаболизма, то их вклад, по-видимому, не существенен, поскольку показатели времени полувыведения и биологической активности циркулирующего в крови лептина у полных и худых людей одинаковы. Более того, применение антилептиновых антител и лептинсвязывающих белков не приводит к инактивации лептина у лиц с ожирением (Мantzoros Ch.S., 1999).
Абсолютный и (или) относительный дефицит лептина включают в организме специфические (пока не идентифицированные) регуляторные механизмы, нацеленные на исправление сниженной эндокринной функции жировой ткани (Панков Ю.А., 2000). Они вызывают ее компенсаторное разрастание с тем, чтобы увеличить секрецию лептина и повысить его содержание в крови вследствие возросшей потребности организма в гормоне, что и приводит в конечном итоге к развитию ожирения. Т.е. происходит гипертрофия жировой ткани аналогично тому, как происходит разрастание щитовидной железы при утрате ее способности секретировать тиреоидные гормоны или при нарушении нормального функционирования таких гормонов (рис. 8).
Подобная гипертрофия жировой ткани, по всей вероятности, происходит при любом нарушении эндокринной функции адипоцитов и вызывает в конечном итоге увеличение веса тела в результате повышенного отложения жира в жировых депо. В том случае, если основу нарушения эндокринной функции жировой ткани составляет неспособность к продукции лептина (абсолютная недостаточность), ее гипертрофия не приводит к устранению дефицита гормона, что проявляется сохраняющейся а(гипо)-лептинемией. При относительной недостаточности гипертрофия жировой ткани сопровождается гиперлептинемией, что и наблюдается в большинстве случаев у лиц, страдающих ожирением. Однако эта гиперлептинемия не компенсирует полностью функциональную несостоятельность лептина и, соответственно, не устраняет механизм патогенеза заболевания.
Фенотип, формирующийся в условиях лептиновой недостаточности, характеризуется рядом негативных проявлений, в том числе повышенным аппетитом и сниженной утилизацией жира в качестве энергетического субстрата. Подобные нарушения способствуют нарастанию гипертрофии жировой ткани, но уже не в качестве компенсаторного механизма, а как проявление нарушенного энергетического обмена.
Таким образом, в соответствие с данной «дефицитной» концепцией пусковым звеном патогенеза ожирения является лептиновая недостаточность, а само ожирение вначале представляет собой компенсаторную реакцию жировой ткани, направленную на ее устранение. Роль повышенного потребления пищевых веществ – вторична и она является следствием нарушенного регуляторного действия лептина.
АБСОЛЮТНАЯ НЕДОСТАТОЧНОСТЬ ЛЕПТИНА
ОТНОСИТЕЛЬНАЯ НЕДОСТАТОЧНОСТЬ ЛЕПТИНА
Мутация
гена ob Мутация
гена db Мутация
гена МК 4 – Р Снижение
транспорта лептина через ГЭБ Дефекты
пострецепторной трансдукции лептинового
сигнала
Дефицит лептина
Несостоятельность биологического
действия лептина



Компенсаторная гипертрофия жировой
ткани, направленная на восполнение
дефицита гормона или устранение его
пониженной биологической активности
Сохраняющаяся а (гипо) лептинемия
Гиперлептинемия, не компенсирующая
пониженную биологическую активность
гормона

Гипертрофия жировой ткани, как
результат повышенного потребления
энергоемких субстратов и нарушенного
метаболизма липидов

Нарушение пищевого поведения и
энергетического обмена в виде повышенного
потребления пищи и снижения утилизации
жира в качестве энергетического
субстрата
Толерантность
к инсулину; гиперинсулинизм; задержка
роста; нарушение репродуктивной функции


Рис. 8. Патогенез ожирения как результата дефицита эндокринной функции жировой ткани (по Ю.А. Панкову).
Иная точка зрения основывается на представлении о первичной роли повышенного потребления энергоемких, легкоусвояемых продуктов питания в развитии неэффективности (недостаточности) регуляторного действия лептина (Акмаев И.Г., Сергеев В.Г., 2002). В этих условиях на фоне низкой физической активности происходит увеличение массы жировой ткани, сопровождаемое гиперсекрецией гормона. Такая хроническая гиперлептинемия нарушает центральные механизмы интеграции лептиновой сигнализации (толерантность к лептину?). Соответственно, проявляются синдромы лептиновой недостаточности, включая изменения пищевого поведения и энергетического обмена. Формируется порочный круг, усугубляющий развитие патологии (рис. 9).
Вместе с тем, эта схема патогенеза не исключает и компенсаторную гипертрофию жировой ткани, направленную на преодоление формирующейся недостаточности биологической активности гормона.
Таким образом, данная концепция в качестве первичного механизма развития ожирения рассматривает избыточное поступление пищевых веществ, обусловливающее срыв липостатической регуляции в виде резистентности ее центральных звеньев к действию лептина. Очевидно, что в этих условиях снижается роль лептина как афферент –
Избыточное поступление в организм
энергоемких, легко усвояемых продуктов
питания на фоне низкой физической
активности

