

Фторазотные катионы
Диоксигенильный ион относится к группе так называемых фторкатионов. В эту группу входит большое число недавно открытых гомополиатомных (т.е. состоящих из нескольких одинаковых атомов) O2+, Сl3+, Br2+, I5+, Хе2+ и т.п. и гетерополиатомных катионов (или просто гетерокатионов) ClO2+, OSF3+, N2F3+ и т.п. Название «фторкатионы» отражает не столько состав, сколько наиболее распространенный метод их получения. Он состоит в действии сильного акцептора фторид-иона на фторсодержащую молекулу
RF + ЭFn-1 R + ЭFn
Молекула RF выступает как основание и в результате реакции превращается в катион, молекула ЭFn-1 действует как кислота и превращается в анион ЭFn. Продуктом реакции кислоты с основанием, естественно, является соль, в данном случае R+ЭFn. По имени создателя одной из теорий кислот и оснований такой процесс называют нейтрализацией по Льюису, а реагенты - кислотой Льюиса и основанием Льюиса.
Рассмотрим теперь подробнее, какие же фторкатионы удалось получить, как они построены и какие свойства характерны для их солей. Начнем с фторкатионов азота. Если подействовать сильной фторкислотой Льюиса, например AsF5, на дифтордиазин, молекула которого имеет формулу F-N=N-F, проще говоря, если сконденсировать эти два газа при температуре жидкого азота, а затем убрать охлаждение и дать им возможность медленно нагреться, то после плавления вещества прореагируют и образуют белую кристаллическую соль N2F+AsF6. При комнатной температуре она устойчива, но если ее нагреть выше 75°С, то начнется возгонка и конденсация N2F+AsF6 на холодных частях прибора. Отсюда следует, что реакция образования гексафторарсената фтордиазония обратима:
N2F2(ГАЗ) + AsF5(ГАЗ) ⇄ N2F+AsF6(КРИСТАЛЛ)
Точно так же соли N2F+ образуются при взаимодействии N2F2 с SbF6, PF5 и BF3. Устойчивость соединений, т.е. минимальная температура, при которой происходит их заметная диссоциация на исходные компоненты, растет в ряду N2F+PF6 < N2F+BF4 < N2F+AsF6 < N2F+SbF6. Такой ряд устойчивости справедлив практически для всех известных сейчас солей фторкатионов. N2F+BF4 и N2F+PF6 имеют значительное давление диссоциации уже при комнатной температуре. Отметим, что первые соли фтордиазония получили в нашей стране (А.В. Панкратов) и в США (Мой и Юнг, 1965 г.).
Другой бинарный фторид азота, тетрафторгидразин, также реагирует с пентафторидами мышьяка и сурьмы и образует при этом соли с катионом N2F3+. Интересно, что в комбинации с N2F3+ полифторанионы не только сурьмы, но и мышьяка имеют более сложный состав [AsF6·AsF5]. Образование многоядерных фторанионов [SbF6·хSbF5] для сурьмы очень характерно. При действии избытка N2F4 на соли с такими анионами они переходят в обычные N2F3+ЭF6.
В отличие от фтористого нитрила и фтористого нитрозила, которые образуют прочные соли с NО2+ и NО+, с большинством фторкислот Льюиса и даже с некоторыми кислородными кислотами, открытый в 1966 году новый оксифторид азота, ONF3, отдает фторид-ион только сильнейшим кислотам. Известны всего три соли с катионом ONF2+: ONF2+SbF6, ONF2+AsF6 и ONF2+BF4. Фтороборат совсем неустойчив - давление диссоциации (на ONF3 и BF3) достигает одной атмосферы при -37°С. Фторарсен имеет давление 10 мм при комнатной температуре, а фторантимонат не обнаруживает признаков диссоциации до 100°С.
Когда от ковалентной молекулы фторида азота или любого другого фторида удаляется ион F, существенно меняется электронная и геометрическая структура заряженного остатка. Так, молекула дифтордиазина имеет следующие геометрические параметры:
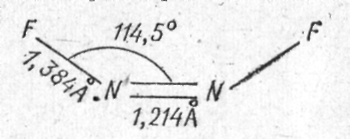
а катион фтордиазония - линейный, [N≡N—F]+, и длина связи азот—азот в нем около 1,10 Å. Валентная силовая постоянная, частота и длина связи N—N в ионе N2F+ очень близки к параметрам связи N≡N в молекуле азота, т.е. связь азот—азот в ионе фтордиазония тройная. Изменение спектральных характеристик связи N—F при переходе от N2F2 к N2F+ также указывает на ее укорочение и упрочнение.
Похожие
изменения происходят и с молекулой
тетрафторгидразина. Превращение ее в
катион N2F3+
сопровождается увеличением кратности
связей, ростом частот и силовых постоянных.
Упрощается и структура иона. В то время
как в молекуле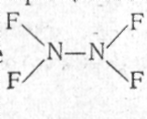 группы
NF2
повернуты
друг относительно друга на 64°, ион N2F3+
плоский. Аналогично и в случае
окситрифторида азота: группа ONF2
пирамидальная,
ион ONF2+
плоский. Связи N—F
и N—О в ионе ONF2+
короче и прочнее, чем в молекуле ONF3.
группы
NF2
повернуты
друг относительно друга на 64°, ион N2F3+
плоский. Аналогично и в случае
окситрифторида азота: группа ONF2
пирамидальная,
ион ONF2+
плоский. Связи N—F
и N—О в ионе ONF2+
короче и прочнее, чем в молекуле ONF3.
Приведенные пока немногочисленные сведения можно обобщить таким образом. Если от молекулы ковалентного фторида оторвать фторид-ион, то оставшаяся часть молекулы как бы сжимается; укорачиваются, упрочняются и становятся более жесткими все связи катионного фрагмента. Такое поведение хорошо согласуется с самыми простыми, школьными представлениями о молекулярных орбиталях и их заселенности. Фтор-катионы образуются из электроноизбыточных (орбитальнодефицитных) молекул. В них заполнены все связывающие орбитали, и часть электронов заселяет несвязывающие и разрыхляющие орбитали. При ионизации электрон удаляется с верхней разрыхляющей орбитали, в результате чего связи становятся более прочными и короткими.
Рассмотрим простейший случай превращения молекулы кислорода в диоксигенильный ион. В нейтральной молекуле О2 ее 12 валентных электронов распределены в следующем порядке: два на s-связывающей молекулярной орбитали (МО), два — на s-разрыхляющей МО, два — на zсв, четыре — на хyсв МО и по одному на двух -разрыхляющих МО — х и у. В целом электронная конфигурация молекулы O2 выглядит так: (sсв)2(sразр)2(zсв)2(х,yсв)4(xразр) (yразр). При ионизации удаляется один электрон с -разрыхляющей орбитали. Этот процесс требует затраты 278,5 ккал/моль. Теперь посмотрим, как сказывается удаление одного электрона с разрыхляющей орбитали на характеристиках связи О—О:
-
О2
О2+
Длина связи, Å
1,2074
1,1227
Энергия разрыва связи, ккал/моль
119,1
153,8
Валентная силовая постоянная, мдин/Å
11,4
16,3
Частота колебаний, см-1
1555
1867
Видно, что связь О—О заметно укоротилась - на 0,074 Å. Не нужно думать, что 0,074 Å - незначительная величина. В масштабах длин связи О—О это совсем немало. Предельная длина О—О около 1,50 Å, так что найденное изменение составляет ~20% от максимально возможного. Энергия связи возросла почти на 30%, силовая постоянная, характеризующая жесткость связи, - более чем на 40%, частота колебаний, выраженная количеством длин волн в 1 см, - на 20%.
Изменение геометрических размеров и энергии связей - не единственное следствие превращения ковалентных молекул в молекулярные ионы. Другой, более важный для синтетической химии результат такого превращения - изменение реакционной способности. Химическая активность RF и ЭFn-1 порознь гораздо ниже, чем продукта их соединения - R+ЭFn. Так, дифтордиазин с водой без восстановителей не реагирует, а соли N2F+ бурно гидролизуются при комнатной температуре. Реакция N2F2 с SO2 и AsF3 не идет до 100°С, a N2F+AsF6 окисляет эти вещества при низкой температуре. N2F4 инертен по отношению к воде, а N2F+Sb2F11 реагирует с водой взрывообразно, а также воспламеняет при контакте ацетон, этанол и другие органические жидкости. Резко повышаются окислительные свойства солей ONF2+ по сравнению со свободным окситрифторидом азота. Для протекания реакции ONF3c органическими веществами или водой требуется активация УФ-облучением или нагреванием. Даже с раствором щелочи NaOH ONF3 реагирует только при температуре выше 90°С, причем очень медленно. А соли ONF2+ окисляют органические вещества со взрывом и бурно разлагаются водой, давая HNO3 и HF. Интересно, что повышение химической активности ONF3 в присутствии фторкислот Льюиса происходит даже тогда, когда равновесная концентрация соли ONF2+ ничтожна. Так, давление диссоциации ONF2+BF4 по схеме ONF2+ВF4(тв) ⇄ ОNF3(гaз) + ВF3(газ) составляет при 25°С около 50 атм, однако введение BF3 в газовую смесь ONF3 + НСl вызывает быструю реакцию с образованием Cl2, NO+BF4 и HF. В отсутствие BF3 газовая смесь ONF3 с НСl вполне устойчива.
Изменение реакционной способности веществ под действием кислот Льюиса широко применяется в синтетической органической химии. В неорганическом синтезе примеров сознательного использования фторкатионов для проведения направленных превращений и синтеза новых соединений пока очень мало. Однако эти реакции могут оказаться весьма перспективными для решения задач, не разрешимых другими способами. Вот один пример использования реакционной способности фторкатионов. Если через трубку с порошком O2+SbF6 пропускать ксенон, то при комнатной температуре довольно быстро протекает реакция
Хе(газ) + 2O2+SbF6(тв) = XeF+Sb2F11(тв) + 2О2.
От 67 до 100% ксенона поглощается твердой солью диоксигенила, и на каждый атом связанного ксенона выделяется две молекулы кислорода. Еще легче в эту реакцию вступает радон. Из солей других фторкатионов связывать ксенон в химическое соединение в мягких условиях способны N2F+SbF6 и Cl2F+AsF6. В последнем случае смесь ClF, AsF5 и Хе конденсировали в сапфировом реакторе при -196°С, давали медленно нагреться до 25°С и выдерживали при этой температуре 1 ч. Количественно проходила реакция Cl2F+AsF6 + Xe = XeF+AsF6 + Cl2, за которой следовала другая, характерная для химии фторидов ксенона, 2XeF+AsF = Xe2F3+AsF6 + AsF5.
