

Wohin stellt man den Kolben? (Wasserbad, lebhaft siedend). 2. Wo löst man Chloressigsäure? (Porzellanschale, ge- Täumig). 3. Wohin wird die ätherische Lösung überführt? (Vakuumdestillationsapparat). 4. Wo wird die untere wäßrige Schicht abgetrennt? (Scheidetrichter). 5. Wo löst man Kupfer- acetat? (Wasser, siedend). 6. Wohin überführt man den Niederschlag? (Scheidetrichter, groß). 7. Wo ist Barbitursäure löslich? (Wasser, heiß). 8. Wohin bringt man absoluten Alkohol? (Rundkolben, trocken). 9. Wo muß der Kolben erwärmt werden? (Wasserbad, siedend). 10. Wohin überführt man die Kristalle?
(Reagenzglas, groß). 11. Wo ist die Aminosäure enthalten? (Eiweißstoffe, fast alle). 12. Wohin spannt man den Kolben? (Reagenzglashalter). 13. Wo wird Äthylalkohol mit Schwefelsäure gemischt? (Rundkolben). 14. Wohin gibt man die Säure? (Alkohol). 15. Wo erwärmt man die Lösung? (auf ... Asbestdrahtnetz). 36. Wohin setzt man einen Stopfen? (auf ... Reagenzglas). 17. Wo kühlt man den Rückstand? (unter Wasser, fließend).
Übung 6. Bejahen Sie die folgenden Fragen mit vollständigen Sätzen, wo Begleiterscheinungen bzw. Bedingungen durch die Präposition "unter" ausgedrückt werden.
Muster I. Es entwickelt sich Äthylen. Beobachten wir dabei starke Schaumbildung? — Ja, Äthylen entwickelt sich unter starker Schaumbildung.
1. Die Reaktion setzt ein. Stellen wir dabei erhebliche Erwärmung fest? 2. Es setzt die Verseifung ein. Entwickelt sich dabei Ammoniak? 3. Die Umsetzung verläuft lebhaft. Entwickelt sich dabei Wärme? 4. Die Reaktion setzt ein. Ist dabei lebhaftes Zischen zu hören? 5. Die Reaktion setzt ein. Ist kräftige Ammoniakentwicklung festzustellen? 6. Es setzt die Diazoniumspal- tung ein. Entwickelt sich dabei Stickstoff?
Muster II. Man versetzt die Lösung mit 300 ml Methanol. Muß man dabei kräftig rühren? — Ja, man versetzt die Lösung mit 300 ml Methanol unter kräftigem Rühren.
1. Man versetzt 200 ml Ammoniak mit 5 g Monochloressig- säure. Muß man dabei ständig schütteln? 2. Man löst die Säure. Soll die Säure gelinde erwärmt werden? 3. Man erhitzt das Gemisch. Soll man dabei öfter schütteln? 4. Wir bereiten die Mischung zu. Sollen wir die erwähnten Vorsichtsmaßnahmen beachten? 5. Das Gemisch wird erhitzt. Wird dabei gut gerührt?
Fragment 3. Man entnimmt etwa 2 ml der Reaktionslösung, setzt 2 g Natriumchlorid zu, und läßt die organische Phase sich abscheiden. Hiervon werden 0,4 bis 1 g in einem 200 ml Erlenmeyerkolben mit Schliffstopfen genau eingewogen. Man setzt 10 ml Essigsäureanhydrid und 1 bis 2 g Kaliumjodid zu und nach 10 Minuten 70 ml Wasser. Dann wird I/2 Minute kräftig umgeschüttelt. Das- ausgeschiedene Jod titriert man mit 0,1 n Natriumthiosulfatlösung und Stärke als Indikator
.
Abb.
2.
Aufsatz
für Normaldruck.
Abb.
1.
Destillieraufsatz
nach Ciaisen mit seitlichem Schliff.
Abb.
3.
