

Глава 7
ПРОЦЕССЫ ЭКСТРАКЦИИ В РАДИОХИМИИ
В радиохимии широко используются экстракционные процессы. Особенно велика их роль в развитии и совершенствовании ядерной технологии.
Достоинствами экстракционных методов являются: 1) универсальность, высокая избирательность и полнота выделения; 2) возможность получения препаратов радиоактивный нуклидов без носителя; 3) большая скорость процесса и простота аппаратурного оформления, позволяющая реализовать эти процессы в технологических схемах с дистанционным управлением.
§ 1. Закономерности и классификация экстракционных процессов
Экстракционные процессы основаны на распределении веществ между двумя несмешивающимися растворителями с последующим разделением фаз*.
Процесс равновесного распределения вещества между двумя несмешивающимися растворителями описывается законом Бертло — Нернста, который является частным случаем закона распределения вещества между двумя фазами (см. гл.5).
Закон Бертло — Нернста может быть сформулирован следующим образом:
при распределении вещества между двумя несмешивающимися жидкими фазами при постоянных давлении и температуре отношение равновесных термодинамических активностей распределяющегося вещества в этих фазах есть величина постоянная, независящая от абсолютного количества вещества и объемов фаз.
Таким образом:
|
(7.1) |
где а и а — термодинамические активности распределяющегося вещества в органической и водной фазах соответственно; РT — термодинамическая константа распределения (константа Бертло — Нернста).
Этот закон применим при наличии термодинамического равновесия, постоянстве состава фаз и одинаковой химической форме распределяющегося вещества в обеих фазах.
Закономерности распределения вещества между двумя фазами не зависят от его концентрации. Однако процесс экстракции веществ, находящихся в растворах в микроконцентрациях (короткоживущих радиоактивных элементов, радиоактивных нуклидов без носителя), имеет ряд особенностей. Прежде всего для таких систем состав органической и водной фаз сохраняется неизменным при изменении концентрации распределяющегося нуклида. Это обусловливает постоянство коэффицентов активности распределяющегося вещества, и уравнение (7.1) может быть записано следующим образом:
~P = m/m, |
(7.2) |
где m и m — концентрации распределяющегося вещества в органической и водной фазах.
В тех случаях, когда распределяющееся вещество находится в органической фазе в виде ассоциатов со степенью ассоциации n или диссоциировано в водной фазе на ионов, уравнение (7.2) принимает соответственно вид:
~P = mnm |
(7.3) |
или
~P = m/m. |
(7.4) |
Следует отметить трудность экспериментального определения термодинамической константы распределения из-за сложности установления химической формы распределяющегося вещества, его концентрации и коэффициентов активности.
Поэтому количественно экстракция обычно характеризуется коэффициентом распределения , равным отношению общих аналитических концентраций распределяющегося элемента независимо от его химической формы:
= mЭ/mЭ. |
(7.5) |
Так, распределение иода в системе водный раствор иодида калия — тетрахлорид углерода ( в водной фазе иод находится в двух химических формах I2 и I-3; экстрагируемая его форма — I2) можно охарактеризовать как термодинамической константой распределения, так и коэффициентом распределения:
 ,
,
где — константа устойчивости иона I-3.
Экспериментальные данные обычно представляют в виде зависимостей m = f(m) или = f(m), носящих название изотерм экстракции.
При экстракционном извлечении вещества из многокомпонентных растворов органическими фазами сложного состава (экстрагент + разбавитель + модификатор**) равновесная концентрация вещества в органической фазе является функцией многих переменных:
m = f (m1, m2, ..., mi, m, mS, mM), где m1, m2, ... , mi;—концентрации неэкстрагируемых компонентов в водной фазе; mS, mM — концентрации экстрагента и модификатора в органической фазе.
Сложность классификации процессов экстракции заключается в большом разнообразии экстракционных систем и отсутствии общей теории этих процессов. Принципы классификации экстракционных процессов могут быть различными. Ниже рассмотрена классификация по типу экстрагента, в основе которой лежит механизм взаимодействия экстрагента с распределяющимся веществом. С этой точки зрения все экстракционные системы можно разделить на две группы. В первой (немногочисленной) группе экстракционных систем имеет место физическое распределение вещества между двумя фазами. В этом наиболее простом случае экстракции взаимодействие распределяющегося вещества с экстрагентом осуществляется за счет сил Ван-дер-Ваальса (или слабого донорно-акцепторного взаимодействия). В общем виде процесс экстракции вещества МА по механизму физического распределения может быть представлен схемой:
MA MA.