Способность диоксигенильной и других солей гетерокатионов реагировать с ксеноном и радоном, несомненно, найдет применение, например, в атомной промышленности для химического связывания радиоактивных изотопов 133Хе, 135Хе и 222Rn, образующихся при некоторых ядерных превращениях. Но сейчас хотелось бы подчеркнуть не эти практические последствия обнаруженного явления, а его принципиальную сторону. Реакцию образования валентного соединения инертного газа, которая еще 17 лет назад казалась невозможной, а десять лет назад требовала для протекания высоких температур и Давлений, сейчас гладко и количественно проводят при комнатной температуре и атмосферном давлении. Это - один из ярких результатов развития химии фторкатионов. Одновременно он показывает, как могут быть далеки от действительности прочно укоренившиеся представления.
ПУТЬ К ТЕТРАФТОРАММОНИЮ
После
того как были получены устойчивые соли
с новыми катионами N2F+,
N2F3+,
SF3+,
ClF2+
и некоторыми другими, после того как
стали известны основные свойства этих
катионов и стала понятной перспектива
их использования в качестве структурных
единиц твердых энергоемких веществ,
перед синтетиками-неорганиками возникло
несколько вопросов. До какого-то времени
все фторкатионы получали одним методом
— реакцией переноса фторид-иона от
основания к кислоте:![]()
Поэтому первый вопрос состоял в следующем: универсальна ли эта реакция? Можно ли, действуя достаточно сильным акцептором фторид-иона на молекулу любого ковалентного фторида, получить соответствующий фторкатион? После ряда безрезультатных проб на этот вопрос пришлось ответить отрицательно. Есть определенное число ковалентных фторидов, которые ни с одной из существующих фторкислот Льюиса ионных комплексов не дают. Например, трифторид азота, дифторид кислорода, перхлорилфторид, FClO3, фторофосген, COF2, гексафторид серы ни с SbF6, ни с AsF5, ни тем более с BF3 и другими полифторидами не реагируют.
Второй вопрос: можно ли получить фторкатионы, которым не соответствует никакая молекула-основание? Иначе говоря, есть ли пути синтеза солей с катионом R+, если молекулы RF не существует? На этот вопрос следует с полной уверенностью ответить положительно. Известно уже несколько фторкатионов и других молекулярных катионов, которым не соответствует какая-либо одна молекула-основание. Способы синтеза таких катионов не просты и не однотипны. Рассмотрим их на примере одного из наиболее интересных фторазотных катионов - тетрафтораммония.
С точки зрения задач синтетической химии твердых энергоемких веществ весьма заманчива мысль заместить все атомы водорода в ионе аммония на атомы фтора и получить, таким образом, соль с катионом NF4+. Вероятно, впервые возможность существования катиона NF4+ теоретически обсуждалась на очередной дискуссии Фарадеевского общества в Великобритании в 1963 году. В отчете о дискуссии помещен доклад Прайса, Пассмора и Росслера. В нем приведены расчеты энергий диссоциации и потенциалов ионизации фторидов и гидридов элементов первых периодов. Поскольку работа эта чисто теоретическая, авторы включили в таблицу на равных правах и существующие и несуществующие соединения. В числе прочих фигурирует и ион NF4+. Базируясь на своих оценках, авторы пришли к такому выводу относительно возможности существования иона NF4+: «Ясно, что его (NF4) потенциал ионизации слишком высок, а его размеры слишком велики, чтобы энергия решетки, в которую он войдет, была достаточна для образования кристаллических солей». Вывод вполне определенный — солей с ионом NF4+ существовать не должно.
Еще раз вопрос о возможности существования солей перфтораммония (ион NF4+ называют также тетрафторнитроний и тетрафторазот) обсуждался на 149-й Национальной конференции Американского химического общества в Детройте в апреле 1965 года. Доклад Дж. Н. Вилсона из Эмервилла, Калифорния, так и назывался «Определение стабильности перфтораммонийного иона и его солей». Опираясь на аналогии с другими тетраэдрическими ионами и молекулами, построенными из легких элементов, используя доступные значения энергии связи и потенциалов ионизации и оценив энергию гипотетических кристаллических решеток по формуле А.Ф. Капустинского, автор рассчитал энтальпию распада четырех солей: NF4+F, NF4+ClO4, (NF4+)2SO42- и NF4+BF4. Оказалось, что разложение первых трех солей должно сопровождаться понижением энтальпии более чем на 60 ккал/моль (соответственно на 67, 70 и 133 ккал/моль), т.е. эти соли при давлении в 1 атм должны быть нестабильны при любой температуре. Для фторобората заключение менее определенно: его энтальпия распада при одном способе расчета оказалась -2±20 ккал/моль, а при другом - -21±20 ккал/моль. Автор учел еще энтропию разложения NF4BF4 и пришел к выводу, что если даже принять энтальпию распада положительной (Н 10 ккал/моль), а не отрицательной, как получилось из его же расчетов, то стандартная свободная энергия разложения все равно будет отрицательной - около -28 ккал/моль, что соответствует давлению разложения порядка 107 атм при комнатной температуре. Меньше 1 атм давление диссоциации будет лишь при температуре ниже 100 K. Так что и этот расчет оставлял мало надежд на получение устойчивых солей с катионом NF4+.
Разумеется, авторитет теоретической химии среди синтетиков очень велик. И неизвестно, сколько лет пришлось бы ждать получения солей тетрафтораммония, если бы к этому времени уже не был накоплен определенный опыт, показавший, что наиболее значительные результаты синтетической химии получаются помимо теории. Так или иначе, только в мартовском номере журнала «Inorganic and Nuclear Chem. Lett.» за 1966 год появились сразу две статьи, в каждой из которых сообщалось об успешном синтеза солей NF4+, в первой статье - NF4+SbF6, во второй - NF4+AsF6. Авторы первой статьи - Толберг, Ревик, Стрингхэм и Хилл, авторы второй - Карл Кристи, Гертин и Павлат.
До знакомства со способами синтеза солей тетрафтораммония полезно поразмыслить над тем, как вообще можно было бы их получить. Это будет, кстати, примером решения проблемы метода синтеза, о которой говорилось в четвертой главе.
Рассмотренные до сих пор фторазотные, кислородазотные и другие катионы получались одним способом - действием акцептора фторид-иона на соответствующий ковалентный фторид. Так, из ONF получают NO+, из O2NF-NO2+, из ONF3-ONF2+ и т.п. Немного иной путь синтеза применен для О2+ - его получали прямым отнятием электрона от молекулы O2 гексафторидом платины. Однако проще получать диоксигенильные соли действием полифторида на радикал O2F, т.е. тем же путем, что и остальные фтор-катионы. Очевидно, что оба эти способа для синтеза солей тетрафтораммония не годятся. Не существует молекулы NF4, которую можно было бы окислить гексафторидом платины или каким-либо другим окислителем с еще большим электронным сродством, если таковой будет найден. Более того, ни разу не удавалось зафиксировать ион NF4+ даже в масс-спектре фторидов азота или любых других соединений. Все остальные перечисленные выше гетерокатионы не только обнаружены в масс-спектрах соответствующих фторидов, но и являются преобладающими - им отвечает наиболее интенсивный пик.
Не существует также молекулы NF5, действуя на которую пентафторидом сурьмы или мышьяка можно было бы получить NF4+SbF6 или NF4+AsF6. Не существует этой молекулы не потому, что синтетики плохо старались, не сумели найти условия ее появления и способа сохранения или не сумели узнать ее среди побочных продуктов. Причины здесь серьезнее. Довольно строгий неэмпирический квантовохимический расчет показывает, что энергия системы NF3+2F на 0,3133 атомных единицы энергии (1 а.ед. энергии = 27,21 эВ) ниже, чем гипотетической молекулы NF5 со структурой тригональной бипирамиды. Именно такую структуру имеет пентафторид фосфора. Анализ причин различия в свойствах соединений азота и фосфора привел авторов обзора (Д. А. Бочвар, Н.П. Гамбарян, Л.М. Эпштейн. О концепции вакантных орбиталей и о причинах различия в свойствах соединений азота и фосфора. - «Успехи химии», 1976, 45, № 7, с. 1316) к выводу, что пяти- и шестикоординационные соединения азота не могут существовать в отличие от соединений фосфора не по причине недоступности d-орбиталей у атома азота, а просто из-за слишком больших пространственных препятствий, т.е. из-за перекрывания электронных оболочек атомов лигандов. Иначе говоря, если разместить пять атомов фтора вокруг атома азота так, чтобы электронные оболочки атомов фтора не перекрывались, то электронная плотность между атомом азота и атомами фтора понизится настолько, что связь N-F фактически исчезнет. А если их сдвинуть к атому азота до образования прочной ковалентной связи N-F, то суммарная энергия пяти связей не компенсирует энергию отталкивания электронных оболочек атомов фтора друг от друга.
Каковы же мыслимые пути синтеза NF4+? Можно было бы попробовать заместить атомы водорода на атомы фтора в ионе аммония, например в NF4+SbF6. Если в качестве фторирующего агента взять элементарный фтор, то реакция будет энергетически выгодной: отрыв первого атома Н от NH4+ потребует около 120 ккал/моль, атомизация F2 - 38 ккал, присоединение атома F к NH3 освободит около 50-70 ккал и образование HF из атомов Н и F даст еще 136 ккал/моль. Общий итог положительный - выделится 30-60 ккал/моль. Близкие числа получатся и для замены последующих атомов. Действительно, фтор реагирует с солями аммония, реакция эта подробно изучена А.В. Панкратовым. При фторировании солей аммония образуются трифторид азота, дифтордиазины, азид фтора, тетрафторгидразин, но ни разу не было отмечено образования иона тетрафтораммония. Забегая вперед, можно сказать, что даже катион монофтораммония, NH3F+, не удается получить фторированием соли аммония. Путь к солям перфтораммония через реакцию замещения в ионе аммония с вероятностью, близкой к единице, бесперспективен.
Другой путь - прямой синтез. Можно было бы попытаться взять трифторид азота, фтор и какую-нибудь кислоту Льюиса, например SbF5 или AsF6, в молярном отношении 1:1:1 и каким-то образом заставить их реагировать между собой. Соль тетрафтораммония могла бы образоваться по суммарному уравнению
NF3 + F2 + SbF5 = NF4+SbF6.