Anschütz-Thiele-Vor-
stoß.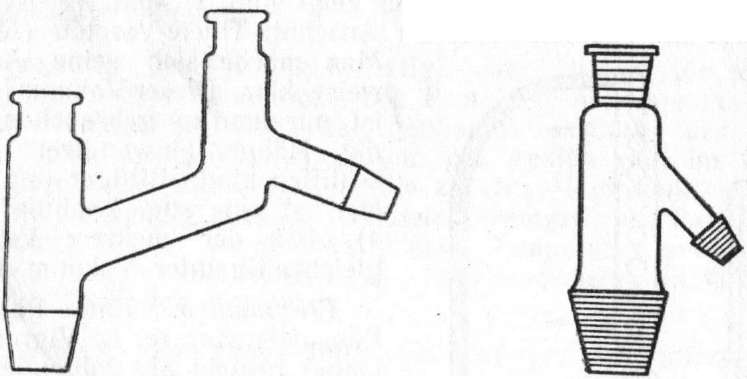
Fragment 4. Als Destillationsblase dient im Laboratorium allgemein der Rundkolben. Zum Beheizen des Kolbens verwendet man Heizbäder. Die Verbindung zwischen Siedekolben und Kühler geschieht durch Destillationsaufsätze. Im Vakuum verwendet man den Claisen-Aufsatz (Abb. 1), der auch bei Destillation unter Normaldruck benutzt werden kann. Einen einfacheren Aufsatz für Normaldruck zeigt Abb. 2. Zum Wechseln der Vorlage im Vakuum dient der Anschütz-Thiele-Vorstoß (Abb. 3).
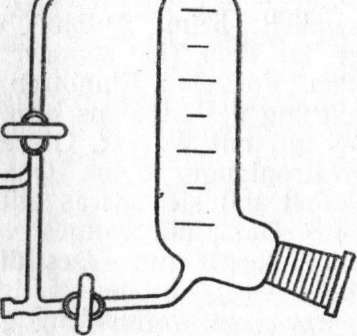
Man mache sich seine Arbeitsweise klar. Dieser VakuumvorstoB ist nur dann zu gebrauchen, wenn die Hähne einwandfrei eing> schliffen sind. Billiger und robuj] ster ist eine sog. "Spinne" (Abb. 4), bei der mehrere Vorlagen gleichzeitig unter Vakuum stehen.
Fragment 5. Eine Rektifikationsapparatur (z. B. Vigreux-Ko- lonne) besteht aus folgenden Teilen (Abb. 5):
1) dem Kolben (Blase) zum Verdampfen der Flüssigkeit ("Sumpf"); 2) der Kolonne;
dem Kolonnenkopf. Hier erfolgt die Messung der Temperatur, die Kondensation des Dampfes und die Teilung des Kondensats in Rücklauf und Destillat;
der Vorlage. Beim Arbeiten inn Vakuum ist eine Einrichtung zum Wechseln der Fraktionen unter Vakuum nötig (Anschütz-Thiele- Vorlage).
Fragment 6. Besondere Auh merksamkeit beim Aufbau der Anlage ist dem Thermometer zu schenken. Vor dem Einführen in die Bohrung des Stopfens benetze man es gut mit Wasser, Glycerin oder Natronlauge. Seine Quecksilberkugel soll sich etwas unterhalb des Dampfüberganges vom Destillierkolben zum Destillierrohr befinden. Um den lästigen Siedeverzug zu vermeiden, gibt man in den Destillierkolben Siedesteinchen.
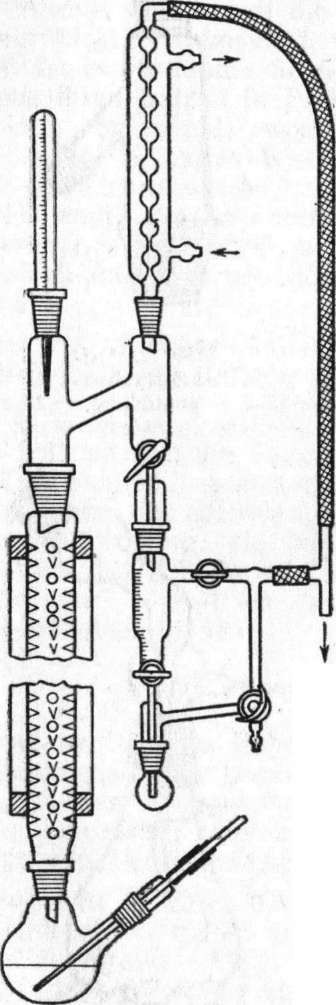
zur
Pumpe
Abb.