Депонирование запасов биологической
энергии: увеличение массы жировой ткани
Хроническая гиперсекреция лептина
(гиперлептинемия)
Нарушение центральных механизмов
лептиновой сигнализации (толерантность
к лептину?)

Повышение аппетита; увеличение
потребления пищи; снижение использования
жиров в энергетическом обмене
Компенсаторная гипертрофия жировой
ткани, направленная на устранение
пониженной биологической активности
лептина
Рис. 9 Патогенез ожирения как следствие функционального срыва центральных механизмов липостатической регуляции при избыточном поступлении нутриентов (по И.Г. Акмаеву, В.Г. Сергееву, 2002).
ного метаболического сигнала жировой ткани (нарушение эндокринной функции жировой ткани), что определяет развитие патологических синдромов и может служить стимулом для ее компенсаторной гипертрофии.
Вероятность подобного развития событий связывается с низкой адаптационной способностью центральных механизмов регуляции обмена веществ к условиям длительного поступления избытка энергии в организм, поскольку в ходе эволюционного развития система гомеостаза формировалась для обеспечения процесса жизнедеятельности в условиях характерной для живой природы нехватки пищевых ресурсов. Поэтому с эволюционной точки зрения переедание является новой ситуацией, для которой не сформированы достаточно надежные механизмы адекватного реагирования (Акмаев И.Г., Сергеев В.Г., 2002).
Таким образом, накопленные данные показывают, что подкожная жировая клетчатка активно участвует в нейроэндокринной регуляции и, тем самым, в реализации различных физиологических функций. Нарушение синтеза лептина адипоцитами или неспособность гормона проявлять биологическое действие (по разным причинам) приводит к разрастанию жировой ткани. Размеры этой ткани можно уменьшить с помощью искусственных подходов, однако в том случае, когда причины, вызвавшие патологию не устраняются, ее гипертрофия, как правило, возобновляется, если после проведенного лечения эндокринная функция жировой ткани не компенсируется заместительной гормональной терапией (Панков Ю.А., 2003).
ФАКТОР НЕКРОЗА ОПУХОЛЕЙ АЛЬФА
Природа и спектр биологической активности
Фактор некроза опухоли альфа (ФНОα) входит в состав семейства факторов некроза опухолей, куда помимо его включены ФНОβ (лифотоксин альфа) и лимфотоксин бета, представленный, в основном, мембранной формой. ФНОα и лимфотоксины представляют собой гликопротеины с М.м, соответственно, 17000 и 20000-25000 кДа.
Гены ФНОα и ФНОβ (генетическое обозначение TNF) локализованы на хромосоме 6 р 21.3 в регионе главного комплекса гистосовместимости (ГКГС) класса III между генами, кодирующими молекулы класса II (HLA-DP, DQ и DR) и класса I (HLA A, B, C). В локусе TNF идентифицированы 5 микросателлитных маркеров (TNFα, TNFβ, TNFc, TNFd и TNFe) каждый из которых имеет 14, 7, 2, 7 и 3 аллеля соответственно. Изучение полиморфизма генов TNF и их связи с генами HLA важно для понимания иммунопатогенеза многих тяжелых заболеваний воспалительной и аутоиммунной природы (Ковальчук Л.В. с соавт., 2001).
Молекулы ФНО гомологичны Fas-лиганду, мембранным молекулам CD 30 и CD 40, фактору роста нервов и образуют единое семейство белков. Их объединение обусловливает общность рецепторов. Известно 2 типа таких рецепторов: gp 55 и gp 75 (М.м 55 и 75 кДа), относящихся к семейству рецепторов фактора роста нервов. В это же семейство входит также Fas-рецептор – мембранная молекула, принимающая сигнал к апоптозу. Это свойство обусловлено присутствием «домена смерти» в цитоплазматической части молекулы данного рецептора. Такой же домен имеется в цитоплазматической части рецептора gp 55, но не gp 75. Соответственно первый, но не второй тип рецептора способный передавать летальный сигнал ФНО внутрь клетки.
Оба типа рецепторов могут высвобождаться с клеточной поверхности в результате протеолитического расщепления и циркулировать в растворимой форме. Уровни циркулирующих рецепторов повышаются при сепсисе, злокачественных опухолях, аутоиммунных заболеваниях, лихорадке, лейкемии, ожирении. Не исключено, что циркулирующие рецепторы могут связывать системный ФНО, представляя таким образом, функциональную буферную систему для этого цитокина (Акмаев И.Г., Сергеев В.Г., 2002).
Наиболее радикально ФНОα и ФНОβ различаются по клеточному происхождению. ФНОα является продуктом моноцитов/макрофагов (в некоторых случаях – активированных Т-лимфоцитов), эндотелиальных, тучных и миелоидных клеток, ЛАК-клеток, клеток нейроглии. Основными продуцентами ФНОβ являются активированные Т-лимфоциты: CD 4+ и CD 8+.
В настоящее время установлено, что ФНОα синтезируется и в адипоцитах. Этот синтез происходит из трансмембранного гормона (26 кДа), который после протеолитического расщепления дает начало растворимой молекуле 17 кДа. «Неожиданная на первый взгляд способность адипоцитов секретировать ФНОα тем не менее согласуется с современными представлениями о существовании единой иммунонейроэндокринной системы регуляции гомеостаза организма, функционирующей на основе общности лигандов и рецепторов, используемых в своей деятельности различными подсистемами» (Акмаев И.Г., Сергеев В.Г., 2002).
Биологическая активность ФНОα включает цитостатическое и флогогенное действие, регуляцию иммунного ответа, гемопоэза и клеточного метаболизма, а также модуляциию нейроэндокринной регуляции (рис. 10).
ФНОα и липидный обмен
Способность адипоцитов к секреции ФНОα, а также его повышенные уровни при ожирении определили интерес к оценке физиологической значимости этого цитокина в метаболизме липидов в организме.
Системное действие ФНОα демонстрирует индукцию им метаболических изменений, известных как кахексия, т.е. состояния при котором наблюдается истощение мышц и
ФНО


