

Аутоиммунная патология
Аутоиммунная патология развивается тогда, когда иммунная система реагирует на собственные, неизмененные антигены. Т.О. – это болезнь иммунной системы, поскольку агрессия развивается против неизмененных АГ. В норме реакция на неизмененные АГ организма со стороны иммунной системы отсутствует. Это связано с существованием иммунологической толерантности.
Иммунологическая толерантность приобретается Т- клетками в тимусе, в процессе эмбриогенеза. В основе лежат процессы «+» селекции клона или клональной экспансии и «-» селекции клона или клональной делеции и клональной анергии.
АПОПТОЗ И ФОРМИРОВАНИЕ ВТОРИЧНОГО КЛОНАЛЬНОГО РЕПЕРТУАРА Т-ЛИМФОЦИТОВ
Первичный (антигенраспознающий) репертуар - результат событий на стадии СД 4- 8- тимоцитов, реализуется на уровне генов и клеточной мембраны и проявляется экспрессией на поверхности клеток Т-клеточного рецептора (ТСR) с индивидуальной специфичностью.
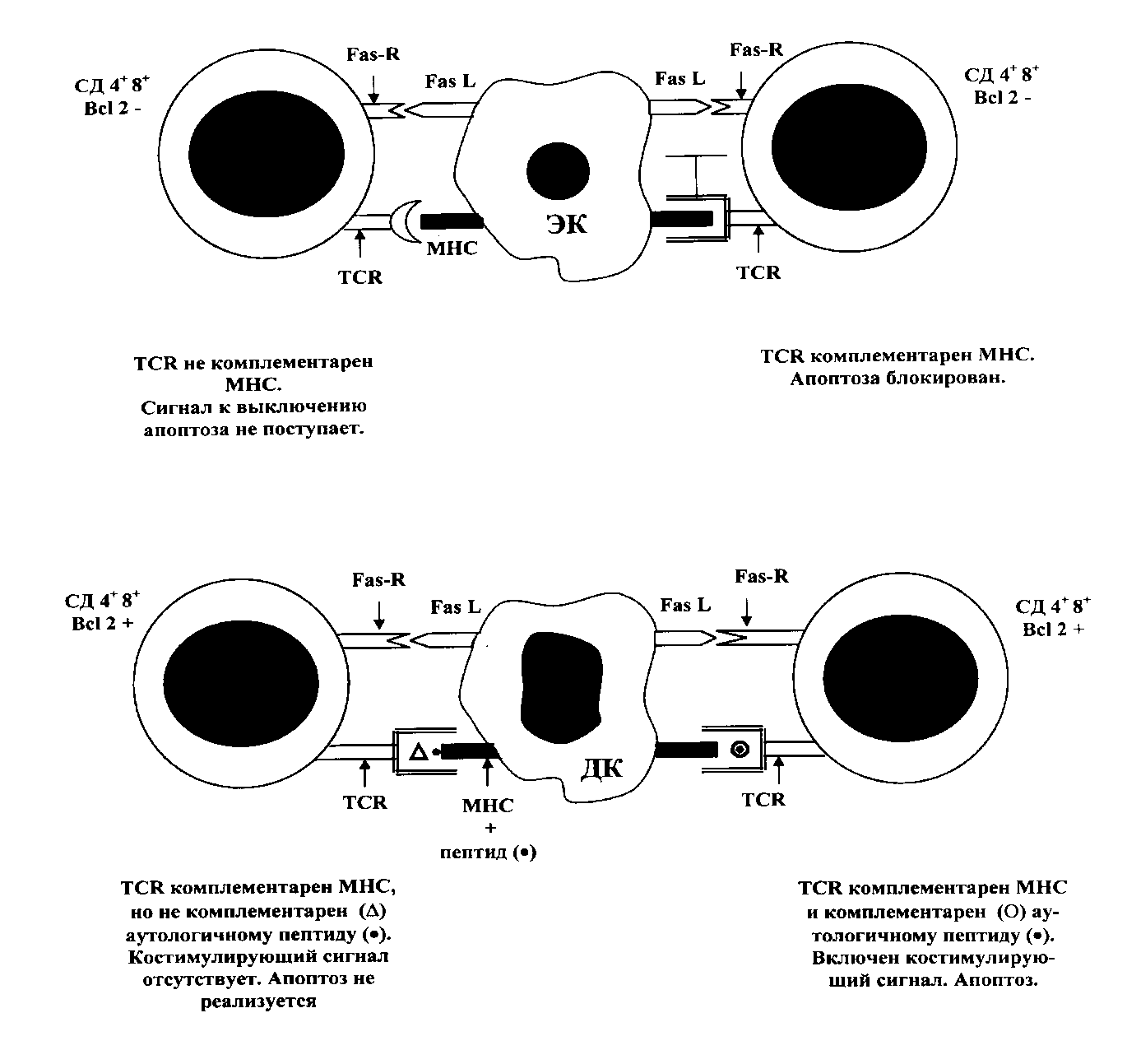
Переход незрелых кортикальных тимоцитов в стадию созревания СД 4+ 8+ сопровождается экспрессией на их поверхности Fas-рецептора (Fas-R), что в сочетании с низкой экспрессией Bcl 2 делает их высокочувствительными к индукторам апоптоза, в частности, к Fas L.
