

IX.1.3.2. Нуклеофильные реакции карбонильной группы карбоновых кислот
Карбонильная группа карбоновых кислот, как и в случае альдегидов и кетонов, способна взаимодействовать с нуклеофильными реагентами. Однако активность карбоновых кислот в таких реакциях значительно ниже, чем альдегидов и кетонов. Это объясняется, по меньшей мере, двумя причинами:
1 .
Гидроксигруппа за счет своего +М-эффекта
заметно уменьшает положительный заряд
на карбонильном атоме углерода:
.
Гидроксигруппа за счет своего +М-эффекта
заметно уменьшает положительный заряд
на карбонильном атоме углерода:
Группа ОН, кроме того, обладает противоположно действующим –I-эффектом, но +М-эффект этой группы намного сильнее, так что в итоге проявляется заметный донорный эффект, понижающий карбонильную активность (стр. 233).
2. Нуклеофилы одновременно являются основаниями; если основность реагента достаточна (а это может быть и основание средней силы), то реагент не атакует карбонильный атом углерода, а просто отрывает протон от группы ОН и образует соль; так, в частности, реагируют аммиак и амины, гидроксиламин, гидразин, цианид-анион и др.:
К арбонильная
активность в солях карбоновых кислот
намного
меньше, чем в самих карбоновых кислотах;
она почти не проявляется. Причина в том,
что анионный фрагмент –О¯ проявляет
сильнейший +М-, а также и значительный
+I-эффекты.
Поэтому после образования солей
нуклеофильные реагенты в большинстве
случаев не реагируют с ними; исключением
являются очень сильные нуклеофилы –
литий- и магнийорганические соединения
и алюмогидрид лития (см. ниже) [имеется
в виду, конечно, избыток нуклеофильного
реагента; его эквивалент, пошедший на
образование соли, теряет свои нуклеофильные
свойства]. Лишь
нуклеофилы, проявляющие слабые основные
свойства (например, спирты) не образуют
солей; однако они являются также и
слабыми нуклеофилами, поэтому без
специальной активации они не способны
присоединяться по карбонильной группе.
арбонильная
активность в солях карбоновых кислот
намного
меньше, чем в самих карбоновых кислотах;
она почти не проявляется. Причина в том,
что анионный фрагмент –О¯ проявляет
сильнейший +М-, а также и значительный
+I-эффекты.
Поэтому после образования солей
нуклеофильные реагенты в большинстве
случаев не реагируют с ними; исключением
являются очень сильные нуклеофилы –
литий- и магнийорганические соединения
и алюмогидрид лития (см. ниже) [имеется
в виду, конечно, избыток нуклеофильного
реагента; его эквивалент, пошедший на
образование соли, теряет свои нуклеофильные
свойства]. Лишь
нуклеофилы, проявляющие слабые основные
свойства (например, спирты) не образуют
солей; однако они являются также и
слабыми нуклеофилами, поэтому без
специальной активации они не способны
присоединяться по карбонильной группе.
Нуклеофильные реакции карбонильной группы карбоновых кислот достаточно эффективно протекают либо при активации реагирующих партнеров (чаще – карбоновой кислоты), либо при высокой температуре, либо с очень сильными нуклеофилами (без активации и в достаточно мягких условиях).
После нуклеофильного присоединения бинарного реагента следует отщепление гидроксильной группы, и конечным итогом реакции оказывается нуклеофильное замещение группы ОН на нуклеофильный фрагмент реагента (Y) по схеме «присоединение – отщепление»:
О тщепление
происходит потому, что промежуточный
продукт присоединения (584) малоустойчив
из-за нахождения двух или даже трех
электроотрицательных групп у одного
атома углерода (три группы в том случае,
если группа Y
электроотрицательна).
Приведенная схема является чисто
формальной; конкретные механизмы могут
заметно отличаться.
тщепление
происходит потому, что промежуточный
продукт присоединения (584) малоустойчив
из-за нахождения двух или даже трех
электроотрицательных групп у одного
атома углерода (три группы в том случае,
если группа Y
электроотрицательна).
Приведенная схема является чисто
формальной; конкретные механизмы могут
заметно отличаться.
Наибольшее число реакций подобного типа приводит к образованию производных карбоновых кислот: сложных эфиров (Y=OR1), амидов (Y= NH2, NHR, NR2), галогенангидридов (Y=Hal), ангидридов (Y=OCOR) и других производных. При взаимодействии с металлорганическими соединениями могут также образовываться кетоны (Y= R1) и продукты их дальнейших превращений. Гидридное восстановление кислот дает первичные спирты.
 1.