Цитостатическое действие -генерация
активных форм кислорода и индукция
апоптоза
Флогогенное действие -усиление
экспрессии на эндотелии молекул адгезии; -стимуляция
ангиогенеза; -усиление
секреции Рg; -хемотаксический
фактор; -стимуляция
синтеза белков острой фазы; -индукция
лихорадки; -активация
нейтрофилов, эозинофилов, моноцитов; -стимуляция
продукции ИЛ-1, ИЛ-6, ФНО
мононуклеарами и другими типами клеток
Регуляция иммунного ответа -стимуляция
пролиферации Т- и В- лимфоцитов; -усиление
антителообразования на Т-зависимые
антигены; -подавление
ГЗТ; -препятствие
формированию иммунологической
толерантности; -увеличение
экспрессии молекул главного комплекса
гистосовместимости на клетках,
инфицированных внутриклеточными
паразитами
Регуляция гемопоэза -угнетение
эритро-, миело-, и лимфопоэза у нормальных
животных; -радиопротекторный
эффект при подавленном кроветворении


Модуляция нейроэндокринной регуляции -усиление
экспрессии гена ПОМК
Регуляция
метаболизма -жирового
обмена; -инсулинрезистентности
Рис. 10 Спектр биологической активности ФНО
(по данным А.А. Ярилина, 1999; Л.В. Ковальчука с соавт., 2001; И.Г. Акмаева, В.Г. Сергеева, 2002)
жировых клеток. Механизмы развития этого истощения разнообразны, но в целом они реализуются в трех направлениях: а) через усиление катаболизма триацилглицеридов (ТАГ) в тканях; б) через подавление эндогенного синтеза триацилглицеридов; в) через снижение поступления в организм нутриентов и обусловленный этим дефицит энергии.
Усиление катаболизма ТАГ, наиболее характерным признаком которого является гиперлипемия, под влиянием ФНОα определяется стимулирующим действием цитокина на ключевые ферменты липолиза (тканевую липазу) через их транскрипционную регуляцию. Вместе с тем, усиливая липолиз в тканях, ФНОα тормозит расщепление ТАГ в составе циркулирующих в крови липопротеидов очень низкой плотности (ЛПОНП) и промежуточной плотности (ЛППП). Это обстоятельство обусловлено его ингибирующим влиянием на липопротеидлипазу, иммобилизованную в стенке сосудов и ответственную за данный процесс. Конкретный механизм такого ингибирующего влияния может быть различным (нарушение синтеза самого фермента, инсулинрезистентность), но в любом случае оно лимитирует возможность использования адипоцитами в жировом обмене транспортных форм липидов, а также способствует дислипопротеидемии и гипертриацилглицеридемии.
Подавление эндогенного синтеза ТАГ под влиянием ФНОα обеспечивается несколькими путями (рис. 11). Во-первых, через угнетение поступления жирных кислот (ЖК) в адипоциты, что связано не только с невозможностью использования ЖК в составе ЛПОНП и ЛППП, но и со снижением экспрессии в адипоцитах транспортеров жирных кислот (FAT и FATP), осуществляющих их перенос через плазматическую мембрану жировых клеток. Во-вторых, через редукцию экспрессии ключевых энзимов липогенеза, в частности, ацетил-коэнзим А-редуктазы и синтетазы жирных кислот, необходимых для их восстановительного синтеза. Этот синтез, в свою очередь, требует значительного расхода энергии, который должен восполняться поступлением в организм энергоемких нутриентов. Однако такая возможность в условиях системного действия ФНОα представляется ограниченной, в связи с подавлением им чувства голода.