Положительная селекция клонов осуществляется в глубоких слоях коры тимуса при взаимодействии тимоцитов с эпителиальными клетками (ЭК), несущими на поверхности молекулы МНС II класса и экспрессирующими Fas L. Контакт Fas L ЭК и Fas-R тимоцитов включает у последних механизм апоптоза, обрекая их на гибель. Процесс может быть остановлен сигналом из участков взаимодействия ТСR с молекулами МНС ЭК. Однако, сигнал поступит лишь в том случае, если ТСR будет комплементарен к этим молекулам, независимо от того, в сочетании с каким (аутологичным и гетерологичным пептидом) они способны его распознавать. Т.о. к дальнейшему развитию будут допущены лишь те клоны тимоцитов, которые обладают той или иной степенью сродства к продуктам аутологичных генов МНС.
Отрицательная селекция осуществляется в мозговом слове и кортико-медуллярной зоне тимуса в процессе взаимодействия тимоцитов с дендритными клетками (ДК), несущими МНС I и II класса. Тимоциты по-прежнему экспрессируют Fas-R, но способны к (слабой) продукции Bcl 2. Поэтому их контакт с ДК, несущими Fas L недостаточен для развития апоптоза. Требуется костимулирующий сигнал. Этот сигнал поступает из участка связывания ТСR с аутологичным антигенным комплексом, состоящим из аутологичного пептида на "своей" молекуле МНС. Такая комплементарность инициирует активацию незрелых тимоцитов, что необходимо для реализации апоптоза ("активационный апоптоз"). В итоге выживают лишь те клоны лимфоцитов, которые распознают комплексы из чужеродных антигенов с аутологичными молекулами МНС. Завершается формирование вторичного клонального репертуара Т-лимфоцитов.
Иммунологическая толерантность формируется не ко всем АГ организма. Существуют т.н. забарьерные АГ, к которым нет толерантности. К забарьерным АГ относятся:
- хрусталик
- сперматозоиды
- коллоид щитовидной железы
- миелин нервных волокон
До тех пор, пока существует физиологический барьер и нет контакта между АГ и лимфоцитами, аутоиммунная патология не развивается.
Н ельзя сказать, что в организме полностью отсутствуют аутореактивные клетки. Они сохраняются, чтобы устранять продукты тканевой деструкции – это аутоиммунные реакции. Выраженность их проявлений лимитируется супресорными механизмами (Тh3 ИЛ-10).
Если организму наносится вред аутоагрессией, то это аутоиммунные заболевания (АИЗ).