Образование сложных эфиров. Наиболее
распространенный вариант образования
сложных эфиров из карбоновых кислот –
взаимодействие карбоновых кислот со
спиртами; в большинстве случаев для
этого требуется катализ
сильными минеральными кислотами
(НС1,
H2SO4,
п-толуолсульфокислота).
Реакция называется этерификацией:
1.
Образование сложных эфиров. Наиболее
распространенный вариант образования
сложных эфиров из карбоновых кислот –
взаимодействие карбоновых кислот со
спиртами; в большинстве случаев для
этого требуется катализ
сильными минеральными кислотами
(НС1,
H2SO4,
п-толуолсульфокислота).
Реакция называется этерификацией:
Если в качестве субстрата рассматривать спирт, то она представляет собой ацилирование спирта карбоновой кислотой (ацилирование спиртов в общем виде представлено в разделе «Спирты»).
Кислотный катализ направлен на активацию либо кислоты (чаще), либо спирта (реже). Известны три механизма взаимодействия карбоновых кислот со спиртами при кислотном катализе.
Первый, самый распространенный механизм, можно представить следующим образом:
Н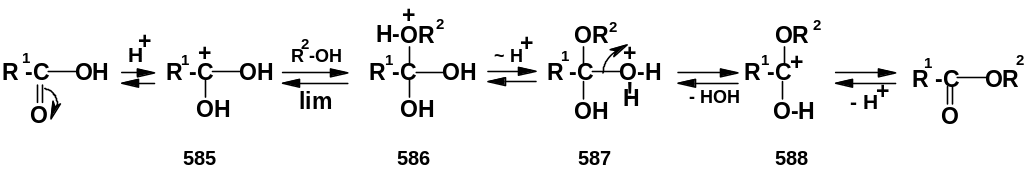 а
первой стадии происходит активация
карбонильной группы путем ее О-протонирова-
ния с образованием интермедиата, который
удобно представить резонансной структурой
(585); эта стадия является быстрой. Далее
следует скоростьопределяющая
(лимитирующая) стадия – собственно
нуклеофильная атака молекулы спирта
на активированную карбонильную группу
(как при образовании полуацеталей, стр.
235). Затем происходит миграция протона
в интермедиате (586) от алкокси- к
гидроксигруппе, после чего от
образовавшегося интермедиата (587)
отщепляется вода («хорошая» уходящая
группа). Образовавшийся при этом
интермедиат (588) – О-протонированный
сложный эфир; на заключительной стадии
происходит депротонирование и образуется
конечный продукт - сложный эфир.
Лимитирующая стадия данного механизма
бимолекулярна,
т.е. и сама реакция является бимолекулярной.
Представленный здесь механизм является
частным случаем механизмов, обозначаемых
как AАС2,
т.е. механизмов кислотно-катализируемых
бимолекулярных реакций ацильных
производных
(А – Acid, Ac – Acyl). Помимо реакции
этерификации, по типу механизмов ААС2
протекает ряд реакций производных
карбоновых кислот; некоторые из них
будут приведены ниже.
а
первой стадии происходит активация
карбонильной группы путем ее О-протонирова-
ния с образованием интермедиата, который
удобно представить резонансной структурой
(585); эта стадия является быстрой. Далее
следует скоростьопределяющая
(лимитирующая) стадия – собственно
нуклеофильная атака молекулы спирта
на активированную карбонильную группу
(как при образовании полуацеталей, стр.
235). Затем происходит миграция протона
в интермедиате (586) от алкокси- к
гидроксигруппе, после чего от
образовавшегося интермедиата (587)
отщепляется вода («хорошая» уходящая
группа). Образовавшийся при этом
интермедиат (588) – О-протонированный
сложный эфир; на заключительной стадии
происходит депротонирование и образуется
конечный продукт - сложный эфир.
Лимитирующая стадия данного механизма
бимолекулярна,
т.е. и сама реакция является бимолекулярной.
Представленный здесь механизм является
частным случаем механизмов, обозначаемых
как AАС2,
т.е. механизмов кислотно-катализируемых
бимолекулярных реакций ацильных
производных
(А – Acid, Ac – Acyl). Помимо реакции
этерификации, по типу механизмов ААС2
протекает ряд реакций производных
карбоновых кислот; некоторые из них
будут приведены ниже.
Все стадии данного превращения обратимы (реакция микроскопически обратима); поэтому в присутствии водных растворов кислот сложные эфиры гидролизуются с образованием исходных кислот и спиртов по тому же механизму ААС2, проходящему в обратном направлении (кислотный гидролиз сложных эфиров). Здесь лимитирующей стадией является присоединение воды к интермедиату (588). Обратимость реакции этерификации по данному механизму выражена весьма заметно, поэтому для того, чтобы сдвинуть равновесие в сторону образования сложного эфира, необходимо удалять образующуюся воду.
