

Свойства карбоновых кислот
IX.1.2. Физические свойства.
К арбоксильная
группа, как и гидроксильная группа
спиртов, способна к образованию водородных
связей, что приводит, во-первых, к
возрастанию температур кипения и,
во-вторых, к увеличению сродства к воде
(гидрофильности). Как и в случае спиртов,
молекулы карбоновых кислот могут
образовывать водородные связи друг с
другом и с молекулами воды (и других
полярных растворителей):
арбоксильная
группа, как и гидроксильная группа
спиртов, способна к образованию водородных
связей, что приводит, во-первых, к
возрастанию температур кипения и,
во-вторых, к увеличению сродства к воде
(гидрофильности). Как и в случае спиртов,
молекулы карбоновых кислот могут
образовывать водородные связи друг с
другом и с молекулами воды (и других
полярных растворителей):
Ассоциаты молекул карбоновых кислот могут иметь как линейную структуру (А), так и циклическую (димерную) структуру (В). Карбоновые кислоты образуют димерные ассоциаты (В) в твердой фазе (кристаллах), в неполярных растворителях и частично даже в газовой фазе; по этой причине температуры кипения карбоновых кислот еще выше, чем спиртов с той же молекулярной массой: например, температура кипения этанола (М=46) равна 78 оС, а для муравьиной кислоты (М=46) она составляет 101 оС. С водой карбоновые кислоты также могут образовывать циклические ассоциаты (С). Низшие монокарбоновые и дикарбоновые кислоты хорошо растворимы в воде.
Карбоксильная группа хорошо обнаруживается методами ИК спектроскопии и спектроскопии ЯМР (как ЯМР 1Н, так и ЯМР 13С). Наличие и характер водородных связей надежно определяются по данным ИК спектров и спектров ЯМР 1Н.
Очень многие карбоновые кислоты безопасны для организма и широко используются в пищевой промышленности (уксусная, лимонная и другие); есть, однако, и весьма серьезные исключения: например, монофторуксусная кислота является смертельным ядом.
IX.1.3. Химические свойства
Химические свойства карбоновых кислот почти полностью определяются карбоксильной группой –С(=О)-ОН – функциональной группой, в которой карбонильная и гидроксильная группы непосредственно связаны и поэтому заметно влияют друг на друга. Вследствие этого карбоновые кислоты, сохраняя определенное сходство с карбонильными соединениями и со спиртами, приобретают весьма заметное своеобразие. Это своеобразие касается, прежде всего, двух аспектов:
А. По сравнению со спиртами и фенолами карбоновые кислоты проявляют гораздо более сильные кислотные свойства.
Б. По сравнению с альдегидами и кетонами карбоновые кислоты труднее присоединяют нуклеофильные реагенты; в большинстве случаев после присоединения происходит отщепление гидроксильной группы.
Карбоксильная группа, подобно некоторым ранее рассмотренным группам, активирует связанное с ней -положение.
Химические свойства карбоновых кислот можно разделить на 4 группы:
I. Реакции с разрывом связи О-Н (кислотные свойства); II. Реакции по карбонильной группе (взаимодействие с нуклеофилами); III. Реакции, сопровождающиеся потерей карбоксильной группы (декарбоксилирование и декарбонилирование); IV. Реакции -положения к карбоксильной группе.
IX.1.3.1. Кислотные свойства.
Карбоновые кислоты проявляют значительно более сильные кислотные свойства, чем спирты и фенолы: если для простых спиртов (алканолов) Ка (константа кислотной диссоциации) в водном растворе имеет размерность порядка 10-15, для простых фенолов (не имеющих акцепторных заместителей) – порядка 10-10, то для простых карбоновых кислот – порядка 10-5. Главной причиной усиления кислотных свойств является достаточно высокая стабильность карбоксилат-аниона, который образуется при кислотной диссоциации карбоновой кислоты:
R
 -COOH
RCOO-
+ H+
-COOH
RCOO-
+ H+
Э то
становится ясным при сравнении электронной
структуры алкоголят-аниона, фенолят-аниона
и карбоксилат-аниона:
то
становится ясным при сравнении электронной
структуры алкоголят-аниона, фенолят-аниона
и карбоксилат-аниона:
В алкоголят-анионе отрицательный заряд не делокализован, в фенолят- и карбоксилат-анионах – делокализован; степень делокализации в карбоксилат-анионе больше, чем в фенолят-анионе: хотя число резонансных структур в карбоксилат-анионе меньше, они идентичны, заряд распределен поровну между двумя атомами кислорода и это приводит к большей стабилизации, чем для фенолят-аниона, где бóльшая часть заряда сосредоточена на одном атоме (кислородном).