МЕХАНИЗМЫ РАЗВИТИЯ АИЗ


Недостаточно сформированная
толерантность
(дефект FAS- зависимого апоптоза) Сформированная
Не сформированная толерантность толерантность
(забарьерные АГ)

Срыв иммунологической толерантности
Нарушение барьеров Механизмы срыва иммунологической толерантности
А нтигенная мимикрия
Поликлональная митогенная активация
Аномальная экспрессия МНСII на неиммуннокомпетентных клетках
Аутоагрессия - Точковые мутации в кодоне клеток
Дефицит Т- супрессорных влияний
В основе аутоиммунных процессов могут лежат клеточные или гуморальные иммунные механизмы, что определяется (как и в случае иммунной защиты) преобладающей пусковой ролью Т-хелперов типов Th1 или Th2. Thl-зависимые аутоиммунные процессы регистрируются особенно часто. Они имеют два основных варианта в зависимости от того, какой тип эффекторных клеток при них активируется — С08+-киллеры или С04+-клетки — продуценты цитокинов. В первом случае включается цитотоксический механизм поражения (например, при инсулинзависимом диабете), связанный с активностью Т-киллеров, во втором — основой патологического процесса является реакция типа ГЗТ (при рассеянном склерозе, ревматоидном артрите), которая осуществляется при участии Т-хелперов и макрофагов. Цитотоксический механизм обусловливает более локализованный тип поражения, например при инсулинзависимом диабете. Напротив, процессы, сопряженные с развитием ГЗТ, вовлекают более значительные массивы тканей (например, при ревматоидном артрите).
Реже встречаются Тп2-зависимые аутоиммунные процессы. Ведущая патогенетическая роль аутоантител показана при системной красной волчанке, аутоиммунной гемолитической анемии, идиопатической тромбоцитопенической пурпуре и ряде других цитопений, тяжелой миастении и др. Для аутоиммунных процессов гуморального типа характерно накопление аутоантител преимущественно lgG-класса, которые обладают способностью вовлекать в процесс другие факторы — гуморальные (комплемент) и клеточные (макрофаги, NK-клетки). Поэтому патогенный эффект аутоантител обычно реализуется с участием цитотоксического механизма (при гемолитической анемии и других аутоиммунных поражениях клеток крови). Реже проявляется стимулирующий эффект аутоантител; при токсическом зобе (Базедова болезнь) и возможно присоединение элементов тироидита.
Возможно также развитие аутоиммунной патологии по иммуноком-плексному типу, например при системной волчанке. Аутоантигенами в этом случае служат широко распространенные молекулы — нативная ДНК, белки межклеточного вещества (особенно коллаген) и т.д., которые взаимодействуют с аутоантителами и формируют иммунные комплексы in situ (фиксированные в тканях). Взаимодействие с иммунными комплексами активирует клетки иммунной системы (макрофаги, NK-клетки и т.д.), что обусловливает развитие локального воспаления и проявление цитотоксичности.
Таким образом, основой аутоиммунных процессов служит развитие иммунного ответа на собственные (аутологичные) антигены организма. Эти процессы возникают при нарушении (часто наследственно обусловленном) центральных или периферических механизмов, обеспечивающих запрет на реакции иммунной системы против аутологичных антигенов. В зависимости от локализации аутоантигенов различают органоспецифи-ческие и системные аутоиммунные процессы, а в зависимости от преобладающих механизмов — клеточно-опосредованные и гуморальные.
ИММУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ
Первичные иммунодефициты
Группу первичных иммунодефицитов образуют заболевания, в основе которых лежит наследственно обусловленная дефектность структуры и функционирования иммунной системы, которая проявляется в нарушении иммунной защиты.
Первичные иммунодефициты — это очень редкие состояния (примерно 1 больной на 1 000 000 человек). Они являются почти исключительно уделом детского возраста, поскольку значительная часть больных с тяжелыми формами иммунодефицитов не доживает до 20 лет, а при более легких формах иммунологические дефекты с возрастом в определенной степени компенсируются.
Как правило, в основе первичных иммунодефицитов лежит генетически обусловленный блок развития клеток иммунной системы или выпадение важных иммунных процессов вследствие дефекта определенных молекул, например ферментов или мембранных структур (схема 1).