5.
Rektifikationsapparatur.
Fragment 8. Im Praktikum fertigt jeder Student für jeden Versuch ein Meßprotokoll an. Die Protokolle sollen enthalten: laufende Nummer des Versuches, genaue Bezeichnung des Versuches, Name des Studenten, Datum der Ausführung, im Kopf des Meßprotokolls alle Angaben, die zu einer Reproduktion des Versuches notwendig sind (verwendete Lösungen und ihre Konzentrationen, Titer, verwendetes Gerät, Temperatur usw.), die Meßergebnisse in tabellierter Form; für die gemessenen Größen werden die Einheiten angegeben.
Fragment 9. Zur Herstellung von Lösungen bekannter Konzentration wird der zu lösende Stoff abgewogen und in einem Meßkolben bei der angegebenen Temperatur auf das geforderte Volumen gebracht. Dabei ist darauf zu achten, daß die abgewogene Menge groß genug ist, um den prozentualen Wägefehler in Grenzen zu halten. Braucht man von einer verdünnten Lösung ein kleines Volumen, so stellt man eine konzentrierte Lösung her, entnimmt mit einer Pipette einen aliquoten Teil und füllt ihn in einem weiteren Meßkolben wieder auf. Dadurch kann man leicht eine Verdünnung auf das Zehn- bis Hundertfache erreichen.
Fragment 10. Eine der wichtigsten elektrochemischen Eigenschaften, die mit der Ionenbildung verbunden ist, ist die Fähigkeit von Elektrolytlösungen, den elektrischen Strom merklich zu leiten. Der Widerstand R oder davon abgeleitete Größen können Aufschluß geben über Dissoziationsgrad, Beweglichkeit und Konzentrationen der Ionen. Der Widerstand von 'Elektrolytlösungen wird wie üblich mit einer Brückenschaltung gemessen. Hier wird das Gefäß mit den Elektroden und der Lösung, ein bekannter fester Widerstand R3 und ein bekannter Widerstand, der durch einen Abgriff in die beiden Teilwiderstände Ri und R2 geteilt werden kann, mit der Stromquelle geschaltet. Nun wird das Verhältnis von Ri zu R2 so lange geändert, bis im Brückenzweig kein Strom mehr fließt bzw. ein Stromminimum vorliegt.
97
Fragment 12. Ein anderes Verfahren, bei dem der Kette prak- ' tisch kein Strom entzogen wird, beruht darauf, daß man der zu messenden EMK eine variable, in jedem Fall genau angebbare Spannung entgegenschaltet und diese so einreguliert, daß in dem betreffenden Stromkreis kein Strom fließt.
Fragment 13. Man bereitet eine Suspension von 2 g Kar-J toffelstärke in 100 ccm Wasser, gibt davon 20ccm in das Ostwald-Viskosimeter und mißt die Durchiaufzeit, indem man häufiger Luft durchbäst, um ein Sedimentie-1 ren zu verhüten.
Man beginnt bei 20°, steigert auf 30°, dann 40° und 50° und mißt jedesmal nach Erreichen der Temperaturkonstanz die Auslaufzeit. Nun steigert man langsamer und erhält bei 62° einen plötzlichen steilen Anstieg der inneren Reibung. Gleichzeitig wird die Suspension heller und durchsichtiger. Die erhaltenen Werte trägt man graphisch auf.
Fragment 14. Um die Ladung! einer Membran zu bestimmen, wird das Gerät der Abb. 6. benutzt. Beil M wird die Membran zwischen diel Glasrohre eingespannt. Die beiden Schenkel des £/-Rohres werden min Wasser oder Elektrolyt gefüllt und an die Platinelektroden P und Q eine Gleichspannung von 50—200 Volt angelegt. Das Wasser wird in dem einen Schenkel steigen. Ist die Membran negativ geladen, so steigt der Elektrolyt im rechten Schenkel bei К. Ist sie positiv geladen, dann steigt das Niveau im linken Rohr.