Но как принудить систему реагировать в желаемом направлении? Ни трифторид азота, ни пентафторид сурьмы порознь с фтором не реагируют и соединений не дают. А нам требуется, чтобы в системе произошли такие превращения: разорвалась связь в молекуле F2 и образовались два атома фтора, один из этих атомов присоединился к молекуле NF3, образовав радикал NF4, другой соединился с молекулой SbF5, образовав радикал SbF6. Затем эти радикалы должны сблизиться и NF4 должен отдать, a SbF6 принять электрон. Образовавшиеся ионы NF4+ и SbF6 объединятся в кристаллическую решетку. Разумеется, приведенная схема отнюдь не претендует на отражение действительного механизма превращений. Возможных путей от системы, состоящей из молекул NF3, F2 и SbF6, к кристаллической соли NF4+SbF6 несколько, они отличаются природой и последовательностью простейших стадий, элементарных актов, составом и реакционной способностью активных промежуточных частиц. В зависимости от внешних условий - температуры, давления, присутствия катализаторов или действия активаторов - природа выберет тот путь, тот канал реакции, который будет связан с наименьшими энергиями активации и наибольшими факторами частоты для всех промежуточных стадий. Гораздо легче эмпирическим путем, т.е. методом проб и ошибок, найти условия получения желаемого вещества, чем измерить все эти кинетические параметры и установить истинный механизм реакции.
Какие же надо создать условия, чтобы прошла реакция прямого синтеза NF4+SbF6? И пойдет ли она вообще? В соответствии с законами термодинамики химическая реакция может протекать слева направо, если в данных условиях свободная энергия (точнее, энергия Гиббса, G) продуктов в правой части уравнения ниже, чем в левой. Реакция идет в сторону уменьшения энергии Гиббса. Изменение этой энергии есть разность Н - TS. Как изменяется энтальпия в приведенной выше реакции, мы не знаем. Неизвестна не только энтальпия образования продукта реакции, но и одного из исходных веществ. До сих пор как следует не измерена энтальпия образования пентафторида сурьмы. Опубликовано, правда, в 1975 году значение -319,54 ккал/моль, но, по мнению специалистов по фторной калориметрии, оно очень ненадежно. Что касается энтропии, то хотя численные значения неизвестны, но очевидно, что реакция будет сопровождаться сильным уменьшением S, поскольку из трех молей газа образуется моль твердого вещества. Энтропийный фактор не благоприятствует протеканию реакции в сторону NF4+SbF6.
Однако не нужно думать, что если бы даже оказалось, что G рассматриваемой реакции положительно, то ее невозможно осуществить. Например, свободная энергия образования окиси азота NO из элементов положительна, G298 = +20,7 ккал/моль, но если предварительно каким-нибудь способом сообщить энергию реагирующим молекулам, скажем, возбудить их или перевести в атомарное состояние в плазме электрического разряда или действием световых квантов, то дальнейшая реакция между атомами или между атомами и молекулами будет идти с понижением энергии Гиббса. Синтез окислов азота из элементов в газоразрядной плазме - хорошо известный процесс. Другой пример - получение диоксидифторида O2F2. Свободная энергия и энтальпия образования его из элементов положительны. Вместе с тем основной препаративный способ его получения - реакция фтора с кислородом в тлеющем разряде, причем стенки разрядной трубки охлаждают жидким азотом.
Если реакция с фтором и пентафторидом сурьмы термодинамически допустима, то целесообразно было бы проводить ее под высоким давлением, поскольку она сопровождается сильным уменьшением объема. Активировать реагенты в этом случае можно было бы умеренным нагреванием. Сильное нагревание опасно, потому что неизвестен предел термической стабильности искомой соли. Если же реакция сопровождается увеличением свободной энергии, то можно попробовать провести ее в электроразряде. При этом нужна большая осторожность, чтобы активация затронула только те молекулы, которые в этом нуждаются, и не разрушила какой-либо из реагентов, например SbF5. Разумеется, разряд можно применить и в том случае, если реакция синтеза идет с понижением энергии Гиббса.
Оба эти метода применили упомянутые выше две группы исследователей, и в обоих случаях результат был положительный. Толберг и сотрудники проводили реакцию в автоклаве из монель-металла объемом 60 см3. В него было помещено по 0,1 моля фтора, трифторида азота и пентафторида сурьмы и добавлено 0,5 моля фтористого водорода в качестве растворителя. Автоклав выдержали три дня при 200°С, при этом давление внутри достигало 185 атм. После вскрытия реактора обнаружилось, что около 40% взятого NF3 превратилось в NF4+SbF6. Вещество было загрязнено твердыми продуктами взаимодействия фтора и SbF5 с материалом автоклава. Аналогично, но с худшим выходом и меньшей чистотой был получен NF4+AsF6. В дальнейшем метод совершенствовался, уточнялись параметры, удален за ненадобностью фтористый водород, но принцип метода сохранился.
Кристи синтезировал соль NF4+ в тлеющем разряде. Смесь NF3 и AsF5 с двукратным избытком фтора при общем давлении 80 мм циркулировала через разрядную трубку, стенки которой охлаждались до -78°С. По мере расходования реагентов давление в системе падало; когда оно опускалось до 10 мм, добавляли исходной смеси. Для получения 1 г NF4+AsF6 потребовалось около 40 ч горения разряда.
Так было получено экспериментальное доказательство существования солей тетрафтораммония. Обе соли оказались очень устойчивыми: разложение их начинается при температуре значительно выше 250°С. Фторантимонат тетрафтораммония имеет температуру плавления 318°С. Плотность NF4AsF6 равна 2,72, a NF4SbF6 - 2,98 г/см3.
Подробные сообщения о синтезе первых двух солей тетрафтораммония появились в 1967 году, а уже в феврале 1970 года в нашей стране было выдано авторское свидетельство на способ получения третьей соли тетрафтораммония - тетрафторобората NF4+BF4. Того самого фторобората, который, по предсказанию Дж. Вилсона, если и может существовать, то при температуре ниже 100 K. NF4+BF4 впервые получен С.М. Синельниковым в Институте новых химических проблем АН СССР в 1969 году.
После получения и оценки термической стабильности первых двух солей тетрафтораммония стало понятно, что предсказание Вилсона насчет NF4+BF4 скорее всего неверно. Соотношение устойчивости солей одного и того же фторкатиона с анионами SbF6, AsF6 и BF4 мало меняется от катиона к катиону. В указанном ряду стабильность уменьшается, но в довольно умеренных пределах. Так, ONF2+AsF6 при 25°С имеет давление диссоциации 10 мм, а над ONF2+BF4 такое же давление достигается при -80°С. Температура, при которой давление диссоциации равно 1 атм, для SF3+AsF6 - 184°C, а для SF3+BF4+ - 53°С. Разница в температурах разложения фторарсенатов и фтороборатов лежит в пределах 100-150°. Зная, что распад NF4+AsF6 начинается при температуре выше 250°С, можно было довольно уверенно предсказать, что NF4+BF4 до 100°С должен быть устойчив.
Приведенные рассуждения - обычный пример того, как неорганики-синтетики решают проблему существования в частном случае. Результат такого рассуждения дает необходимые надежды на успех в синтезе. Такого типа предсказания не имеют никаких теоретических оснований, они насквозь эмпиричны, но в большинстве случаев сбываются. Не составил исключения и случай с фтороборатом тетрафтораммония. После ряда прикидок и манипуляций давлением, температурой, силой разрядного тока, скоростью подачи реагентов и т.п. синтез его свелся к простой операции. В небольшой U-образной разрядной трубке, погруженной в жидкий азот, намораживали на стенки смесь NF3 с BF3, напускали фтор до давления 50 мм и включали разряд. Через 5 мин давление падало до 1-2 мм. В дальнейшем фтор напускали непрерывно при включенном разряде. Фтор подавали с такой скоростью, чтобы давление в системе оставалось постоянным, т.е. чтобы скорость подачи и расходования на реакцию с NF3 и BF3 была одинакова. За час в трубке объемом 50 см3 удается получить несколько граммов NF4+BF4. Выход его близок к количественному.
Стоит обратить внимание на разительное отличие в скорости синтеза NF4BF4 и NF4AsF6. Оба вещества получают в тлеющем разряде из однотипных реагентов, однако для получения 1 г NF4AsF6 требовалось около 40 ч, а на получение 1 г NF4BF4 - всего несколько минут. Главная причина этого - низкая температура стенок разрядной трубки при синтезе NF4BF4.
В той же лаборатории, где С.М. Синельников разрабатывал способ получения NF4BF4, примерно в то же время И.В. Никитин закончил подробное исследование синтеза и распада трифторида азота в тлеющем разряде. Оказалось, что даже при небольшой мощности разряда NF3 быстро и почти полностью распадается на фтор и азот. Синтез NF3 из элементов в тлеющем разряде возможен, но требует больших удельных энергий и успешно идет только при вымораживании продукта жидким азотом. Если стенки разрядной трубки охладить не жидким азотом (-196°С), а твердой углекислотой (-78°С), как это было в опытах Кристи, то образования NF3 в плазме почти не наблюдается. Этим, видимо, объясняется неэффективность синтеза NF4AsF6 в разряде.
Другая причина - неустойчивость пентафторида мышьяка в разряде. Среди сильных фторкислот Льюиса лишь трифторид бора отличается очень высокой стабильностью. Он может длительно находиться в плазме тлеющего разряда, не подвергаясь деструкции. Вместе с высокой летучестью (температура кипения -100°С) и высоким сродством к фторид-иону (~90 ккал/моль) это свойство делает трифторид бора весьма ценным веществом для синтеза фторкатионов в разряде. Если в низкотемпературной плазме образуется неустойчивая фторсодержащая молекула, способная быть донором фторид-иона, то стабилизировать ее можно либо путем закалки, т.е. конденсации и быстрого охлаждения на стенке, либо путем химического связывания в нелетучее или малолетучее соединение с кислотой Льюиса. Трифторид бора - незаменимый реагент для такрго связывания. Так, вводя в разряд кислород, фтор и BF3, можно непосредственно получать O2+BF4, а из азота, фтора и BF3 в разряде образуется NF4BF4.
Фтороборат тетрафтораммония оказался весьма термически устойчивым веществом. Он превосходит по температуре разложения большинство фтороборатов фторкатионов. Среди солей NF4+ только фторарсенат и фторантимонат разлагаются при более высокой температуре, чем NF4+BF4.
В условиях непрерывного нагревания NF4+BF4 распадается на NF3, BF3 и F2 в интервале 240-350°С с значительным поглощением тепла. Следовательно, синтез его из тех же веществ - реакция экзотермическая. Скорее всего образование его из NF3, BF3 и F2 сопровождается не только понижением энтальпии, но и уменьшением изобарного потенциала (энергии Гиббса), а разряд действует в данном случае как своеобразный катализатор, увеличивающий скорость термодинамически допустимой реакции.
В последующие годы интерес химиков-синтетиков к этому необычному классу соединений не только не ослаб, но, скорее, усилился. За семь лет, с 1971 по 1978 год, было получено еще 11 солей тетрафтораммония с различными анионами. Вот реестр известных сейчас (лето 1978 года) солей NF4+ (в порядке увеличения номера центрального атома аниона):
NF4BF4 (NF4)2NiF6 NF4SnF6
NF4PF6 NF4GeF6 (NF4)2SnF6
(NF4)2TiF6 (NF4)2GeF6 NF4SbF6
NF4Ti2F9 NF4AsF6 NF4BiF6
NF4Ti3F13 NF4NbF6
Bсe они - гигроскопичные кристаллические вещества, вполне устойчивые в отсутствие влаги и восстановителей. Хранить их можно в металлической посуде (сталь, никель, монель, медь) или в контейнерах из тефлона.