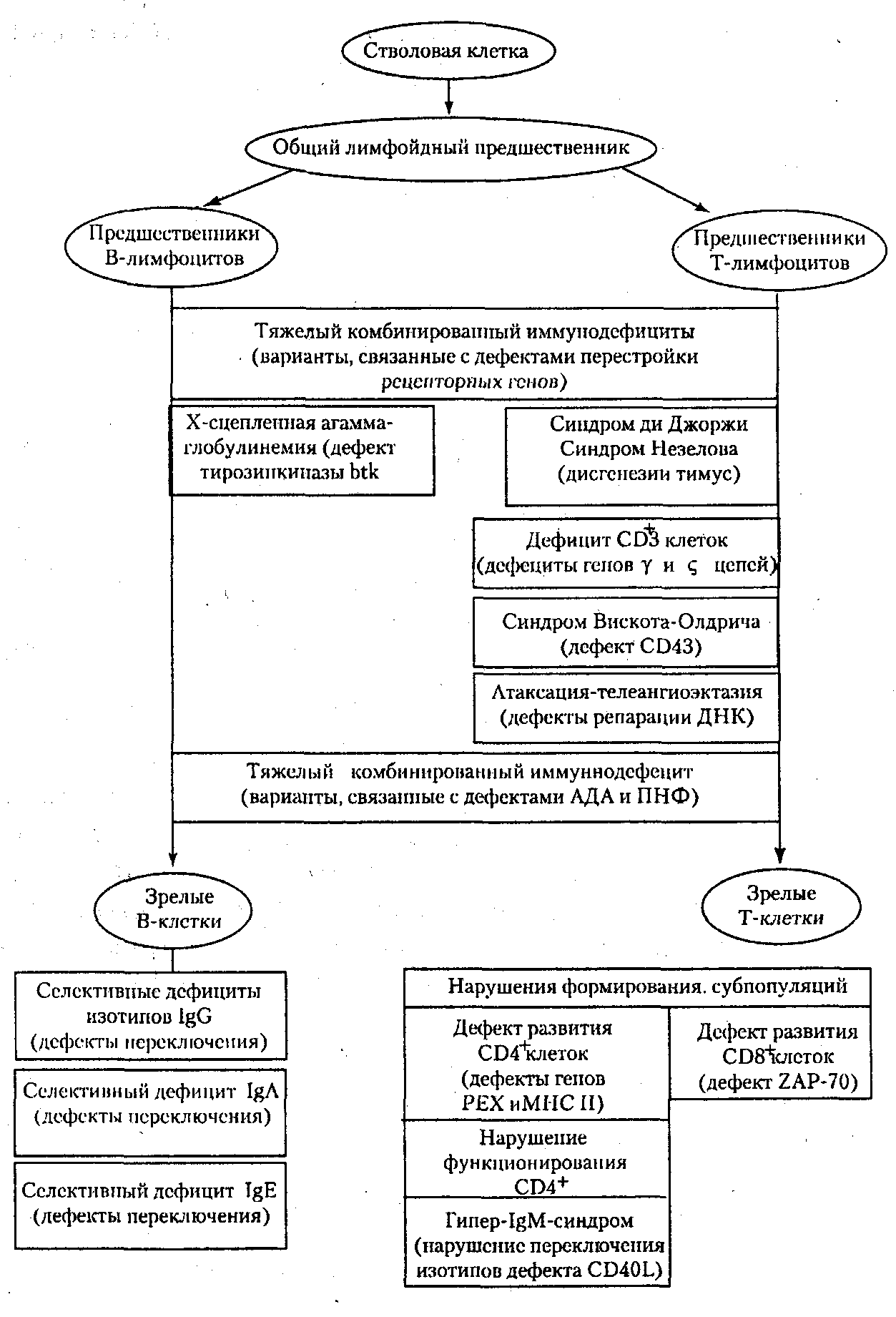
Первичные иммунодефициты можно разделить по преобладающему типу поражений звеньев иммунной системы на 3 типа:
— комбинированные иммунодефициты;
— иммунодефициты с преимущественным поражением клеточного иммунитета;
— преимущественно гуморальные иммунодефициты.
К первым относят заболевания, в основе которых лежат генетические дефекты, затрагивающие различные линии дифференцировки лимфоцитов, а также ранние этапы их развития, общие для Т- и В-линий. Во вторую группу входят иммунодефициты, при которых нарушается развитие Т-клеток и страдают опосредуемые ими реакции клеточного иммунитета; к этой же группе относятся дефекты фагоцитирующих клеток. В группу гуморальных иммунодефицитов включают патологию, в основе которой лежит нарушение развития В-клеток и Т-хелперов гуморального ответа, а также патологию компонентов комплемента.