TEXTE
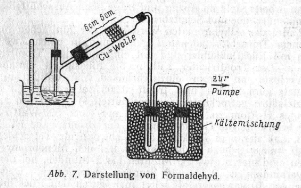
TEXT 1. DARSTELLUNG VON FORMALDEHYD (METHANAL)
Chemikalien
50 ml Methanol Kupferwolle oder -draht Eis-Kochsalz-Mischung
Reaktion: CH3OH+1/202 → H*CH0+H20.
Kupfer dient der Dehydrierung; der Wasserstoff wird sofort zu Wasser oxydiert.
Durchführung: Der Aufbau der Apparatur ist aus Abb. 7 ersichtlich. Man verwendet einen 100-ml-Destillierkolben, dessen seitliches Ansatzrohr im Winkel nach oben gebogen und zu einer Spitze von 1 ... 1,5mm ausgezogen wird. Sie ragt in ein Verbrennungsrohr, das eine 5 ... 6cm lange Schicht Kupferwolle oder ein Kupferdrahtnetz enthält. Es soll sich etwa 6 cm hinter der Spitze befinden. Als Vorlage dienen große Reagenzgläser, die man, wie im Bild gezeigt, mit einer Wasserstrahlpumpe verbindet und in eine Eis-Kochsalz-Mischung stellt. In den Destil lierkolben gibt man die angegebene Menge Methanol, erhitzt im Wasserbad, dessen Temperatur ständig zwischen 46 ... 47°C gehalten wird, und saugt einen mäßigen Luftstrom durch den Alkohol. Dann erhitzt man die Kupferwolle zur beginnenden Rotglut, bis ein Aufglühen den Beginn der Reaktion anzeigt. Weiteres Erwärmen ist unnötig, weil die Umsetzung unter starker Wärmeentwicklung verläuft. Der Luftstrom ist so zu regulieren, daß das Kupfer in ständiger Rotglut bleibt. Die Reaktionsprodukte sammeln sich in den Vorlagen. Man erhält eine wäßrige Lösung des Formaldehyds, die 30 ... 32%ig ist. Zur Beendigung der Reaktion löst man zunächst die Verbindung zum ersten Reagenzglas und stellt dann die Wasserpumpe ab.
Ausbeute: 45 ... 55 ml Formaldehydlösung; Dauer: 30 . .. 50 Minuten.
Daten
Molekulargewicht Dichte, g/ml F, °C Kp, °C
Methylalkohol . . . 32,04 0,7923 -97,1 64,7
Formaldehyd . . . 30.03 0,815 -92 -21
Charakteristik: Formaldehyd ist ein stechend riechen- j des Gas, das sich leicht in Wasser löst — ein Liter Wasser löst bis zu 400 Liter gasförmigen Formaldehyd. Der handelsübliche I Formaldehyd, auch Formalin genannt, enthält 30... 40%, des j Gases. Maximal kann man eine 55% ige Lösung herstellen. Form-1 aldehyd ist ein wichtiger Grundstoff der chemischen Industrie, | Man benötigt ihn für die Gewinnung von Kunststoffen und Arz- I neimitteln.
TEXT 2. ALLGEMEINE ARBEITSVORSCHRIFT FÜR DIE PHOTOBROMIERUNG VON ALKYLAROMATEN IN DER SEITENKETTE
Man arbeite stets im Abzug und trage beim Ausschütteln usw. jj Gummihandschuhe und Schutzbrille.