Появились новые методы получения солей NF4+. Наиболее интересный из них - низкотемпературный УФ-фотолиз. Для получения NF4BF4 этим методом смесь NF3, BF3 и F2 в стальном реакторе с сапфировыми окнами облучают светом ртутно-кварцевой лампы. Металлические стенки реактора охлаждают жидким азотом. При мощности лампы 900 Вт в небольшом реакторе удается получить до 3 г NF4BF4 за 1 ч. Способ пригоден и для синтеза других солей тетрафтораммония - фторарсената, фторофосфата, фторогерманата. Исследование фотолитической реакции между трифторидом азота, фтором и фторкислотой Льюиса позволило понять механизм образования иона NF4+. Под действием квантов света происходит ионизация молекул NF3 с образованием нестабильного катион-радикала NF3+, а реакция этого радикала с атомарным фтором дает NF4+. Весьма вероятно промежуточное образование NF3+ и при синтезе солей NF4+ другими методами, и при их термическом распаде.
Соли тетрафтораммония уникальны по своему составу. Ни среди органических, ни среди неорганических соединений нет твердых веществ с таким высоким содержанием фтора. Например, NF4BF4 на 86% состоит из фтора. Можно было бы сказать даже так: NF4BF4 - это твердый фтор, загрязненный 14% бора и азота. Правда, такое утверждение более образно, нежели точно. Эти 14% бора и азота прочно химически связаны с фтором, и осуществить весьма заманчивую реакцию превращения «твердого фтора» в газообразный:
NF4BF4 = BN + 4F2
не удается. При таком гипотетическом распаде на нитрид бора и элементарный фтор фтороборат тетрафтораммония давал бы 507 л фтора с. килограмма твердого вещества.
Несмотря на крайнюю «молодость» солей тетрафтораммония, с момента синтеза большинства из них не прошло и десяти лет, выдано уже несколько патентов на их использование. Соли NF4+ в смеси с политетрафторэтиленом предложено применять в качестве окислителей твердых ракетных топлив высокой плотности. Правда, единичный импульс таких топлив невысок: большая часть фтора в солях тетрафтораммония энергетически неактивна. Более перспективно их применение в качестве источников фтора для химических лазеров на HF и DF. Атомарный фтор может генерироваться по реакции
2NF3 + F2 = N2 + 8F.
Необходимое для реакции тепло поставляет экзотермическое взаимодействие NF4ЭF6 с тефлоном:
NF4ЭF6 + (CF2)n = CF4 + NF3 + ЭF5.
Ион NF4+ имеет структуру правильного тетраэдра, так же, как и изоэлектронная ему молекула CF4. Точно так же изоструктурны и изоэлектронны NH4+ и СН4. Существуют молекулы всех трех промежуточных составов между четырехфтористым углеродом и метаном: CF4 - CF3H - CF2H2 - CFH3 - CH4. Возникает вопрос: могут ли существовать ионы, промежуточные по составу между NF4+ и NH4+, т.е. можно ли последовательно замещать атомы водорода в ионе NH4+ на атомы фтора, получая после каждой замены устойчивый ион NH3F+, NH2F2+ и NHF3+? Аналогия с системой СН4 - CF4 подсказывает положительный ответ. Но метод аналогий, как уже говорилось в начале этой работы, ненадежен, предсказания по аналогии в химии легких элементов часто не оправдываются. В данном случае действительность соответствует предсказанию довольно хорошо: из трех предсказанных форм две получены. Не выделены только соли с катионом NF3H+. Попытка их синтеза по реакции
NF3 + HF + SbF5 = NHF3+SbF6
не привела к появлению каких-либо твердых веществ, устойчивых при температурах выше -78°С. Не получив соединения, трудно объяснить, почему оно не существует. Во всяком случае причина не в отсутствии сродства молекулы NF3 к протону. Присоединение Н+ к NF3 в газовой фазе с образованием NF3H+ сопровождается выделением 150 ккал/моль. Для сравнения вспомним, что сродство NH3 к протону 207 ккал/моль - величина соизмеримая.
Соли дифтораммония легко образуются в среде жидкого фтористого водорода из дифторамина и кислоты Льюиса
HNF2 + HF + SbF5 = NH2F2+SbF6
в виде кристаллических веществ, хорошо растворимых в HF. При комнатной температуре они быстро экзотермически распадаются.
Все рассмотренные нами фторсодержащие катионы, а также все еще нерассмотренные, обладают одним общим свойством - они устойчивы только в виде солей с полифторанионами типа ЭFn и разрушают (фторируют) любые другие анионы. Исключение составляет монофтораммонийный катион NH3F+. Он получен в виде солей с кислородными кислотами - хлорной, NH3F+ClO4, метилсерной, NH3F+CH3SO3, и трифторметилсерной, NH3F+CF3SO3. Для синтеза использованы приемы органической химии: и одно их исходных веществ было органическим - алкилфторкарбамат. Перхлорат получен по реакции
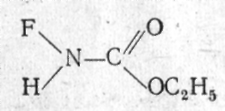 +
2НСlO4
= NH3F+ClO4
+ СО2
+ C2H5OClO3.
+
2НСlO4
= NH3F+ClO4
+ СО2
+ C2H5OClO3.
Соли NH3F+ довольно устойчивы. Перхлорат и метилсульфат распадаются при температуре около 100°С, с сильным тепловыделением, а трифторметилсульфат плавится с частичным разложением при 165°С. Таким образом, в ряду NH4+, NH3F+, NH2F2+, NHF3+, NF4+ устойчивость падает слева направо, достигает минимума на NHF3+ и резко растет к NF4+.
Подведем итоги. К настоящему времени известно шесть фторсодержащих катионов азота:
-
Год открытия
Количество полученных солей
N2F3+
1965
2
N2F+
1965
4
NF4+
1966
14
ОNF2+
1966
3
NH3F+
1968
3
NH2F2+
1975
2
Пять из них появились за очень короткий срок - всего три года, от 1965 до 1968. Вспомним, что к моменту открытия NF4+ катионы NО+ и NО2+ были известны уже более 50 лет, а со времени открытия NF3 Руффом прошло около 40 лет. Причины того, что столь интересные соединения появились не в тридцатых годах, а в шестидесятых, связаны не только с техническими и экспериментальными трудностями, но и с мировоззрением, с господствовавшими в химии представлениями. Возможность существования таких катионов представлялась химикам невероятной. После того как стало ясно, что существование фторкатионов возможно, синтетики проявили массу изобретательности и нашли пути получения множества молекулярных катионов, построенных из атомов неметаллов. В течение менее чем десяти лет были получены соли десятков новых фторкатионов с центральными атомами азота, фосфора, серы, хлора, селена, брома, криптона, теллура, йода, ксенона. С фторкатионами хлора мы познакомимся в следующем разделе.
ФТОРКАТИОНЫ И ОКСИФТОРИДЫ ХЛОРА
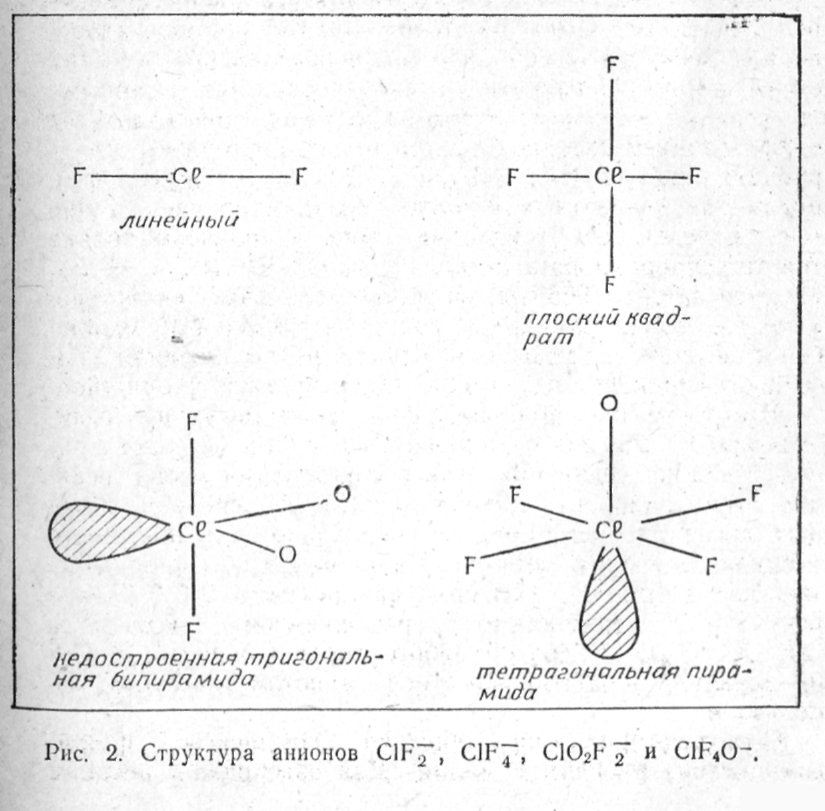
Валентные и координационные возможности хлора больше, чем азота. Атом азота имеет пять валентных электронов, и его предельное известное координационное число четыре. У атома хлора семь валентных электронов, а его координационное число достигает шести. Несмотря на это, хлор, как и азот, образует только восемь молекулярных катионов со связью Cl-F и/или Сl-О. Вот их формулы: Сl3+, Cl2F+, ClF2+, ClF4+, ClOF2+, СlO2+, ClO2F2+, ClF6+. Некоторые из них по составу аналогичны азотным катионам, например, Cl2F+ и N2F+, ClF4+ и NF4+, СlO2+ и NO2+, ClOF2+ и NOF2+. Однако в геометрической структуре их мало общего. Симметрия катионов хлора ниже, чем однотипных соединений азота. Это вполне понятно. В пятивалентном состоянии у азота нет неподеленных электронных пар и атомы-лиганды располагаются вокруг азота самым симметричным образом - тетраэдрически в NF4+, по вершинам треугольника в ONF2+, линейно в NO2+. У пятивалентного хлора есть одна неподеленная пара электронов, она в соответствии с правилом Гиллеспи - Найхолма занимает свое место в координационной сфере хлора; в результате чего ClF4+ не тетраэдр, а тригональная бипирамида, у которой одно из трех экваториальных мест занято парой электронов, не участвующей в связи. По той же причине в ионе СlO2+ угол О-Сl-О не 180°, как у NO2+, а 122°. В отличие от однотипного фторазотного иона нелинеен катион Cl2F+. Лишь у производных семивалентного хлора, где нет неподеленных электронных пар, ионы имеют высокую симметрию: ClF6+ - октаэдрическую, ClO2F2+ и СlO4 - тетраэдрическую.