В последние годы выясняются молекулярные основы поражения при первичных иммунодефицитах. Одной из первых была расшифрована природа комбинированных иммунодефицитов, связанных с недостаточностью ферментов пуринового метаболизма. Известны варианты таких дефектов, обусловленные мутациями генов, кодирующих аденозиндезаминазу и пуриннуклеотидфосфорилазу. Основой другой формы тяжелого комбинированного иммунодефицита, затрагивающего Т- и В-ростки лимфопоэза, служит дефект процесса перестройки генов антигенраспознающих рецепторов, связанный с отсутствием ферментов рекомбиназ, которые катализируют этот процесс.
Очень разнообразен спектр генетически обусловленных нарушений выработки антител. Их причиной может быть как поражение В-лимфоцитов (их развития или экспрессии генов иммуноглобулинов), так и дефектность Т-клеток (ослабление хелперной активности). Примером первого рода может служить агаммаглобулинемия Брутона, сцепленная сХ-хро-мосомой. Ее основой являются мутации гена, детерминирующего фермент тирозинкиназу btk, которая связана с антигенраспознающим рецептором В-лимфоцитов. Отсутствие этой тирозинкиназы делает невозможным развитие В-лимфоцитов уже на самых ранних стадиях.
В основе другого первичного иммунодефицита — гипер-IgМ-синдрома лежит дефект CD154 — молекулы, появляющейся на поверхности Т-клеток при их активации; в результате ее взаимодействия с молекулой CD40 поверхности В-лимфоцитов в эти клетки передается сигнал, обеспечивающий их дифференцировку в антителообразующие клетки, а также переключение изотипов секретируемых антител. В отсутствие этого сигнала происходит синтез иммуноглобулинов только одного изотипа — IgM, что сопровождается ослаблением гуморального иммунного ответа. Существуют формы гуморальных иммунодефицитов, при которых нарушено образование иммуноглобулинов какого-либо одного изотипа. Среди таких селективных дефектов наиболее частым является дефицит IgA.
При нем присутствуют В-лимфоциты, несущие мембранный IgA, однако не образуются плазматические клетки, секретирующие IgА-антитела.
Ряд комбинированных иммунодефицитов возникает при локализованных дефектах генов мембранных молекул адгезии. Следствием таких мутаций является нарушение миграции клеток, в первую очередь нейтрофилов и моноцитов/макрофагов, а также их взаимодействий с клетками других типов. Примером могут служить сходные поражения, развивающиеся как результат наследственных дефектов экспрессии (Р2-интегринов и углеводных детерминант, распознаваемых селектином L Эти поражения представляют собой два варианта LAD-синдрома — дефицит адгезии лейкоцитов, признаком которого является ослабление функции нейтрофилов, и повышение чувствительности к гнойным инфекциям.
Дефекты компонентов комплемента представлены вариантами с поражением практически всех основных факторов классического и альтернативного путей активации комплемента. Как правило, выпадение единичных компонентов системы комплемента проявляется в умеренном снижении устойчивости к некоторым возбудителям. Лишь дефицит ингибитора С1 q сопровождается развитием ангионевротического отека, обусловленного накоплением вазоактивных пептидов С5а и СЗа.
Иммунодефициты, в основе которых лежит дефект генов цитокинов, немногочисленны, что связано с «избыточностью» системы цитокинов, которая обусловлена взаимозаменяемостью их функций. Лишь когда генетический дефект затрагивает функцию многих цитокинов, это проявляется в тяжелых расстройствах иммунитета, что происходит, например, при дефекте гена уцепи, общей для рецепторов интерлейкинов 2, 4, 7, 13 и 15.
В результате дефекта, затрагивающего ген мембранного сиалопротеина CD43, развивается синдром Вискотта—Олдрича, о чем свидетельствует тромбоцитопения с геморрагическим синдррмом в сочетании с экземой и комбинированным иммунодефицитом. При этом заболевании аномально функционирует цитоскелет, что отражается на подвижности клеток и межклеточных взаимодействиях, важных для осуществления иммунных процессов.
При атаксии-телеангиэктазии наблюдается поражение различных функций, обусловленное слабостью аппарата репарации ДНК и нестабильностью хромосом, а также дефектами клеточного цикла. Это дает неожиданное сочетание симптомов: комбинированный иммунодефицит (недоразвитие вилочковой железы, дефицит Т-клеток и иммуноглобулинов «поздних» изотипов — IgG2, lgG4, IgE, IgA), неврологические отклонения (атаксия), поражение сосудистой стенки (телеангиэктазии), нарушение пигментации.