0,2 Mol des Alkylaromaten werden in der fünffachen Menge ] trockenem Tetrachlorkohlenstoff gelöst und in einen Zweihals- I kolben mit Rückflußkühler und gut eingespanntem Tropftrichter | gegeben. Der Tropftrichter soll in die Flüssigkeit eintauchen, um Verluste an Brom einzuschränken. Man erhitzt auf dem Drahtnetz zum Sieden und tropft 0,205 Mol vorher mit konz. j Schwefelsäure getrocknetes (ausgeschütteltes) Brom pro zu er-'i setzendes H-Atom zu. Dabei wird mit einer 500-Watt-Photo-B lampe bestrahlt. Die Geschwindigkeit der Bromzugabe ist so zu j regeln, daß der vom Rückflußkühler abtropfende Tetrachlorkoh-Я lenstoff immer nahezu farblos bleibt. Bei den Monobromverbin-J düngen sind hierzu etwa 30 Minuten bis 2 Stunden, bei den Di-J bromverbindungen 2 bis 10 Stunden erforderlich. Den ent-Я weichenden Bromwasserstoff leitet man durch den Rückflußkühe ler, der einen durchbohrten Stopfen mit einem Glasrohr und Gummischlauch trägt, in einen zur Hälfte mit Wasser gefüllten Erlenmeyer-Kolben. Das Einleitungsrohr soll dabei nicht ein-B tauchen, sondern etwa 1 cm über der Wasseroberfläche enden (warum?). Die entstehende verdünnte Bromwasserstoffsäure wird durch Destillation über eine kurze Kolonne gewonnen: Man fängt die bei /СР.7бо126°С azeotrop siedende 48%ige Bromwas-* serstoffsäure auf. Nach Beendigung der Reaktion unterbricht man die Bestrahlung. Sofern ein festes Produkt zu erwarten ist, gießt man die heiße Lösung sofort in einen Erlenmeyer-Kolben, läßt .auskristallisieren, wenn nötig im Kühlschrank, und reinigt durch Umkristallisation.
TEXT 3. ALLGEMEINE ARBEITSVORSCHRIFT FÜR DIE ADDITION j VON BROM AN OLEFINE
In einem Dreihalskolben mit Rührer, Tropftrichter und Innenthermometer wird das Olefin in der 2- bis 3-fachen Menge Tetra«
chlorkohlenstoff oder Chloroform auf 0°C gekühlt. Bei 0 bis 5°C tropft man die äquimolare Menge Brom in etwa dem doppelten Volumen des gleichen Lösungsmittels unter gutem Rühren so zu, daß die Temperatur in den angegebenen Grenzen gehalten wird und keine größere Konzentration unverbrauchten Broms auftritt (Farbe!). Das Additionsprodukt fällt aus und wird abgesaugt, andernfalls destilliert man das Lösungsmittel ab und reinigt den Rückstand durch Destillation oder Umkristallisation.
TEXT 4. DARSTELLUNG VON BENZOPHEN'ON
In einen 1-1-Dreihalskolben mit Rührer, Tropftrichter und Kühler mit Chlorcalziumrohr bringt man 1,5 Mol trockenen Tetrachlorkohlenstoff und 0,3 Mol Aluminiumchlorid guter Qualität. Man kühlt auf 10 bis 15 °C und gibt von einer Gesamtmenge von 0,7 Mol Benzol 2 ml (auch bei größeren Ansätzen nicht mehr) auf einmal zu. Nachdem die Reaktion auf diese Weise zum Anspringen gebracht wurde, kühlt man auf 5 bis 10 °C und tropft den Rest des Benzols bei dieser Temperatur zu (genau einhalten!). Nach Beendigung der Zugabe wird noch drei Stunden bei 10°C gerührt und dann über Nacht bei Raumtemperatur stehen gelassen.
Der Rückflußkühler wird durch einen absteigenden Kühler ersetzt. Dann gibt man durch den Tropftrichter vorsichtig 250 ml Wasser zu, wobei nicht gekühlt zu werden braucht, da die Halogenverbindung ohnehin verseift werden soll. Der überschüssige Tetrachlorkohlenstoff wird abdestilliert und anschließend 30 Minuten Wasserdampf durch die Lösung geleitet, wobei das Dihalogenid verseift wird. Nach dem Abkühlen wird die organische Schicht abgetrennt, die wäßrige Schicht nochmals mit Benzol extrahiert, die vereinigten Schichten werden mit Wasser gewaschen und über Magnesiumsulfat getrocknet. Nach Abde- stillieren des Lösungsmittels fraktioniert man i. V. Kp.15 190°C; F. 48 °C; Ausbeute 65%.