Как и в случае катионов азота, большая часть солей фторкатионов хлора получена действием сильных акцепторов фторид-иона на соответствующие фториды или оксифториды хлора. Так, из трифторида хлора получены соли с ионом ClF2+, из пентафторида - с ионом ClF4+, из окситрифторида - соли с ионом OClF2+, а из FClO2 - соли хлорила СlO2+. Катион F3ClO2+ тоже можно получить совмещением SbF5, AsF5 или BF3 с диокситрифторидом хлора O2ClF3, однако исторически все сложилось иначе. Сначала была получена соль с катионом ClO2F2+, а потом из нее довольно сложным путем выделен новый оксифторид хлора ClO2F3.
Молекулы высшего фторида хлора ClF7 не существует, так же, как и не существует высшего фторида азота. Так что получить ион ClF6+ реакцией переноса фторида невозможно. К синтезу гексафторхлорного катиона приступили тогда, когда уже был накоплен значительный опыт в синтезе солей NF4+. Можно было думать, что для решения внешне однотипных задач будут пригодны одни и те же приемы. Если из NF3, F2 и SbF5 (или AsF5, или BF3) под действием высокой температуры и давления, или под действием облучения, или в плазме разряда получается соль NF4+, то естественно ожидать, что при аналогичных воздействиях на смесь ClF5, F2 и SbF5 образуется ClF6+SbF6. Или, если взять другие кислоты Льюиса, можно будет получить ClF6+AsF6, ClF6+BF4 и т.п. Такие опыты были сделаны. Смесь ClF5, F2 и AsF5 в различных молярных отношениях нагревали в течение 5-10 дней до 125-145°С. Давление при этом достигало 70 атм. При разгрузке автоклава ничего, кроме исходных продуктов, найдено не было. Нагревание до 160°С приводило к частичному распаду ClF5 до ClF3. Ничего нового не получилось также из смеси ClF5, F2 и BF3 после выдерживания в автоклаве при 95°С в течение 8 дней. Когда вместо AsF5 и BF3 взяли сильнейшую из кислот Льюиса SbF6, увеличили время контакта до 40 суток и температуру до 225°С, в продуктах реакции были найдены соли ClF2+, ClF4+, Ni2+, Cu2+ с анионом SbF6·xSbF5 и никаких признаков катиона ClF6+. Пропускание смеси ClF5, F2 и BF3 в отношении 1:4,2:1 через тлеющий разряд, охлажденный до -78°С, привело к образованию только ClF2+BF4. И в этом случае ClF6+ найдено не было.
Следует подчеркнуть, что когда эта работа проводилась, автор ее, Карл Кристи, уже знал спектр и другие свойства катиона ClF6+. Если бы в каком-то из опытов образовались даже небольшие количества соли ClF6+, она не укрылась бы от исследователя.
Таким образом, методы, разработанные для синтеза солей фторкатиона азота высшей валентности, не привели к успеху в синтезе высшего фторкатиона хлора. Можно было бы подробно проанализировать причины этого факта. В самом общем виде они заключаются в несходстве химии фторидов азота и фторидов хлора. Однако сейчас нам важно отметить, что это общее явление. Метод аналогий в тонком неорганическом синтезе в общем случае не работает. Получение новых соединений требует новых методов синтеза.
Каким же образом удалось все-таки получить катион ClF6+? Для его синтеза был использован тот же реагент, что и за десять лет до этого для синтеза первой соли O2+ - гексафторид платины. У гексафторида платины есть два свойства, которые делают его чрезвычайно ценным веществом в синтезе фторкатионов. Во-первых, PtF6 является сильнейшим окислителем. Оценка электронного сродства молекулы PtF6 дает величину более 6,8 эВ. Это одно из самых высоких значений электронного сродства, известных в настоящее время для молекул каких-либо химических соединений. Для сравнения укажем, что электронное сродство радикала СlO4 5,8 эВ, радикала NO3 - 3,77 эВ, атома Сl - 3,61 эВ, атома F - 3,45 эВ. Сравнимое с PtF6 сродство к электрону могли бы иметь радикалы SbF6 и AsF6. Однако реальность их пока еще требует доказательств. Во-вторых, гексафторид платины после присоединения электрона превращается в анион пятивалентной платины PtVF6, который стабилизирует образовавшийся катион в кристаллической решетке в такой же степени, как анионы SbF6 или AsF6. То есть PtF6 совмещает в себе и свойства окислителя, и свойства сильной кислоты Льюиса и при этом не дает побочных продуктов реакции. Это идеальное сочетание для синтеза фторкатионов элементов в высшей степени окисления.
При исследовании взаимодействия гексафторида платины с FClO2 было замечено, что если смесь двух веществ быстро нагреть от -196 до -78°С и закончить процесс при этой температуре, реакция протекает по такому уравнению:
6FClO2 + 6PtF6 = 5СlO2+PtF6 + ClF6+PtF6 + O2.
Желаемая соль ClF6+PtF6 составляет лишь одну шестую часть в смеси твердых продуктов реакции. Разделить такую смесь и выделить ClF6+PtF6 затруднительно. Зато удалось достигнуть гораздо более высокого содержания искомой соли, заменив FClO2 на ClF5. Взаимодействие ClF5 с PtF6 идет при комнатной температуре под действием света. Если облучать реакционную смесь ртутно-кварцевой лампой сквозь пирексо-водяной фильтр в течение двух недель, протекает следующая реакция:
2ClF5 + 2PtF6 = ClF4+PtF6 + ClF6+PtF6.
Гексафтороплатинат - единственная известная соль катиона ClF6+, но и она не получена в индивидуальном состоянии, но лишь в смеси с гексафтороплатинатами других хлорсодержащих катионов. Эта соль имеет желто-канареечный цвет, устойчива при комнатной температуре в отсутствие влаги, обладает свойствами сильного окислителя - с органическими веществами и водой взрывает.
В настоящее время известны соли высших фторкатионов трех галогенов ClF6+, BrF6+ и IF6+. Синтез солей IF6+ принципиальных трудностей не представлял, поскольку было доступно исходное соединение - гептафторид йода. Реакция IF7 с фторкислотами Льюиса дает устойчивые соли IF6+SbF6, IF6+AsF6 и т.п. Сложнее обстояло дело с BrF6+. Патент на способ получения гептафторида брома был выдан в США в 1971 году (приоритет от 28 апреля 1965 года), однако, скорее всего, авторы патента что-то напутали, и существование BrF7 нельзя считать доказанным. Во всяком случае Гиллеспи и Шробильген в 1974 году не смогли воспользоваться гептафторидом брома для синтеза солей BrF6+ и действовали так, как будто BrF7 не существует.
Хлор и бром - элементы-аналоги, сходства в их химии гораздо больше, чем в химии азота и хлора, но опять для решения внешне одинаковой синтетической задачи - получения BrF6+ при известном способе синтеза ClF6+ - пришлось применять разные методы. Окислительное фторирование BrF5 дo BrF6+ провели не с помощью PtF6, а посредством солей другого фторкатиона - Kr2F3+, взятого в виде Kr2F3+SbF6. Действие солей этих катионов в какой-то степени напоминает действие PtF6. Катион выполняет окислительную функцию и одновременно служит фторирующим агентом, а анион SbF6 стабилизирует новый катион в кристаллической решетке. Единственный побочный продукт - элементарный криптон. Освобождение от него никаких специальных мер не требует. Для получения BrF6+SbF6 к раствору BrF4+Sb2F11 в BrF5 добавляли KrF2 при -196°С и нагревали смесь до 25°С, а затем удаляли все летучие примеси в вакууме при 50°С. Состав соли был промежуточным между BrF6+Sb2F11 и BrF6+SbF6.
Незадолго до того как был впервые получен ClF6+, удалось синтезировать еще один катион семивалентного хлора - ClO2F2+. Способ его получения очень мало отличается от способа получения ClF6+ из FClO2. Кстати говоря, целью исследования реакции между FClO2 и PtF6 был именно синтез ClO2F2+. Если смесь FClO2 с PtF6 в стальном цилиндре медленно нагреть от -196 до 25°С и выдержать при этой температуре три дня, то полученные продукты указывают на протекание следующей реакции
2FClO2 + 2PtF6 = ClO2+PtF6 + ClO2+F2+PtF6.
Оксифторид хлора, соответствующий катиону ClO2F2+, в то время получен еще не был. Для его выделения на смесь ClO2+PtF6 и ClO2F2+PtF6 действовали фтористым нитрилом. Как более сильное основание Льюиса NO2F вытеснил ClO2F и ClO2F3 из их солей. Полученную смесь трех газов частично разделили фракционированной конденсацией. Получилась смесь ClO2F и ClO2F3. К ней добавили избыток трифторида бора - оба основания опять превратились в соли ClO2+BF4 и ClO2F2+BF4. По счастью, оказалось, что ClO2+F2BF4 не имеет заметного давления диссоциации при комнатной температуре, а давление диссоциации ClO2+BF4 достигает 180 мм уже при 22°С. Это позволило полностью освободить ClO2F2+BF4 от ClO2+BF4. На полученный чистый СlO2F2+BF4 опять подействовали фтористым нитрилом (строго дозированным количеством!) и получили свободный от примесей диокситрифторид хлора. Выход его составил около 10%. Ни один из оксифторидов галогенов не стоил исследователям стольких усилий и не требовал такого тонкого и утомительного эксперимента, как ClO2F3. Трудно найти в неорганической химии легких элементов еще пример такой изощренной сложности синтеза нового соединения.
Таким образом, сейчас известно два фторкатиона семивалентного хлора - ClO2F2+ и ClF6+. Основание, отвечающее первому, выделено и исследовано, фторид, сопряженный катиону ClF6+, выделить не удалось. При действии сильных оснований Льюиса на ClF6PtF6 даже при низкой температуре, до -78°С, выделяющиеся газы состоят из ClF5.+ F2 и не содержат ClF7. Если ClF7 и может существовать, что весьма сомнительно, то при -78°С он неустойчив.
Возможен ли синтез новых, еще неизвестных молекулярных катионов семивалентного хлора? Будем рассматривать только катионы, в которых хлор служит центральным атомом, а кислород и фтор - лигандами. Запишем все возможные промежуточные формы от высшего окисла до высшего фторида хлора:
Сl2O7 ClO3F ClO2F3 [ClOF5] [ClF7][ClO3] ClO2F2+ [ClOF4+] ClF5+
Во второй строке помещены формулы соответствующих катионов. В квадратные скобки заключены неизвестные сейчас соединения. Заключение в квадратные скобки катионов означает, что они не получены в виде солей. В масс-спектре положительных ионов ClO3F, Сl2O7 или НСlO4 катион СlO3+ преобладает.