 Второй
механизм
встречается значительно реже – в случае
пространственно затрудненных кислот,
таких, как о,о-дизамещенные
ароматические карбоновые кислоты.
Реакция по механизму ААС2
в этом случае затруднена, т.к. ключевой
интермедиат (586) пространственно напряжен
(тетракоординированный атом углерода,
при котором находится объёмный
заместитель). В этом случае реакцию
проводят в сильнокислой
среде:
например, растворяют
карбоновую
кислоту в конц. H2SO4
и осторожное выливают раствор в спирт;
механизм при этом следующий:
Второй
механизм
встречается значительно реже – в случае
пространственно затрудненных кислот,
таких, как о,о-дизамещенные
ароматические карбоновые кислоты.
Реакция по механизму ААС2
в этом случае затруднена, т.к. ключевой
интермедиат (586) пространственно напряжен
(тетракоординированный атом углерода,
при котором находится объёмный
заместитель). В этом случае реакцию
проводят в сильнокислой
среде:
например, растворяют
карбоновую
кислоту в конц. H2SO4
и осторожное выливают раствор в спирт;
механизм при этом следующий:
После О-протонирования гидроксигруппы на скоростьопределяющей стадии образуется ключевой интермедиат – ацил-катион (ацилий-катион) (589); в этом интермедиате атом углерода двухкоординирован, поэтому даже самый разветвленный радикал R1 не создает пространственного напряжения. Далее (после выливания в спирт) следует нуклеофильная атака молекулы спирта, и после депротонирования образуется конечный продукт.
Поскольку лимитирующая стадия мономолекулярна, этот механизм обозначают как ААС1. Реакция и здесь микроскопически обратима; подобным образом проводят кислотный гидролиз пространственно затрудненных эфиров.
Третий механизм имеет место только в тех случаях, когда спирт R2ОН может образовать достаточно устойчивый карбокатион (третичные спирты, спирты с бензильными или аллильными радикалами). В этом случае кислотный катализ направлен не на карбоновую кислоту, а на спирт, из которого генерируется карбокатион (590):
Д анный
механизм – кислотно-катализируемое
SN1-
замещение группы ОН в спиртах; роль
нуклеофила выполняет карбоновая кислота.
Реакцию обозначают как АAL1
(АL
– алкил); действительно, в двух предыдущих
механизмах разрывается связь О-ацил, а
в данном – связь О-алкил. В механизмах
ААС2
и ААС1
нуклеофилом является спирт, электрофилом
– карбоновая кислота; в механизме ААL1
ситуация противоположная.
анный
механизм – кислотно-катализируемое
SN1-
замещение группы ОН в спиртах; роль
нуклеофила выполняет карбоновая кислота.
Реакцию обозначают как АAL1
(АL
– алкил); действительно, в двух предыдущих
механизмах разрывается связь О-ацил, а
в данном – связь О-алкил. В механизмах
ААС2
и ААС1
нуклеофилом является спирт, электрофилом
– карбоновая кислота; в механизме ААL1
ситуация противоположная.
Иногда вместо минеральных кислот в качестве катализатора этерификации используют кислоту Льюиса – эфират трифторида бора. Некоторые, относительно сильные кислоты (муравьиная, щавелевая) могут этерифицироваться и в отсутствие катализатора.
 Для
получения сложных эфиров может
использоваться взаимодействие солей
карбоновых кислот с галогенпроизводными,
тозилатами и другими соединениями с
хорошими нуклеофугными группами:
Для
получения сложных эфиров может
использоваться взаимодействие солей
карбоновых кислот с галогенпроизводными,
тозилатами и другими соединениями с
хорошими нуклеофугными группами:
Это – обычные реакции нуклеофильного замещения. Карбоксилат-анион – довольно слабый нуклеофил (делокализация заряда) и обычно используется в реакциях SN1.
Чрезвычайно широко используются для получения сложных эфиров производные карбоновых кислот, прежде всего хлорангидриды и ангидриды; эти производные значительно активнее самих карбоновых кислот в реакциях с нуклеофилами. Данные реакции будут описаны при рассмотрении этих производных.
 Специфический
вариант – образование циклических
сложных эфиров;
эти эфиры в большинстве случаев образуются
из гидроксикислот. Если такие эфиры
содержат 5-
и 6- членные циклы,
они образуются легче,
чем другие типы сложных эфиров. Можно
выделить два основных типа циклических
сложных эфиров - лактиды
и лактоны:
Специфический
вариант – образование циклических
сложных эфиров;
эти эфиры в большинстве случаев образуются
из гидроксикислот. Если такие эфиры
содержат 5-
и 6- членные циклы,
они образуются легче,
чем другие типы сложных эфиров. Можно
выделить два основных типа циклических
сложных эфиров - лактиды
и лактоны:
Лактиды образуются при нагревании -гидроксикислот; катализ здесь не требуется:
 Вначале
происходит межмолекулярная,
а затем внутримолекулярная
этерификация. Лактиды не особо устойчивы,
их роль в органическом синтезе пока
довольно ограничена.