Помимо рассмотренных «точечных» поражений иммунной системы известны первичные иммунодефициты, развитие которых обусловлено множественными дефектами, затрагивающими формирование в эмбриогенезе различных органов, включая органы иммунной системы. Так, наследственный порок, приводящий к нарушению развития у эмбрионов человека производных 3 и 4 жаберных щелей, служит основой синдрома Ди Джорджи с дефектом развития вилочковой железы (она не заселяется предшественниками Т-клеток, развитие которых прерывается на костномозговой стадии) и гистогенетически родственных органов (паращитовидных желез и т.д).
Основным симптомокомплексом, отражающим нарушение иммунной защиты при первичных иммунодефицитах, является инфекционный синдром, т.е. понижение резистентности к инфекционным агентам, в том числе сапрофитным (Pneumocystis carinii, Candida, цитомегаловирус, некоторые энтеровирусы). Характер нарушений иммунной защиты определяется локализацией поражения в иммунной системе. Так, при блокаде процесса перестройки рецепторных генов отсутствуют как Т-, так и В-клетки и не развиваются ни клеточные, ни гуморальные формы иммунного ответа. При селективных дефектах определенных классов лимфоцитов, а также их субпопуляций выпадают именно те иммунологические функции, за которые ответственны поражаемые типы клеток. При блокаде развития В-клеток развивается агаммаглобулинемия с нарушением гуморальной защиты от внеклеточных бактерий и их токсинов, а при дефицитах Т-лимфоцитов страдает клеточная защита от вирусов и микобактерий. При некоторых формах первичных иммунодефицитов (атаксия-телеангиэкста-зия, синдром Вискотта-Олдрича и т.д.) значительно повышается риск развития злокачественных опухолей (до 10—15 %). Нередко нарушения иммунологических функций регистрируются при нормальной численности соответствующих клеток.
Клинико-иммунологическое обследование дает четкие результаты лишь при тех формах первичных иммунодефицитов, при которых точно локализован дефект. Так, при тяжелом комбинированном иммунодефиците отсутствуют как Т-, так и В-клетки, при синдроме Ди Джорджи резко снижено содержание Т-лимфоцитов, а при агаммаглобулинемиях — В-лимфоцитов. По изменению концентрации иммуноглобулинов в сыворотке или компонентов комплемента различных изотипов может быть установлена локализация дефекта в системе гуморального иммунитета. Все большую диагностическую значимость приобретает определение конкретных мембранных маркеров клеток иммунной системы (молекул адгезии, CD154, С043ит.д.), а также методы, позволяющие выявить мутации конкретных генов.
Вторичные иммунодефициты
Вторичные, или приобретенные, иммунодефициты определяют как нарушение иммунной защиты организма, развивающееся в пост-натальном периоде вследствие действия внешних или внутренних факторов, непосредственно не связанных с генетическим аппаратом.
Фактически эти иммунодефициты лишены самостоятельности и рассматриваются как состояния, сопутствующие известным заболеваниям или действию повреждающих факторов.
Роль наследственного фактора в развитии вторичных иммунодефи-цитов не исключается, поскольку чувствительность иммунной системы к действию факторов, вызывающих формирование иммунодефицитных состояний, варьирует часто и зависит от наследственности. Однако наследственные факторы сами по себе, без действия индуктора, недостаточны для проявления вторичного иммунодефицита.
Вторичные иммунодефицитные состояния чрезвычайно широко распространены: в большей или меньшей степени отклонения в иммунной системе сопутствуют всем заболеваниям, особенно вирусным, ряду эндокринных и метаболических поражений и т.д. Они проявляются при действии большинства экстраординарных внешних агентов (классический пример — пострадиационный иммунодефицит), в том числе неблагоприятных экологических факторов. Известны физиологические иммунодефи-циты, свойственные раннему постнатальному и старческому возрастам, а также иммунодефициты, связанные со стрессом. Главное проявление вторичных (как и первичных) иммунодефицитов состоит в понижении устойчивости к инфекционным агентам, в частности к оппортунистическим, со склонностью к хронизации воспалительных процессов. В ряде случаев регистрируется повышение частоты развития злокачественных опухолей. Основой многих проявлений вторичных иммунодефицитов является гибель клеток иммунной системы, которая может реализоваться в форме некроза (гибель вследствие нарушения целости мембраны) или апоптоза (гибель в результате деградации ДНК, обусловленной собственными ферментами клетки).