После успешного синтеза ClO2F (Шумахер, 1942 г.), ClO3F (Энгельбрект, 1952 г.), ClOF3 (Пилипович, 1965 г.; Бугон, 1970 г.), ClO2F8 (Кристи, 1972 г.) стало казаться, что последний оксифторид семивалентного хлора, ClOF5 будет вот-вот получен. На IV Европейском симпозиуме по химии фтора (Югославия, 1972 г.) было даже представлено сообщение о синтезе ClOF5, а немного спустя вышла статья тех же авторов в журнала «Angewandte Chemie».
Однако тщательная проверка их результатов показала, что авторы поторопились и приняли желаемое за сущее. Очень похожая история произошла и с катионом СlO3+.
Неоднократные попытки получить OClF5 и соли с катионами ClOF4+ и СlO3+ к успеху не привели, и нет оснований ожидать, что будут когда-либо получены стабильные соединения, отвечающие этим трем формулам.
Выпишем теперь формулы возможных оксифторидов и фторида пятивалентного хлора, а также формулы соответствующих катионов:
ClO2F ClOF3 ClF5 СlO2+ ClOF2+ ClF4+
Ни одно из соединений нет нужды заключать в квадратные скобки, все они синтезированы и исследованы.
Хлорилфторид, ClO2F - первый ставший известным оксифторид хлора. Он получен во время второй мировой войны в Германии низкотемпературным фторированием двуокиси хлора. Хлорилфторид - сильное основание Льюиса. Он образует соли СlO2+ не только с обычными полифтор-анионами – SbF6, BF4, PF6, но и с анионами более слабых кислот – SnF6, SiF62-, SO3F и даже с бесфторными анионами. Недавно удалось доказать, что шестиокись хлора Сl2O6, сильно отличающаяся по своей летучести и некоторым другим свойствам от остальных окислов хлора, в кристаллическом состоянии построена из ионов СlO2+ и СlO4. В этом отношении она является аналогом азотного ангидрида. Как показала в 1973 году З.К.Никитина, комплекс безводного перхлората алюминия с молекулой шестиокиси хлора Al(СlO4)3·Сl2О6, полученный реакцией Сl2О6 с хлоридом алюминия, тоже построен как соль хлорильного катиона СlO2+[Аl(СlO4)4]. Спустя несколько лет было получено аналогичное соединение галлия СlO2+[Ga(СlO4)4]. В составе и способе синтеза этих комплексов продолжается сходство между Сl2О6 и N2O5. Азотный ангидрид также реагирует с хлоридами алюминия и галлия, образуя соли NO2+[Al(NO3)4] и NO2+[Ga(NO3)4].
Казалось бы, скорее следует ожидать подобия в свойствах высших окислов хлора и азота, тем более что их молекулы построены однотипно:

Однако в химическом поведении Сl2O7 и N2O5 есть много различий. Структура молекул хлорного ангидрида одинакова и в газообразном, и в жидком, и в твердом состояниях. Во всех агрегатных состояниях это ковалентное соединение без каких-либо признаков поляризации по типу ClO3+СlO4. Азотный же ангидрид, как уже было сказано, в твердом состоянии имеет ионное строение.
Пентафторид хлора получен через год после открытия фторидов ксенона. На автора, работавшего с фторидами хлора, повлияло именно то, что гексафторид ксенона - инертного газа! - уже известен, а у хлора - чрезвычайно реакционноспособного элемента - не выделен даже пентафторид. Для синтеза он применил те же условия, что и при получении XeF6, -350°С и 250 атм. Таким способом из смеси ClF3 и F2 (1:14) получен с небольшим выходом ClF5. Впоследствии были разработаны гораздо более эффективные промышленные методы его синтеза.
Кристаллические солеобразные аддукты ClF4+ЭF6 пентафторид образует лишь с сильнейшими акцепторами фторид-иона - SbF5, AsF5 и PtF5. В системе ClF5 - BF3 соединений не образуется. На диаграмме плавкости двойной системы ClF5 – SbF5 есть три открытых максимума, отвечающих соединениям ClF4+SbF6 (т. пл. 120°С), ClF4+Sb2F11 (т. пл. 64°С) и ClF4+Sb4F21 (т. пл. 62°С), [В.Ф. Суховерхов, 1975 г.]. Давление диссоциации ClF4+AsF6 уже при комнатной температуре составляет несколько миллиметров.
Первое сообщение о синтезе нового оксифторида хлора ClOF3 пришло в 1970 году из Франции. Бугон и его сотрудники в течение 45 ч облучали ртутно-кварцевой лампой газовую смесь ClF3 и OF2 при 25°С. В продуктах реакции им удалось идентифицировать OClF3 наряду с ClF5, фтором и кислородом. Через два года вышла серия статей сотрудников американской фирмы «Рокетдайн» (корпорация «Рокуэл», Калифорния), из которых стало известно, что окситрифторид хлора был получен ими еще в 1965 году, но результаты не публиковались из-за секретного характера работы. Независимо от этих двух групп ClOF3 был получен в нашей стране С.М. Синельниковым в 1970 году. Американские исследователи применили для получения ClOF3 реакцию фторирования ковалентных гипохлоритов, т.е. соединений с концевой группой –О-Сl, например, Сl2O и ClONO2. Синельников получал ClOF3 в тлеющем разряде из других оксифторидов хлора или прямо из элементов в присутствии BF3.
Окситрифториду хлора свойственно переходить в ионное состояние: при растворении в жидком фтористом водороде даже в отсутствие кислот Льюиса он образует, ион ClOF2+. Соли этого катиона получены с большим числом полифторанионов – BF4, AsF6, SbF6, PF6, UF6, BiF6, VF6, NbF6, TaF6, SiF62-.
Оксифторид трехвалентного хлора должен иметь состав OClF. Несмотря на многочисленные попытки соединения с такой формулой долгое время выделить не удавалось. Лишь в 1972 году было доказано, что молекулы OClF образуются в качестве нестабильного промежуточного продукта при реакции ClF3 с водой в проточном газовом реакторе. Время полураспада OClF при комнатной температуре составляет всего 25 с. В конденсированном состоянии OClF не существует. Во всех случаях, когда OClF образуется как промежуточный продукт, он быстро диспропорционирует на FClO2 и ClF. К длительному существованию он способен лишь в матрице, из твердого аргона при температуре ниже 15 K. Для матричной изоляции OClF подвергают фотолизу смесь ClF и O3 в твердом аргоне. Молекула оксимонофторида хлора изогнутая с углом О-Cl-F около 120°. Все попытки получить соли типа ClO+ЭF6 окончились неудачей. Использованная ранее форма вывода для фторида и оксифторида трехвалентного хлора будет выглядеть следующим образом:
OClF ClF3 [СlO+] ClF2+
Если теперь, основываясь на том, что мы узнали о синтезе солей фторкатионов, поставить вопрос, как будет реагировать с кислотами Льюиса монофторид хлора, то ответ на него должен был бы выглядеть примерно так: либо никакого взаимодействия не будет, либо образуется соль Cl+ЭF6. В действительности ни того, ни другого не происходит. Природа выбрала более сложный путь.
Первые исследователи реакции ClF с BF3 и AsF5 заметили образование кристаллических соединений и приписали им структуру Cl+BF4 и Cl+AsF6. Ошибка их была вскоре обнаружена. Состояла она в неправильном определении состава образующихся твердых соединений. До сих пор не получено ни одной соли, катионом в которой был бы атом легкого элемента - неметалла. Все известные катионы, построенные из атомов этой группы элементов, многоатомны, минимум двухатомны, как О2+. Не является исключением и катион, образующийся при реакции монофторида хлора с кислотами Льюиса. Низкотемпературное взаимодействие ClF с AsF5 и BF3 ведет к образованию кристаллических соединений состава 2:1, 2ClF·BF3 и 2ClF·AsF6, независимо от того, какой из двух реагентов берется в избытке. Анионы в этих аддуктах обычные - BF4 или AsF6, а катион имеет состав Cl2F+ и структуру
![]()
Исходный монофторид хлора не димеризован, так что связь Cl-Сl возникает в ходе реакции. Можно представить, что присоединение атома фтора к кислоте Льюиса и оттягивание электронной плотности от атома Сl к F в молекуле ClF создает благоприятные условия для сольватации Сl+ другой молекулой ClF:
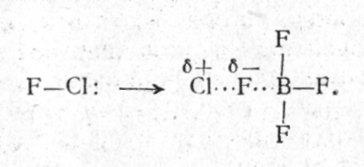
Стрелкой
обозначена координационная связь. В
дальнейшем оказалось, что сольватация
может происходить не только молекулой
ClF.
Если на эквимолярную смесь Сl2
и
ClF
подействовать пентафторидом мышьяка,
образуется соль с катионом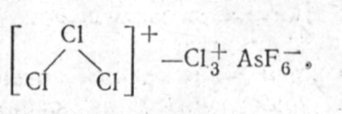
С таким явлением, когда многоатомный катион «монтируется» в ходе реакции из двух разных молекул, мы уже сталкивались во фторидах азота: HNF2 + HF + SbF6 = H2NF2+SbF6. Аналогично из двух молекул HF и молекулы SbF5 образуется соль H2F+SbF6, а из раствора, содержащего HF, Н2O и SbF5, кристаллизуется H3O+SbF6. Можно привести еще примеры образования более сложного гетерокатиона путем сольватации катиона малого размера нейтральной молекулой. В системе XeF2 - SbF6 наряду с соединением XeF+SbF6 кристаллизуется [XeF·XeF2]+SbF6, записываемое обычно как Xe2F+SbF6. Точно так же существуют катионы KrF+ и Kr2F3+. Одна молекула может вытеснять другую из комплексного катиона. Так, молекула OSF2 вытесняет ClF из Cl2F+AsF6, давая гексафтороарсенат с таким катионом
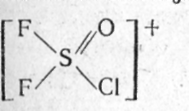
Скорее всего именно такой путь, когда катион возникает в результате объединения двух или более молекул под влиянием отрыва от одной из них фторид-иона, будет одним из основных методов синтеза новых гетерокатионов в ближайшие годы.
ЭНЕРГОЕМКИЕ АНИОНЫ
Практический интерес к молекулярным катионам, построенным из атомов кислорода, фтора, азота и хлора в различных сочетаниях, объясняется низкой энергией связи между образующими их атомами. Синтезировано уже довольно много катионов, которые с точки зрения запасенной в них энергии вполне подходят как структурные единицы твердых энергоемких соединений. К сожалению, вторая неизбежная составная часть любого ионного кристалла - анион - почти во всех рассмотренных случаях был просто энергетическим балластом. Все преимущества энергетически активного фтора, с таким трудом достигнутые в ходе синтеза новых фторкатионов, оказываются обесцененными плохой энергетикой анионов.