Степень диссоциации карбоновых кислот в значительной степени зависит от растворителя: она наибольшая в водных растворах и снижается по мере уменьшения полярности растворителя; в бензольном растворе карбоновые кислоты практически не диссоциируют.
 Кислотные
свойства карбоновых кислот проявляются
в типичных для кислот реакциях - с
активными металлами, основными оксидами,
основаниями (гидроксидами, аммиаком,
аминами).
Кислотные
свойства карбоновых кислот проявляются
в типичных для кислот реакциях - с
активными металлами, основными оксидами,
основаниями (гидроксидами, аммиаком,
аминами).
В результате этих реакций образуются соли, называемые карбоксилатами (анион RCOO‾ - карбоксилат-анион). Названия солей производят от названия кислот с прибавлением окончания «ат» (ацетат натрия, R=СН3, бензоат аммония, R=С6Н5 и т.д.).
 Для
аминокислот, содержащих как кислотную,
так и основную группы (т.е. являющихся
амфотерными соединениями), характерно
образование внутренних
солей (биполярных
ионов, цвиттер-ионов):
Для
аминокислот, содержащих как кислотную,
так и основную группы (т.е. являющихся
амфотерными соединениями), характерно
образование внутренних
солей (биполярных
ионов, цвиттер-ионов):
Наиболее известные аминокислоты – природные аминокислоты – в кристаллическом виде представляют собой именно цвиттер-ионы; этим и объясняются их высокие температуры плавления. В водных растворах аминокислот, в средах, близких к нейтральным (в так называемых изоэлектрических точках), также преобладают цвиттер-ионы.
Хотя кислотные свойства карбоновых кислот проявляются вполне отчетливо, большинство из них значительно слабее сильных минеральных кислот – об этом свидетельствуют приведенные выше величины Ка (порядка 10-5). Поэтому соли многих карбоновых кислот в водных растворах частично гидролизованы; сильные минеральные кислоты вытесняют карбоновые кислоты из растворов их солей.
Образование солей карбоновых кислот с аминами как способ расщепления рацемических кислот или аминов на энантиомеры.
Как уже упоминалось, соединения, состоящие из хиральных молекул, часто существуют в виде рацемических смесей или рацемических соединений. Может возникнуть потребность разделить рацемат на составляющие его энантиомеры (или, по крайней мере, выделить хотя бы один энантиомер). Такая потребность возникает, например, при исследовании биологической активности соединений, т.к. энантиомеры различаются по биоактивности, причем порой весьма существенно. Для синтеза природных соединений также требуются оптически чистые энантиомеры (например, чистые энантиомеры аминокислот для синтеза пептидов и белков). Вместе с тем, задача разделения рацематов на энантиомеры изначально требует решения проблем, связанных с тем, что большинство свойств энантиомеров идентичны.
Известно несколько способов расщепления рацемических смесей: ферментативное разделение, хроматографическое разделение на хиральных носителях; в отдельных случаях энантиомеры кристаллизуются в виде зеркально подобных кристаллов, которые можно механически разделить (именно этим путем Л.Пастер в 1848 г. осуществил первое расщепление рацемата). Но пока наиболее универсальным способом является временное превращение рацемической смеси в смесь двух диастереомеров; такую смесь разделить гораздо легче, т.к. диастереомеры отличаются по свойствам в гораздо большей степени, чем энантиомеры. Для этого рацемическую смесь вводят в реакцию с оптически чистым реагентом (т.е. с чистым энантиомером реагента); при этом необходимо получать такие продукты реакции, из которых после разделения можно легко вернуться к исходным соединениям.