Влияние ФНОα на пищевое поведение, выражающееся подавлением аппетита может опосредоваться его способностью усиливать (как и лептин) экспрессию генов ПОМК в гипоталамусе. Снижение пищевой активности обусловливает дефицит энергетических субстратов, что с одной стороны стимулирует активность катаболических процессов, а с другой - существенно уменьшает эффективность процессов биосинтеза, включая биосинтез ТАГ.
В совокупности представленные метаболические нарушения и составляют основу для снижения массы жировой и мышечной ткани, т.е. для развития кахексии.
ФНОα и резистентность к инсулину при ожирении
Ожирение характеризуется преобладанием процессов депонирования ТАГ в жировой ткани и инсулиновой резистентностью тканей организма. В настоящее время получены доказательства того, что в процессы развития инсулиновой резистентности активно вовлечены цитокины и пептиды, синтезируемые адипоцитами. Среди них особое место отводится ФНОα.
Участие ФНОα в формировании инсулинрезистентности, вероятно определяется несколькими механизмами, среди которых могут быть выделены (Акмаев И.Г., Сергеев В.Г., 2002): а) снижение сигнальной способности инсулиновых рецепторов; б) экранирование рецепторов на клеточных мембранах избытком жирных кислот; в) подавление экспрессии гена Glut 4 и тем самым – снижение инсулинстимулируемого транспорта глюкозы в клетки. Помимо адипоцитов ФНОα - индуцированная инсулиновая резистентность обнаруживается в фибробластах, миелоидных клетках и мышечных клетках крыс. ФНОα придается важное значение в развитии инсулинрезистентности у больных диабетом 2 типа, у которых уровни его экспрессии обнаруживают строгую корреляцию с уровнем гиперинсулинемии и снижением чувствительности ткани к инсулину.
В процессы модуляции активности инсулиновых рецепторов под влиянием ФНОα может быть вовлечен и лептин. Оба они демонстрируют параллели в своей биологии: корре-
ФНО


Активация липолиза в тканях за счет
транскрипционной регуляции его ключевых
ферментов
Ингибирование липопротеидлипазы
сосудистой стенки (инсулинзависимой)
Снижение экспрессии транспортеров
ЖК в адипоцитах (FAT
и FATP)
Редукция экспрессии ключевых энзимов
липогенеза: Ац–КоА-редуктазы и синтетазы
ЖК
Подавление аппетита


Высвобождение ЖК и их поступление в
циркуляцию
Подавление эндогенного синтеза ЖК
Нарушение гидролиза ТАГ в ЛПОНП и
ЛППП
Угнетение захвата ЖК адипоцитами из
циркуляции
Снижение поступления нутриентов


+
-


Гиперлипемия
Дислипопротеидемия
+