Идеальным твердым энергоемким соединением был бы ионный кристалл, построенный из катионов типа NF4+, ONF2+, ClF6+ и т.п. и анионов, состоящих из тех же легких атомов, соединенных такими же слабыми связями. Подходящими анионами могли бы быть СlO4 и NO3. Однако они не «уживаются» в одной кристаллической решетке с подавляющим большинством фторкатионов. Реакционная способность катионов NF4+, N2F+, ClF2+ и т.д. столь высока, что при контакте, например, с анионом СlO4 происходит его фторирование с выделением ClO3F. Так, при попытке получить N2F+СlO4 уже при низкой температуре полностью проходит такая реакция:
![]()
В аналогичных условиях из нитрат-иона под действием фторсодержащего катиона образуется NO2F.
Так что анионы кислородных кислот для создания твердых фторсодержащих окислителей не подходят. Для этой цели необходимы полифторанионы. Однако обычные полифторанионы - SbF6, BF4, PF6 - в данном случае также не подходят. Фтор, входящий в их состав, энергетически пассивен. Энергия связи Э-F в анионах ЭFn почти так же высока, как и в продуктах сгорания. Например, средняя термохимическая энергия связи в молекуле BF3 153 ккал/моль, в анионе BF4 - 138 ккал/моль, в молекуле PF5 - 109 ккал/моль. Возникает вопрос: нельзя ли синтезировать полифторанионы с высоким запасом химической энергии? Нельзя ли, например, вместо центральных атомов бора, фосфора или мышьяка использовать атомы хлора, азота или кислорода? Тогда энергия связи в анионах была бы того же порядка, что и в соответствующих фтор-катионах или ковалентных фторидах. Попытки синтезировать энергоемкие полифторанионы делались неоднократно, но достигнутые результаты пока очень скромны.
В шестидесятых годах вполне реальным казалось получение солей с анионом NF2. Действительно, радикал NF2 во многих соединениях ведет себя подобно атому галогена. Это хорошо видно, если сгруппировать некоторые производные NF2 с соответствующими производными хлора и фтора:
F-F Н-F CF3-F F-F
F-NF2 Н-Cl CF3-Cl Сl-Сl
Cl-F Н-NF2 CF3-NF2 F2N-NF4
Cl-NF2
Галогенам и псевдогалогенам свойственно образовывать соли М+Х. Как будто нет явных причин, препятствующих радикалу NF2 присоединить электрон и войти в качестве анионной составляющей в ионную решетку M+NF2+. Радикал NF2 имеет ненулевое электронное сродство. Оно невелико, того же порядка, что и у молекулы O2, около 0,5 эВ. Молекуле кислорода такого сродства к электрону вполне достаточно, чтобы образовывать довольно стабильные надперекиси Cs+O2, Rb+O2, K+O2, Na+O2. Но ни одной соли с анионом NF2 получить не удалось. Более того, до настоящего времени не получено вообще ни одного аниона со связью N-F. Сообщалось об идентификации анион-радикала ONF3 в низкотемпературной матрице, но как структурные элементы какой-либо кристаллической решетки анионы со связью азот - фтор неизвестны. Трудно сказать, существует ли общая причина этого явления, и, следовательно, попытки синтезировать такие анионы безнадежны или в каждом отдельном случае причины неустойчивости различны и какие-то анионные формы со связью N-F существовать могут. Во всяком случае если у атома азота в каком-либо его соединении и есть способность быть акцептором фторид-иона, то до сих пор эту способность реализовать не удалось.
Не лучше обстоит дело и в группе О-F-соединений. Единственный стабильный фторид кислорода OF2 в отличие от своего хлорного аналога при реакции с водой кислоты, ионизованной в растворе, не образует. До недавнего времени соединения с формулой HOF ни в свободном состоянии, ни в виде солей известно не было. Лишь в 1971 году HOF была получена в виде неустойчивого газообразного вещества пропусканием фтора через реактор, заполненный тефлоновыми кольцами Рашига с нанесенной на них влагой. Давление паров HOF достигает 5 мм при -64°С, температура плавления -117°С. Хотя протон в HOF обладает кислыми свойствами, что видно, например, из наблюдавшегося быстрого обмена протона на дейтерий в присутствии D2О, никаких соединений с анионом OF не получено. Довольно многочисленная группа FO-производных, которые условно можно назвать гипофторитами, FONО2, FOClО3, FOCF3, FOSF5 и т.п., являются летучими ковалентными веществами.
Для того чтобы молекула какого-либо соединения, в нашем случае фторида, окисла или оксифторида азота, кислорода или хлора, превратилась в фторсодержащий анион, надо, чтобы реакция
M+F + ЭFn-1Om = M+[ЭFnOm]-
сопровождалась понижением свободной энергии Гиббса, т.е. надо, чтобы молекула имела сродство к фторид-иону. Путь получения фторанионов, выраженный приведенным уравнением, не единственный, но наиболее распространенный, Как мы видели, ни фториды, ни оксифториды, ни окислы азота не способны к присоединению фторид-иона. То же можно сказать и о фторидах кислорода. Из трех выбранных нами элементов только хлор в некоторых соединениях способен образовывать анионные фторкомплексы, т.е. быть акцептором фторид-иона.
Два из трех бинарных фторидов хлора, ClF и ClF3, образуют полифторанионы ClF2 и ClF4. Получить их можно совмещением фторида хлора с фторидом щелочного металла. Реакция протекает с трудом из-за того, что поверхность фторида металла покрывается твердым фторо-хлоратом, препятствующим контакту между реагентами. Приходится нагревать и перемешивать реакционную массу под давлением, брать большой избыток фторида хлора, но все равно добиться полного превращения MF в MClFn очень трудно. К тому же далеко не со всеми фторидами эта реакция возможна. Монофторид хлора присоединяется с образованием дифторохлоратов только к фторидам цезия, рубидия и калия. Ни с LiF, ни с NaF, ни тем более с фторидами двухвалентных металлов реакция не идет. Точно так же ведет себя трифторид хлора - получены только три тетрафторохлората металла: CsClF4, RbClF4 и KClF4. Имеется патент (1965 год) на способ получения гексафторо-хлоратов цезия, рубидия и калия из ClF5 и MF, однако более поздние исследования Кристи не подтвердили способности пентафторида хлора акцептировать фторид-ион.
Некоторые оксифториды хлора также могут присоединять ион F образуя с фторидами щелочных металлов кристаллические соединения. Как и с фторидами хлора, реакция очень капризна, требует длительного времени, высокого давления оксифторида и специально подготовленного фторида щелочного металла. Например, чтобы фторид цезия был достаточно активным для реакции с ClO2F, его получают термическим разложением комплекса CsF·OC(CF3)2. В другой работе хорошо очищенный CsF проплавляли в платиновом тигле и потом тщательно измельчали.
Окситрифторид хлора образует комплексы с фторидами цезия, рубидия и калия. Для завершения реакции требуется выдерживание MF в избытке ClOF3 в течение месяца при комнатной температуре. Все три аддукта построены ионно: M+ClF4O. Структура аниона ClOF4 показана на рис. 2. Хлорилфторид также способен присоединяться к фториду цезия с образованием Cs+ClO2F2. С фторидами других металлов реакция не исследовалась. Оксифториды семивалентного хлора аддуктов с ионными фторидами не образуют. Все соли с фторохлоратными анионами очень гигроскопичны и бурно, иногда со взрывом, реагируют с водой. Термическая стабильность M+ClF4O и M+ClO2F2 невысока, М+ClF4 более устойчив.
Структура фторохлоратных анионов хорошо подчиняется модели локализованных электронных пар. Не участвующие в связи неподеленные электронные пары занимают места в координационном многограннике атома хлора. В ионе ClF2 в плоскости, перпендикулярной к линии F—Cl—F, расположены три неподеленные пары, так что в целом пять электронных пар - две, образующие связи с атомами фтора, и три неподеленные - направлены к вершинам тригональной бипирамиды. Две неподеленные пары электронов в ClF4 расположены над и под плоскостью иона, дополняя окружение хлора до октаэдрического. Положение неподеленных пар в ионах ClO2F2 и ClF4O показано на рис. 2-й в комментариях не нуждается. Тем же закономерностям подчиняется структура фторкатионов, фторидов и оксифторидов хлора и азота.
Какую же пользу принес синтез четырех фторанионов хлора для создания твердых энергоемких соединений? Возможно ли совмещение в одной кристаллической решетке этих энергетически активных анионов с фтор-катионами азота и хлора? На этот вопрос в подавляющем большинстве случаев приходится ответить отрицательно. Если бы, например, катион ClF4+ давал устойчивую ионную решетку с анионами ClF4+, то трифторид хлора был бы в обычных условиях не летучей жидкостью, а кристаллическим веществом. В действительности равновесие самоионизации в жидком трифториде хлора практически полностью сдвинуто в сторону ковалентной формы:
2ClF3 ClF2+ + ClF4
Электропроводность жидкого ClF3 имеет ничтожную величину ~10-9 Ом-1·см-1. Подобным же образом обстоит дело и в случае ClF, ClO2F и ClOF3. Нет надежды построить ионный кристалл из фторазотных катионов и фторхлорных анионов. Причина здесь достаточно простая. Будучи очень слабыми основаниями, фториды азота и фториды хлора (имеются в виду соединения со связью N-F и Cl-F) образуют устойчивые ионные соли только с сильнейшими кислотами Льюиса. С другой стороны, фториды и оксифториды хлора - очень слабые кислоты Льюиса и дают более или менее стабильные соли только с самыми сильными основаниями Льюиса - CsF, RbF, KF. Попытка создать устойчивую соль из катиона слабого основания и аниона слабой кислоты к успеху обычно не ведет.
Из всех известных фторидов и оксифторидов азота наибольшей склонностью отдавать фторид-ион обладает фтористый нитрозил. Катион NO+ образует устойчивые соли не только с большинством полифторанионов, но и с многими кислородными кислотами. Некоторый хотя и незначительный успех достигнут и в синтезе солей NO+ с фторохлоратными анионами. Незначителен он потому, что стабильность полученных солей крайне низка. Они существуют лишь при температуре ниже 0°С. Фтористый нитрозил реагирует только с ClF и ClF3, с пентафторидом и оксифторидами хлора взаимодействия не обнаруживается. В отличие от щелочных фторидов NOF соединяется с ClF и ClF3 просто при совместной конденсации при достаточно низкой температуре. Обе соли распадаются по равновесным реакциям:
NO+ClF2 (КРИСТ) ⇄ NOF(ГA3) + ClF(ГАЗ); РДИС = 1 атм, при -22°С;
NO+ClF4 (КРИСТ) ⇄ NOF(ГАЗ) + ClF3(ГАЗ); РДИС = 1 атм, при -5°С.
Энтальпия присоединения NOF к фториду хлора в обоих случаях составляет около 15 ккал/моль. В аналогичных условиях фтористый нитрил NО2F ни с одним фторидом хлора аддуктов не дает.