 Наиболее
легко этот метод осуществляется при
образовании солей из карбоновых кислот
и аминов. Допустим, необходимо разделить
на энантиомеры рацемическую карбоновую
кислоту. Получают соль этой кислоты с
оптически
активным амином;
можно использовать первичные, вторичные
и третичные амины
( для простоты
возьмем пример, когда кислота и амин
имеют по одному асимметрическому атому
углерода):
Наиболее
легко этот метод осуществляется при
образовании солей из карбоновых кислот
и аминов. Допустим, необходимо разделить
на энантиомеры рацемическую карбоновую
кислоту. Получают соль этой кислоты с
оптически
активным амином;
можно использовать первичные, вторичные
и третичные амины
( для простоты
возьмем пример, когда кислота и амин
имеют по одному асимметрическому атому
углерода):
При взаимодействии рацемической (R-, S-) кислоты с чистым энантиомером амина (имеющего, допустим, R-конфигурацию) получается смесь равных количеств двух диастереомерных солей (R-, R- и S-, R-). Эту смесь разделяют, например, дробной кристаллизацией (диастереомеры часто различаются по растворимости); далее, разлагая R-, R- соль подкислением, получают R-кислоту, аналогично из S-, R- соли получают S-кислоту.
Разумеется, ситуация сохраняется при любом числе асимметрических атомов углерода как в кислоте, так и в амине. Для разделения рацемических кислот используют природные амины – алкалоиды, которые всегда находятся в оптически активной форме; используют морфин, эфедрин (стр. 229), бруцин, хинин и другие соединения.
Этот метод можно аналогично использовать и для разделения рацемических аминов, получая их соли с оптически активными кислотами.
Зависимость силы карбоновых кислот от характера заместителей. Уравнение Гаммета. Кислотность карбоновых кислот зависит от природы радикала, связанного с карбоксильной группой. Донорные группы уменьшают кислотность, акцепторные – увеличивают; это объясняется тем, что первые дестабилизируют карбоксилат-анион, вторые – стабилизируют. Этот вопрос уже затрагивался ранее (стр. 39); на силу карбоновых кислот оказывают влияние только индуктивные эффекты заместителей, и влияние пропорционально величине эффекта.
А. Алифатические кислоты. Донорными заместителями в таких кислотах, как правило, являются алкильные группы; их +I- эффект невелик, поэтому они лишь в малой степени понижают кислотность; например, уксусная, пропионовая и 2,2-диметилпропановая кислоты почти не отличаются по величинам рКа (соответственно 4,75, 4,87, 5,03). Напротив, ряд акцепторных заместителей обладает значительными по величине –I-эффектами; такие заместители могут заметно повышать кислотность. Хлоруксусная кислота сильнее уксусной на два порядка, дихлоруксусная – на три, а трихлоруксусная – на четыре порядка (рКа соответственно 2,85, 1,48, 0,70); еще большую силу имеет трифторуксусная кислота (рКа = 0,2) – это уже кислота, сравнимая по силе с минеральными. [Напомним: рКа – отрицательный логарифм константы кислотной диссоциации в воде (Ка); чем меньше величина рКа, тем сильнее кислота]. Относительно высокую кислотность имеет щавелевая кислота НООС-СООН [рКа(1)=1,27, но рКа(2)=4,27!].