Уравнение (7.1) для экстракционных систем этой группы имеет вид:
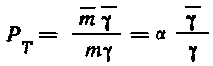 ,
где
и
— коэффициенты активности распределяющегося
вещества в органической и водной фазах.
,
где
и
— коэффициенты активности распределяющегося
вещества в органической и водной фазах.
Такой механизм процесса экстракции наиболее характерен для нейтральных неполярных органических растворителей, например бензола, октана, тетрахлорида углерода и др. По этому механизму экстрагируются слабо гидратированные, практически недиссоциирующие в водной фазе соединения, например вещества в свободном состоянии, оксиды, ковалентные галогениды, слабые органические и неорганические кислоты или основания. Механизм физического распределения лежит в основе извлечения таких важных продуктов деления, как иод (в форме молекулярного иода), рутений и осмий (в форме тетраоксидов) и др.
Большинство известных экстракционных систем относится ко второй группе, для которой характерно химическое взаимодействие экстрагируемого соединения с экстрагентом. Для этих систем процесс экстракции может быть представлен уравнением химической реакции:
![]() ,
где
S — экстрагент; q — сольватное число
(число молекул экстрагента, входящих в
состав экстрагируемого соединения).
,
где
S — экстрагент; q — сольватное число
(число молекул экстрагента, входящих в
состав экстрагируемого соединения).
В этом случае процесс экстракции как химическая реакция, подчиняющаяся закону действующих масс, может быть охарактеризован константой равновесия, называемой константой экстракции. Следует различать термодинамическую константу экстракции (КT), рассчитываемую с учетом коэффициентов активности компонентов в обеих фазах, концентрационную константу экстракции (К) и эффективную константу экстракции (К), отнесенную к водной фазе определенного состава (ионная сила раствора I = const). Термодинамическая константа экстракции рассматриваемого процесса может быть представлена следующим выражением:
|
(7.6) |
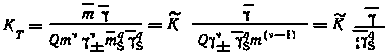 ,
,где m и m — концентрации распределяющегося вещества соответственно в органической и водной фазах; = + + - — общее число ионов, на которые распадается молекула МеА; Q = ++ - -— так называемый фактор валентности; mS — концентрация экстрагента в органической фазе (mS = m0 — qm, где m0 — исходная концентрация экстрагента, q—сольватное число); ±, , s —соответственно коэффициенты активности распределяющегося вещества в водной и органической фазах и экстрагента в органической фазе.
Коэффициент распределения для этого случая экстракции имеет вид:
|
(7.7) |
Из соотношений (7.6) и (7.7) имеем
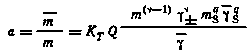 ,
где
KT — термодинамическая константа
экстракции.
,
где
KT — термодинамическая константа
экстракции.
Знание константы экстракции в сочетании с уравнением математической модели
= f(KT, m1, m2, ..., mi, mS) позволяет определять стехиометрию экстракции и получать информацию о процессах комплексообразования, ассоциации и других явлениях, происходящих в системе***.
Рассматриваемая група экстракционных систем, для которой характерно химическое взаимодействие, может быть подразделена по типу экстрагента на три подгруппы: экстракцию нейтральными, кислыми и основными экстрагентами.
В качестве нейтральных экстрагентов обычно используются кислородсодержащие реагенты (простые и сложные эфиры, спирты, фосфорорганические соединения), обладающие высокой сольватирующей способностью по отношению к неорганическим веществам. Механизм процесса экстракции нейтральными экстрагентами зависит от соотношения между изобарно-изотермическим потенциалом гидратации извлекаемого вещества (Gr) и взаимодействия его с молекулами экстрагента (Gвз). При условии, что Gr < Gвз, реализуется координационный (сольватационный) механизм. При экстракции по этому механизму имеет место образование координационной связи непосредственно между функциональной группой экстрагента и ионом экстрагируемого металла (или протоном кислоты). Образующиеся при этом соединения (аддукты) называются сольватами.
Примером экстракции по сольватному механизму является экстракция нитратов U(VI), Th(IV), трансурановых и редкоземельных элементов нейтральными фосфорорганическими соединениями.