Дифторохлорат и тетрафторохлорат нитрозила, так же, как и перхлорат монофтораммония, можно рассматривать как модель или прообраз твердых энергоемких фторсодержащих соединений. Однако свойства этих веществ, особенно термическая стабильность, пока еще сильно отличаются от того, что требует техника. Предстоит еще длительный синтетический поиск, пока удастся решить одну из труднейших проблем тонкого неорганического синтеза - создать стабильные энергоемкие фторанионы.
ФТОРКАТИОНЫ УГЛЕРОДА, ФОСФОРА, СЕРЫ И БРОМА
Картина достижений тонкого неорганического синтеза в рассматриваемой области будет неполной, если хотя бы кратко не упомянуть о фторкатионах, содержащих серу, фосфор, бром и некоторые другие неметаллы. Интересно также выяснить, какие еще фторсодержащие катионы образуют или могут образовать неметаллические элементы первого восьмичленного периода.
Известны ли фторкатионы углерода? Ни один из простейших фторуглеродов и фторуглеводородов даже с сильнейшими акцепторами фторид-иона ионных соединений не образует. Неоднократно изучалось поведение фтористого метила в сверхкислых средах - растворах SbF5 в SO2 или в смеси SO2 и HSO3F. Явных указаний на присутствие ионов СН3+ в таких растворах не найдено. Не образуется катионов и при действии кислот Льюиса на оксифтор углерода - фторофосген. Лишь в аргоновой матрице при температуре ниже 15 K в результате фотоионизации молекул CF3X, X = Cl, Br, I, Н, в инфракрасном спектре зафиксированы ионы, отнесенные к CF3+.
Несколько солей фторкатиона углерода совершенно неожиданного состава было получено совсем недавно, в 1974 году. Оказалось, что под действием окислителя в присутствии сильной кислоты Льюиса молекула гексафторбензола отдает один электрон, превращаясь в катион C6F6+. В качестве окислителя для этого синтеза использовали диоксигенильную соль O2+AsF6. Реакция между C6F6 и O2AsF6 протекает в среде жидкого гексафторида вольфрама около 0°С и сопровождается выделением кислорода и образованием светло-желтой соли C6F6+AsF6, плавящейся при 69°С с разложением. Более стабильна C6F6+SbF6. По способу образования катион C6F6+ подобен диоксигенильному: оба получают окислением исходной нейтральной молекулы того же состава, что и катион. Казалось бы, проще всего получить соль C6F6+ действием PtF6 на гексафторбензол, т.е. так же, как в свое время впервые был получен O2+PtF6. Именно так и попытался первоначально получить его один из сотрудников Бартлета, однако гексафторид платины не окислил, а профторировал C6F6, катиона при этом не образовалось.
Недавно появилось сообщение о получении соли C8+AsF6 действием O2+AsF6 на графит. Там же говорится, что при обработке графита перекисным соединением FO2SOOSO2F образуется соль C12+SO3F, а в аналогичных условиях из нитрида бора и S2O6F2 - соль (BN)4+SO3F.
Среди рассмотренных катионов встречались молекулярные структуры со связями N-F, О-F, С-F, но не было ни одного со связью О-F. При действии фторкислот Льюиса на диоксиполифториды O2F2, O4F2 и вообще OnF2n1 образуется катион O2+, не содержащий фтора, a OF2 с кислотами Льюиса либо не реагирует, либо в более жестких условиях дает тот же O2+. Если три хорошо известных иона расположить в таком порядке:
NH4+ OН3+
NF4+ ?
то на месте вопросительного знака следовало бы записать катион OF3+. Возможность его синтеза не представляется безнадежной. Экспериментальные попытки получить соли с таким катионом делались, но к успеху не привели.
По-видимому, у многих фторазотных катионов могут существовать хлорные аналоги. В 1977 году получен первый из них – ONCl2+. Синтез его более сложен, чем ONF2+ поскольку молекулы ONCl2F или ONCl3 неизвестны. Для синтеза был взят трихлорид азота и хлористый тионил в качестве донора атома кислорода:
NCl3 + SOCl2 + SbCl5 –(CCl4) ONCl2+SbCl6 + SCl2.
Соль устойчива до 145°С, при вакуумном пиролизе выделяет хлор и переходит в NO+SbCl6.
Гетерокатионов фосфора известно очень немного, гораздо меньше, чем гетерокатионов азота. Вообще сходства в химии этой группы соединений азота и фосфора почти нет. Для фосфора характерен катион РСl4+, твердый пентахлорид фосфора построен ионно PCl4+PCl6, у азота аналогичного катиона нет. Окситрифторид фосфора, так же, как и окситрифторид азота, образует твердые соединения с BF3, AsF5 и SbF6, однако структура их в отличие от соответствующих производных ONF3 не ионная. Они образованы за счет донорно-акцепторных связей через атом кислорода: F3PO:AsF5. Не известны фосфорные аналоги катионов N2F+, N2F3+, NH2F2+ NH3F+, NO+, NO2+. Лишь ион тетрафтораммония имеет фосфорный аналог. Несмотря на то что синтез солей PF4+ и в техническом, и в теоретическом отношении проще, чем солей NF4+, первая соль тетрафторфосфония была получена на семь лет позже, чем соль NF4+. Комплекс PF4+Sb3F16 образуется просто при совмещении пентафторида фосфора с избытком SbF6. Комплекс очень неустойчив, при комнатной температуре давление его диссоциации достигает 130 мм.
Как и NF4+, ион PF4+ тетраэдрический. Известны также метил- и этилзамещенные тетрафторфосфония: (CH3)3PF+(CH3)2PF2+, CH3PF3+, (C6H5)3PF+. Соли их более стабильны, чем PF4+.
По обилию, разнообразию состава и сложности структуры гетеро- и гомополиатомных катионов сера превосходит все остальные неметаллы. Начнем с ее простейших фторкатионов. Тетрафторид и окситетрафторид серы образуют соли с катионами SF3+ и OSF3+ при действии AsF5, SbF5 и других кислот Льюиса. Ион SF3+ пирамидальный с углом F-S-F около 97°, ион OSF3+ псевдотетраэдрический с углами F-S-F 102° и О-S-F 115°. Известно несколько производных иона SF3+, в которых часть или все атомы фтора замещены на другие атомы или радикалы: CF3SF2+, F2SNCO+, Cl2SNCO+, CH3SCl4+, (CH3)2SCl+, SBr3+. Получено несколько солей хлорзамещенного OSF3+: OSClF2+PF6, OSClF2+AsF6 и OSClF2+SbF6, а также более сложные производные:
![]() ,
HCH3NSOF4+,
OCNSOF2+.
,
HCH3NSOF4+,
OCNSOF2+.
При действии пентафторида сурьмы на раствор CH3F в жидкой двуокиси серы можно выделить кристаллическое вещество состава CH3F·SO2·2SbF5. Оно построено из анионов Sb2F11 и катионов CH3OSO+, имеющих плоскую цис-структуру:

Первый член ряда сера-азотных катионов, двухатомный NS+, образуется при реакции фторкислот Льюиса с циклотиазилфторидом S4N4F4 или прямо с NSF. Весьма интересен более сложный сера-азотный катион S3N2+. Он имеет один неспаренный электрон, парамагнитен, ярко окрашен. Это один из немногих неорганических радикал-катионов, устойчивых в кристаллической решетке. Соли его не могут быть получены простым отщеплением фторид-иона, поскольку не существует исходного фторида N2S3F. Единственная известная соль этого катиона, красно-коричневый S3N2+AsF6, получена действием избытка AsF5 на раствор S4N4 в жидкой SO2. В ходе реакции происходит окислительное фторирование и деструкция молекулы S4N4. Катион S3N2+ имеет плоское циклическое строение:
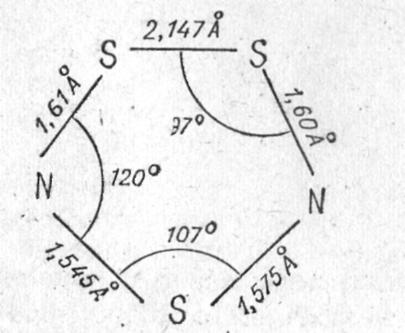
Если подействовать хлоросульфоновой кислотой на S3N2Cl, образуется соль с катионом S3N42+. Он построен из двух плоских колец S3N2, лежащих в параллельных плоскостях и слабо связанных черездва атома серы.
Известен также бициклический катион S4N52+, моноциклические: S4N3+ и S3N22+.
Недавно открыто несколько циклических двухзарядных гомополиатомных катионов серы, таких, как S42+, S82+, S162+. При действии элементарного йода на S162+(SbF6)2 получена соль с катионом S7I+. В тех же условиях из S162+(AsF6)2 получен S8I+.
Фторкатионы брома по составу и структуре подобны фторкатионам хлора. Вот их формулы: Br3+, BrF2+, BrF4+, BrF6+, BrOF2+, BrO2+; выделены также соли типа Br2+ЭF6, аналогов которых нет у хлора. Делались попытки получить соль с катионом BrО2F2+, но вместо него образовывался только BrOF2+. Не получен и соответствующий оксифторид BrО2F3. Трудно сказать, связано ли это с невозможностью их существования или с неудачно подобранными методами синтеза. Предсказывать что-либо в химии бромкислородных соединений рискованно. Вспомним историю перброматов.
Перхлораты - наиболее стабильные и наиболее распространенные соли хлоркислородных кислот. Их естественные аналоги в химии брома - перброматы - долгое время синтезировать не удавалось. Экспериментаторы перепробовали множество способов, использовали самые сильные из имевшихся в их распоряжении окислителей - перекиси и надперекиси металлов, висмутат натрия, персульфаты, хлор, фтор, озон - и все безрезультатно. Проблема перброматов рассматривалась теоретически, и было предложено несколько объяснений причин, препятствующих их существованию. По существу, была предсказана невозможность их синтеза. И вот когда химики стали уже привыкать к мысли, что перброматов существовать не должно, Аппельман в США получил пербромат натрия окислением бромата дифторидом ксенона в водном растворе при комнатной температуре. Работа опубликована в 1968 году. В последующие годы были получены и довольно подробно исследованы перброматы всех щелочных металлов, аммония и некоторые другие. Сейчас они вполне доступны. Кажется удивительным, что окислить бромат до пербромата удалось не только водным раствором XeF2, но и фтором и озоном, т.е. теми самыми окислителями, которыми раньше, до первого синтеза МВrO4, этого сделать не удавалось. Впрочем, удивляться тут нечему. Это обычное явление в синтетической химии. Когда экспериментатор заранее знает, что целевое вещество существует, и знает, как оно выглядит (точнее, как выглядят его спектры), вероятность успеха в синтезе возрастает во много раз.