Б. Ароматические кислоты. Простые ароматические (арилкарбоновые) кислоты несколько сильнее простых алифатических (алкилкарбоновых), т.к. простые арильные радикалы обладают –I-эффектом; например, для бензойной кислоты рКа=4,17, в то время как для уксусной кислоты рКа= 4,75. Заместители, стоящие в ароматическом ядре, влияют на кислотность; при этом необходимо учитывать не только индуктивные, но и мезомерные эффекты этих заместителей, потому что не только I-, но и М-эффекты заместителей влияют на электронную плотность при атоме углерода, непосредственно связанном с карбоксильной группой, а, следовательно, на индуктивный эффект арильного радикала. Заместители, стоящие в мета-положении к карбоксильной группе, оказывают только индуктивное влияние на этот атом; заместители, стоящие в орто- и пара-положениях – и индуктивное и мезомерное влияние; влияние мезомерного эффекта проиллюстрировано на схеме:
Н апример,
если имеется +М- заместитель (ОМе), то
резонансная структура (580) проявляет
+I-эффект
по отношению к карбоксилат-аниону;
напротив, при наличии –М-заместителя
(NO2),
резонансная структура (581) проявляет
–I-эффект;
естественно, вклад этих резонансных
структур влияет на общий индуктивный
эффект арильного радикала.
апример,
если имеется +М- заместитель (ОМе), то
резонансная структура (580) проявляет
+I-эффект
по отношению к карбоксилат-аниону;
напротив, при наличии –М-заместителя
(NO2),
резонансная структура (581) проявляет
–I-эффект;
естественно, вклад этих резонансных
структур влияет на общий индуктивный
эффект арильного радикала.
Влияние мезомерных эффектов заместителей в ароматическом ядре на кислотность, разумеется, намного меньше, чем их влияние на стабильность ионных интермедиатов (например, -аддуктов), но оно все же заметно. Если сравнить кислотность мета- и пара-метоксибензойных кислот, то первая из них сильнее бензойной (рКа=4,07), а вторая - слабее (рКа=4,45), потому что мета-ОМе-группа проявляет только –I-эффект, а пара-ОМе-группа проявляет –I- и +М-эффекты, и донорный мезомерный эффект оказывает большее влияние, чем акцепторный индуктивный. пара-Нитробензойная кислота (рКа=3,36) сильнее мета-нитробензойной (рКа=3,46), потому что в пара-изомере к сильному –I- эффекту группы NO2 прибавляется –М-эффект; разумеется, обе эти кислоты заметно сильнее бензойной.
Сравнение степеней диссоциации замещенных бензойных кислот позволяет количественно оценить электронное влияние мета- и пара- заместителей в бензольном ядре на протекание различных реакций производных бензола. Электронное влияние орто-заместителей оценить намного труднее, т.к. эти заместители находятся рядом с реакционным центром и оказывают пространственное влияние; разделить электронное и пространственное влияние весьма сложно. Количественная оценка электронных эффектов заместителей весьма важна, т.к. позволяет рассчитывать скорости реакций или состояния их равновесия (если оно достигается); она позволяет также сравнивать разные реакции по их чувствительности к электронным эффектам.
![]() Для
количественной оценки (точнее для
количественного сравнения) влияния
электронных эффектов мета-
и
пара-заместителей
на равновесия или скорости реакций
используется уравнение
Гаммета (L.
Hammett,
точнее
читается как Хэммет):
Для
количественной оценки (точнее для
количественного сравнения) влияния
электронных эффектов мета-
и
пара-заместителей
на равновесия или скорости реакций
используется уравнение
Гаммета (L.
Hammett,
точнее
читается как Хэммет):
где К0 – константа равновесия какой-либо реакции для незамещенного производного бензола, К – константа равновесия той же реакции для производного, имеющего мета- или пара-заместитель; k0 и k – аналогичные обозначения констант скорости какой-либо реакции; - константа, характеризующая электронное влияние заместителя на протекание реакции; она носит название константы заместителя; - константа, характеризующая чувствительность реакции к электронному влиянию заместителей – константа реакции.
Если заместитель является донором, константа имеет знак минус, если акцептором – знак плюс; чем сильнее донорное или акцепторное влияние, тем больше абсолютная величина -константы.
Если равновесие смещается вправо или реакция ускоряется донорными заместителями, -константа имеет знак минус, если акцепторными – знак плюс; чем более чувствительна реакция к электронным влияниям заместителей, тем больше абсолютная величина константы.