Наиболее хорошо изучена экстракция уранилнитрата трибутилфосфатом, которая может быть представлена уравнением
![]() ,
где
ТБФ — трибутилфосфат (C4H9O)3PO.
,
где
ТБФ — трибутилфосфат (C4H9O)3PO.
Гидратно-сольватный механизм экстракции нейтральными реагентами имеет место, если Gr > Gвз. В общем виде процесс может быть представлен схемой:
![]() ,
где
h, q — соответственно гидратное и
сольватное числа.
,
где
h, q — соответственно гидратное и
сольватное числа.
Для экстракции по гидратно-сольватному механизму характерна преимущественная гидратация катионной части образующегося гидратосольвата и присоединение экстрагента через координационно связанную воду.
Примером процессов экстракции по гидратно-сольватному механизму может служить экстракция спиртами актиноидных элементов, полония и др.
В качестве кислых
экстрагентов используются органические
кислоты, которые можно разделить на две
группы. К первой относятся органические
кислоты, образующие с экстрагируемыми
катионами соединения с ионной связью,
не осложненной дополнительным
взаимодействием (карбоновые, нафтеновые
кислоты, фенолы и, с некоторой оговоркой,
фосфорорганические кислоты). Ко второй
группе относятся двухосновные органические
кислоты, которые содержат кроме кислотной
функциональной группы
![]() еще
одну функциональную группу
еще
одну функциональную группу
![]() ,
образующую координационную связь с
катионом экстрагируемого металла с
образованием внутримолекулярных
соединений (хелатов). Примером экстрагентов
этого класса являются 8-оксихинолин,
ацетилацетон, теноилтрифторацетон и
др. В общем виде экстракция металлов
органическими кислотами состава HR может
быть представлена как реакция присоединения
или реакция катионного обмена:
,
образующую координационную связь с
катионом экстрагируемого металла с
образованием внутримолекулярных
соединений (хелатов). Примером экстрагентов
этого класса являются 8-оксихинолин,
ацетилацетон, теноилтрифторацетон и
др. В общем виде экстракция металлов
органическими кислотами состава HR может
быть представлена как реакция присоединения
или реакция катионного обмена:
 .
.
По этому механизму экстрагируются, например, актиноидные элементы алкилфосфорными кислотами и такими реагентами, как теноилтрифторацетон, 8-оксихинолин и др.
К подгруппе основных экстрагентов относятся органические основания различного строения общей формулы RnЭ и их соли RnЭА, где R — органические радикалы или атомы водорода, Э — азот, фосфор, мышьяк, сера и т. д. Из экстрагентов этой группы лучше других изучены амины и соли четвертичных аммониевых оснований.
В основе процессов экстракции указанными экстрагентами лежит их взаимодействие с кислотами и солями металлов по схеме:
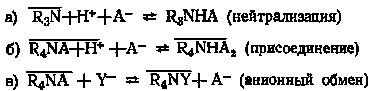 ,
где
A- Y- — экстрагируемый анион
кислоты или ацидокомплекса состава
[MeAz+q]q-.
,
где
A- Y- — экстрагируемый анион
кислоты или ацидокомплекса состава
[MeAz+q]q-.
Принимая во внимание, что прочность ацидокомплексов в водной фазе велика, процесс экстракции этих ионов чаще представляют в виде реакций присоединения.
Примером экстракции основными экстрагентами может служить процесс извлечения шестивалентного урана из сернокислых сред раствором третичного амина в малополярном растворителе, что можно представить следующими уравнениями:
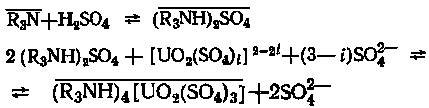 .
.
По этому же механизму трансурановые элементы в степени окисления 3+ экстрагируются аминами из нитратных и сульфатных растворов.
Рассмотренная классификация процессов экстракции удобна для изучения изотерм экстракции. Следует отметить, однако, что она весьма условна. Так, органические кислоты могут вести себя как нейтральные экстрагенты при экстракции металлов из кислых растворов.
Для систем, содержащих распределяющееся вещество в микроконцентрациях, необходимо учитывать такие явления, как синергизм и способность к соэкстрагированию. Синергизм (синергетический эффект) имеет место при экстракции смесями экстрагентов и заключается в том, что коэффициент распределения при использовании смеси экстрагентов оказывается большим, чем его значение, рассчитанное по правилу аддитивности. Количественной оценкой этого эффекта служит синергетный коэффициент Sk****:
 ,
где
— коэффициент
распределения при экстракции смесью
экстрагентов; (1,
2 — коэффициенты
распределения при экстракции каждым
из экстрагентов в отдельности.