Апоптоз лимфоцитов развивается при действии многих лечебных химиопрепаратов, облучения, а также кортикостероидов, уровень которых существенно повышается при стрессовых состояниях. Другой механизм инактивации клеток иммунной системы состоит в их функциональной блокаде, достигаемой связыванием с поверхностью клетки или накоплением внутри клетки агентов, ингибирующих их активность. Их роль могут выполнять аутоантитела, цАМФ, простагландины и другие медиаторы воспаления, некоторые цитокины, супрессорные и «блокирующие факторы» опухолей, ингибирующие продукты патогенов.
Наконец, основой вторичных иммунодефицитных состояний может стать дисбаланс клеток иммунной системы — эффекторных и супрессор-ных клеток, субпопуляций CD4+- и СР8+ Т-лимфоцитов, Т-хелперовТп1- и Тп2-типов. Снижение соотношения CD4+/CD8+, иногда обозначаемого как иммунорегуляторный индекс, регистрируется при ряде заболеваний; это может быть обусловлено как снижением численности С04+-хелперов вследствие ослабления функции вилочковой железы и других причин, так и повышением содержания С08+-лимфоцитов, которое может сопровождаться превалированием супрессорной активности. Дисбаланс хелперов типов Тп1 и ТИ2 приводит к преобладанию гуморального или клеточного типа иммунной защиты, который может оказаться неадекватным при конкретной форме инфекции, о чем уже говорилось выше. Среди заболеваний, которые особенно часто сопровождаются развитием вторичного иммунодефицита, следует назвать хронические неспецифические заболевания легких, некоторые эндокринопатии, ожоговую болезнь, хроническую почечную недостаточность. Из инфекционных агентов, вызывающих поражение иммунной системы, на первом месте стоят вирусы, которые часто проявляют тропность к Т-лимфоцитам.
Вирусную этиологию имеет синдром приобретенного иммунодефицита (СПИД) — единственная самостоятельная нозологическая форма вторичных иммунодефицитов. Его вызывает лентивирус — вирус иммунодефицита человека, ВИЧ-1 (HlV-1 —от англ. Human immunodeficiency virus). Основой СПИДа является поражение клеток, несущих на своей поверхности молекулу CD4, — Т-хелперов, макрофагов, дендритных клеток. Эта избирательность обусловлена сродством белка др 120 оболочки ВИЧ-1 к молекуле CD4. Гибель клеток связана не только с непосредственным цитопатогенным действием вируса, но и с индукцией апоптоза клеток вследствие перекрестного связывания CD4 белком др 120. При СПИДе прогрессивно снижаются все формы иммунной защиты, в первую очередь клеточной.
Среди экзогенных факторов, способных вызывать вторичный иммунодефицит, наиболее важны ионизирующая радиация и цитотоксические препараты, используемые в химиотерапии. В обоих случаях основной мишенью повреждающего действия являются лимфоциты. Под влиянием высоких доз этих факторов (при радиационных катастрофах, лечебном использовании высоких доз облучения или химиопрепаратов) развивается комбинированный иммунодефицит, который может привести организм к гибели вследствие развития инфекционных осложнений.
При длительном действии малых доз ионизирующей радиации, химических агентов, загрязняющих среду обитания человека (пестицидов, отходов промышленных предприятий) также развивается иммунодефицит. Он может быть выражен слабо, и его природа не всегда ясна. Эта форма иммунодефицита близка по генезу и проявлениям к возрастным иммунодефицитам: радиация и другие неблагоприятные экологические факторы ускоряют процесс старения иммунной системы.
Таким образом, иммунологическая недостаточность может развиваться как вследствие генетических дефектов, затрагивающих формирование и функционирование иммунной системы (первичные иммунодефициты), так и под влиянием внешних и внутренних факторов, действующих на сформировавшийся организм (вторичные иммунодефицитные состояния).