Вначале
происходит межмолекулярная,
а затем внутримолекулярная
этерификация. Лактиды не особо устойчивы,
их роль в органическом синтезе пока
довольно ограничена.
Лактоны – продукты внутримолекулярной этерификации гидроксикислот:
И звестны
лактоны с различными размерами циклов,
начиная с 4-х-членного (n=2);
наиболее широко представлены 5- и
6-членные лактоны (n=3,4);
они образуются при нагревании
игидроксикислот
без катализа, а довольно часто образуются
и без нагревания, т.е. происходит
самопроизвольная циклизация
гидроксикислоты. Такой самопроизвольной
циклизации подвергается, например,
кумариновая кислота (591); образуется
лактон (кумарин) (592):
звестны
лактоны с различными размерами циклов,
начиная с 4-х-членного (n=2);
наиболее широко представлены 5- и
6-членные лактоны (n=3,4);
они образуются при нагревании
игидроксикислот
без катализа, а довольно часто образуются
и без нагревания, т.е. происходит
самопроизвольная циклизация
гидроксикислоты. Такой самопроизвольной
циклизации подвергается, например,
кумариновая кислота (591); образуется
лактон (кумарин) (592):
Примеры легкого образования лактонов уже упоминались в разделе «Углеводы» (стр. 292, 293).
Лактоны с циклами, бóльшими, чем 6-членные, менее доступны; тем не менее, в природе встречаются соединения, содержащие 12-, 14- и 16-членные лактонные циклы – так называемые макролидные антибиотики, к которым, в частности, относятся эритромицины. Вообще лактоны достаточно широко распространены в природе; природные лактоны обладают высокой биологической активностью. Лактоны широко используются в органическом синтезе.
 2.
Образование амидов. Как
уже упоминалось, карбоновые кислоты
при взаимодействии с аммиаком образуют
соли. При нагревании этих солей до
достаточно высокой температуры (выше
150 оС)
происходит их дегидратация с образованием
амидов:
2.
Образование амидов. Как
уже упоминалось, карбоновые кислоты
при взаимодействии с аммиаком образуют
соли. При нагревании этих солей до
достаточно высокой температуры (выше
150 оС)
происходит их дегидратация с образованием
амидов:
В начале
аммонийная соль диссоциирует на исходные
карбоновую кислоту и аммиак, а затем
аммиак нуклеофильно присоединяется по
карбонильной группе кислоты:
начале
аммонийная соль диссоциирует на исходные
карбоновую кислоту и аммиак, а затем
аммиак нуклеофильно присоединяется по
карбонильной группе кислоты:
Присоединение становится возможным благодаря жестким условиям; к тому же аммиак заметно более сильный нуклеофил, чем спирты. Все же данный вариант – далеко не главный способ получения амидов; гораздо чаще их получают из хлорангидридов и ангидридов кислот, а иногда и из сложных эфиров (будет рассмотрено позднее).
Имиды и циклические амиды. При образовании амидов так же, как и при образовании сложных эфиров, в некоторых случаях возможно образование циклических структур.
 A.
Имиды
образуются при нагревании аммонийных
солей некоторых двухосновных кислот,
например, янтарной:
A.
Имиды
образуются при нагревании аммонийных
солей некоторых двухосновных кислот,
например, янтарной:
При образовании имидов происходит двойное ацилирование аммиака (или первичного амина); оно происходит легко в тех случаях, когда образуются 5- или 6-членные циклы. Особенно легко образуются имиды из циклических ангидридов дикарбоновых кислот (см. ниже).
 Б.
Дикетопиперазины
образуются
при нагревании аминокислот:
Б.
Дикетопиперазины
образуются
при нагревании аминокислот:
В. Лактамы образуются при нагревании иаминокислот (n=3,4):
О бразование
дикетопиперазинов и лактамов вполне
аналогично образованию лактидов и
лактонов, рассмотренному выше. Более
распространенными являются лактамы;
известны лактамы с разными размерами
циклов, начиная с 4-членного. Известны
природные
лактамы; в частности, 4-членный лактамный
цикл входит в структуры антибиотиков
пенициллинов.
бразование
дикетопиперазинов и лактамов вполне
аналогично образованию лактидов и
лактонов, рассмотренному выше. Более
распространенными являются лактамы;
известны лактамы с разными размерами
циклов, начиная с 4-членного. Известны
природные
лактамы; в частности, 4-членный лактамный
цикл входит в структуры антибиотиков
пенициллинов.