,
где
— коэффициент
распределения при экстракции смесью
экстрагентов; (1,
2 — коэффициенты
распределения при экстракции каждым
из экстрагентов в отдельности.
В основе синергетического эффекта лежит образование в органической фазе смешанных аддуктов (сольватов). Например, при экстракции урана (VI) смесью ТТА и ТБФ в органическую фазу переходит соединение состава UO2(TTA)2-TБФ.
Процессы соэкстракции сопровождаются образованием нового экстрагента, экстракционная способность которого выше, чем исходного. Примером процесса соэкстракции может служить экстракция катионов металлов растворами Д2ЭГФК при введении в водные растворы соединений циркония или гафния. В этом случае в органической фазе образуется соединение Zr(Hf)R42HR, причем степень диссоциации координированных молекул HR в этом комплексе увеличивается.
Экстракция является процессом массопередачи, происходящим в результате существования разности химических потенциалов распределяющегося вещества в органической и водной фазах. Скорость экстракции характеризуется изменением во времени концентраций распределяющегося вещества в органической или водной фазах (dc/dt). Как правило, процесс массопередачи при экстракции сопровождается химическими реакциями, которые могут протекать в объемах фаз, в слоях, непосредственно прилегающих к границе раздела, и на самой границе раздела фаз. При этом скорость .процесса экстракции определяется самой медленной стадией*****.
Различают три режима экстракции. В диффузионном режиме скорость экстракции зависит только от скорости массопередачи. Химическая реакция протекает очень быстро. В кинетическом режиме скорость экстракции определяется скоростью химических реакций. Смешанный или промежуточный режим экстракции имеет место, когда скорости массопередачи и химических реакций сравнимы.
Скорость процесса экстракции зависит от интенсивности перемешивания и величины поверхности раздела фаз, причем характер этих зависимостей определяется режимом процесса. Для одной и той же системы увеличение интенсивности перемешивания и поверхности раздела фаз может привести к изменению режима, например, к переходу из диффузионного режима в смешанный, а затем в кинетический.
Скорость процесса экстракции определяет необходимое время контакта фаз на стадии их смешения и разделения. В радиохимической технологии сокращение времени контакта особенно важно, так как позволяет не только увеличить производительность процесса, но и уменьшить единовременную загрузку радиоактивного материала и тем самым понизить радиационное воздействие его на экстрагент.
* Основы жидкостной экстракции /Под ред. Г. А. Ягодина. М., Химия, 1981. ** Модификаторами называют вещества, добавляемые в органическую фазу с целью предупреждения образования третьей фазы. *** Эта информация может быть получена на основании отклонения от распределения, предсказываемого законом действующих масс. **** При больших концентрациях распределяющего вещества, когда емкость экстрагента в значительной степени использована, имеет место антагонистический эффект (Sk<0). ***** См.: Фомин В.В. Кинетика экстракции. М., Атомиздат, 1978. § 2. Основы экспериментальных методов исследования
Практическое осуществление процессов экстракции определяется задачей исследования. Это может быть изучение закономерностей самого процесса экстракции радиоактивных элементов или использование экстракции как метода разделения радиоактивных элементов или радиоактивных нуклидов.
Исследование закономерностей экстракции радиоактивных элементов включает в себя определение состава экстрагируемых соединений, количественное описание экстракционного равновесия, в том числе определение константы экстракции, установление вида математической модели = f(K, m1, m2, mi, mS ) или корреляционных зависимостей константы экстракции от различных параметров, характеризующих строение экстрагента, природу разбавителя, природу катиона или аниона распределяющегося вещества.
Исследование кинетики процесса экстракции состоит в установлении режима процесса. Для этого исследуется зависимость скорости экстракции (dc/dt) от интенсивности перемешивания (1), величины поверхности раздела фаз (2), а также от концентраций компонентов системы (3). Ниже представлены зависимости скорости экстракции от этих факторов для различных режимов процесса.