 Уравнение
Гаммета – типичное корреляционное
уравнение; оно оперирует не абсолютными,
а относительными (сравнительными)
величинами констант заместителей и
констант реакций. Для того, чтобы
определить величины -констант,
необходимо выбрать стандартную
(эталонную) реакцию; в качестве такой
реакции была выбрана диссоциация
замещенных бензойных кислот в воде при
25 оС
(Z –
мета- и
пара-заместители):
Уравнение
Гаммета – типичное корреляционное
уравнение; оно оперирует не абсолютными,
а относительными (сравнительными)
величинами констант заместителей и
констант реакций. Для того, чтобы
определить величины -констант,
необходимо выбрать стандартную
(эталонную) реакцию; в качестве такой
реакции была выбрана диссоциация
замещенных бензойных кислот в воде при
25 оС
(Z –
мета- и
пара-заместители):
Этой реакции была приписана величина , равная 1 (величина, разумеется, положительная, т.к. диссоциация кислот облегчается акцепторами). Тогда
lg К/К0 = ; = lgK – lgK0 = 4,17 – (pKа)z,
где 4,17 – рКа незамещенной бензойной кислоты (Z=H), (рКа)z – рКа кислоты, имеющей заместитель Z.
Поскольку константы диссоциации кислот легко определяются, нахождение величин -констант не составило труда. В частности, (м-ОМе) = +0,10 (акцепторный индуктивный эффект); п-ОМе) = - 0,28 (суммарный донорный эффект); (м-NO2) = +0,71; (п-NO2)= +0,81.
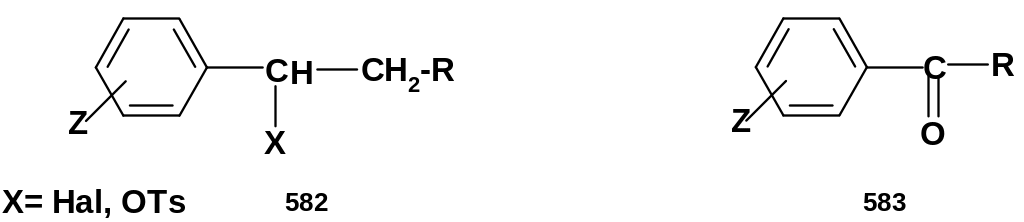 Найденные
значения -констант
применимы к
большому числу разнообразных реакций
субстратов, содержащих арильные
фрагменты, например, бензильных соединений
типа (582)
или ароматических кетонов типа (583):
Найденные
значения -констант
применимы к
большому числу разнообразных реакций
субстратов, содержащих арильные
фрагменты, например, бензильных соединений
типа (582)
или ароматических кетонов типа (583):
Скорости бимолекулярных реакций нуклеофильного замещения (SN2) или бимолекулярного элиминирования (Е2) для субстратов (582) и реакций нуклеофильного присоединения к субстратам (583) прекрасно коррелируют с данными значениями -констант; это позволяет рассчитывать скорости реакции для достаточно большого ряда субстратов, если измерена скорость для двух членов ряда – незамещенного и одного замещенного.
Значения -констант, определенные по величинам констант диссоциации бензойных кислот, применимы, однако, не ко всем реакциям. Для ряда реакций наблюдается корреляция скоростей с другими значениями -констант, которые определяются на базе других стандартных реакций.
Корреляции Гаммета позволяют не только рассчитывать скорости реакций, но и делать заключения о механизмах реакций. Эти корреляции применимы не только к химическим, но и к некоторым физическим свойствам; например, частоты поглощения в ИК спектрах и химические сдвиги сигналов в спектрах ЯМР ароматических субстратов во многих случаях коррелируют с величинами -констант заместителей. Уравнение Гаммета весьма широко используют в органической химии. [Некоторым неудобством при первоначальном знакомстве с уравнением Гаммета является то, что знаки - и-констант противоположны знакам I- и М-эффектов; например, для донорных заместителей -константа имеет знак минус, а электронные эффекты – знак плюс. К этой ситуации надо привыкнуть].
Более подробно варианты корреляционных уравнений и их использование в органической химии рассматриваются в специальных курсах.