Режим процесса |
Критерии установления режима |
Диффузионный ..................... |
|
Кинетический с химической реакцией в объеме фаз ..................... |
|
Кинетический с химической реакцией на поверхности раздела фаз ..................... |
|
При использовании процесса экстракции как метода выделения и разделения радиоактивных элементов или радиоактивных нуклидов задача исследования сводится к определению оптимальных условий процесса. Эффективность извлечения элемента в органическую фазу для одностадийного процесса экстракции характеризуется коэффициентом распределения , коэффициентом извлечения или величиной извлекаемой доли Е:
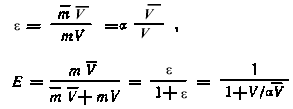 ,
где
V, V — объем водной
и органической фаз соответственно. При
определении коэффициента распределения
радиометрическим методом измеряются
объемные активности исходного раствора
и равновесных водной и органической
фаз (AV и АV).
Расчет производится
по формуле
,
где
V, V — объем водной
и органической фаз соответственно. При
определении коэффициента распределения
радиометрическим методом измеряются
объемные активности исходного раствора
и равновесных водной и органической
фаз (AV и АV).
Расчет производится
по формуле
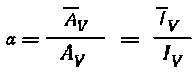 ,
где
IV — регистрируемые скорости счета
при условии, что плотности органической
и водной фаз одинаковы. В случае
существенного различия плотностей фаз
коэффициент распределения может быть
рассчитан по формуле:
,
где
IV — регистрируемые скорости счета
при условии, что плотности органической
и водной фаз одинаковы. В случае
существенного различия плотностей фаз
коэффициент распределения может быть
рассчитан по формуле:
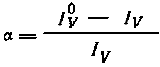 ,
где
I0V — скорость счета исходного
раствора при условии, что состав водной
фазы в процессе экстракции не меняется.
В противном случае необходимо количественно
выделять распределяющееся вещество из
органической и водной фаз.
,
где
I0V — скорость счета исходного
раствора при условии, что состав водной
фазы в процессе экстракции не меняется.
В противном случае необходимо количественно
выделять распределяющееся вещество из
органической и водной фаз.
Эффективность разделения двух веществ за одну стадию процесса экстракции при условии, что 1 > 2 определяется значением коэффициента разделения (1/2 = степенью очистки 1-го вещества в органической фазе S и степенью очистки 2-го вещества в водной фазе S:
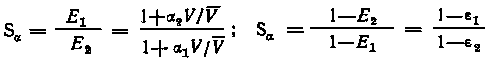 .
.
При небольших значениях a извлекаемая доля возрастает при условии, что V >> V. В атом случае выгоднее проводить многократкную экстракцию, например экстракцию перекрестным током, схема которой представлена на рис. 10.
 Рис.
10. Схема
процесса экстракции с перекрестным
током жидкости:
Э1,
Э2,
Э3
- экстракты; Р1,
Р2,
Р3
- рафинаты.
Рис.
10. Схема
процесса экстракции с перекрестным
током жидкости:
Э1,
Э2,
Э3
- экстракты; Р1,
Р2,
Р3
- рафинаты.
Тогда неизвлекаемая доля за одну ступень процесса:
![]() и
за n ступеней:
и
за n ступеней:
![]() .
.
Общая извлекаемая доля за n последовательных экстракций:
![]() .
.
Таким образом, задаваясь величиной Еn, можно рассчитать число ступеней экстракции n для данных значений V и V.
Следует отметить, что при многостадийном процессе экстракции удается добиться большей полноты извлечения, чем за однократную экстракцию тем же общим объемом экстрагента.
Степень очистки 1-го вещества в органической фазе (при 1 > 2) и степень очистки 2-го вещества в водной фазе при n последовательных экстракциях могут быть рассчитаны по формулам:
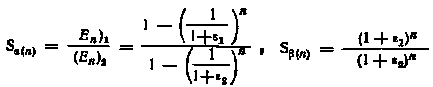 .
.
При 1 > 2 с увеличением числа ступеней процесса экстракции S(n) возрастает, a S(n) снижается. Таким образом, нельзя одновременно увеличивать Еn и S(n) в экстракте.
Для увеличения степени очистки S(n) в органической фазе при многостадийном процессе экстракции осуществляют периодическое промывание органической фазы после одной или нескольких экстракций с целью удаления менее экстрагируемого вещества.
Кинетический режим с химической реакцией в объеме фаз позволяет осуществлять процесс экстракции при минимальном времени контакта фаз для этого экстракцию необходимо проводить в условиях интенсивного перемешивания и большой поверхности раздела фаз.