TEXT 5. NEUTRALISATIONSENTHALPIE
A u f g a b e n. a) Bestimmen Sie experimentell:
die Temperatur der Kalorimeterflüssigkeit in Abhängigkeit von der Zeit bei Zufuhr einer bestimmten Wärmemenge auf elektrischem Wege;
die Temperatur der Kalorimeterflüssigkeit in Abhängigkeit von der Zeit bei der Neutralisation von 10 ml 1„ Salzsäure.
b) Berechnen Sie:
die Wärmekapazität С des Kalorimeters;
die Neutralisationsenthalpie für die Neutralisation von "Salzsäure mit Natronlauge.
Grundlagen des Versuches:
Zur Bestimmung von Enthalpieänderungen in Lösungen werden offene Kalorimeter verwendet, die in der Regel aus einem- Dewargefäß oder aus zwei durch einen Luftmantel getrennten Bechergläsern, ferner einem Beckmann-Thermometer und einem- Rührer bestehen. Die Reaktionsenthalpie für die Reaktion
NaOH + HCl NaCl + H30 wird als Neutralisationsenthalpie ЛДН bezeichnet.
Bei der Neutralisation starker Basen mit starken Säuren- findet man für ArH praktisch immer den gleichen Wert von 13,7 kcal/mol, wenn die gemessenen Werte um die Verdünnungsenthalpie korrigiert sind. Die Neutralisationsenthalpie ist also> -von der Natur der reagierenden Stoffe unabhängig. Der gemessene Wert entspricht der Reaktion der Wasserstoff- und der Hydroxidionen zu Wasser:
H+ + OH" -> H20; Д/?Н = —13,7 kcal/mol, worin das Wesen jeder Neutralisation besteht.
Der genannte Wert wird nur bei Reaktionen zwischen Säuren> und Basen gemessen, die vollständig dissoziiert sind. Bei schwachen Säuren bzw. Basen setzt sich der experimentell bestimmte- Wert additiv aus der Neutralisationsenthalpie und den Enthalpieänderungen zusammen, die sich aus der Änderung des Disso- ziations-, Assoziations- und Hydratationsgrades der Moleküleergeben.
Jeder Enthalpiebestimmung geht die Eichung des verwendeten Kalorimeters voraus. Hierbei wird die vom Kalorimeter aufgenommene Wärmemenge q berücksichtigt und die Wärmekapazität С des Kalorimeters bestimmt.
? = С.АГ.
Die Wärmekapazität С erhält man in dem vorliegenden Versuch, indem man dem Kalorimeter auf elektrischem Wege Wärmezuführt und die dazugehörige Temperaturänderung AT mißt. Die zugeführte Wärmemenge ergibt sich bei Kenntnis des Widerstandes R des Heizdrahtes und der Stromstärke / bzw. der Spannung U und der Stromstärke / sowie der Betriebszeit t der Heizung aus dem Produkt der elektrischen Arbeit und dem elektrischen Wärmeäquivalent nach
q — 0,239t/-l-t bzw. q = 0,239/? P t.
3. Durchführung des Versuches
Geräte: PVC-Behälter für das Dewargefäß, Dewargefäß (600 ml), Bechmann-Thermometer, Lupe zum Ablesen des Thermometers, Heizung (Widerstandsdraht, R — 3,5 Ohm, Zuleitungen aus Kupfer), Rührer, Stromversorgungsgerät, 2 Vielfachmeßgeräte, Einlaufpipette, 210-ml-Pipetten.
Reagenzien: In Salzsäure, In Natronlauge.
Arbeitsablauf. a) Eichung des Kalorimeters. Das Kalorimeter wird nach Abb. 8 zusammengesetzt und:

das Dewargefäß mit 300 ml Wasser gefüllt. Danach werden die Heizung, der Schalter und das Amperemeter in Reihe geschaltet und an eine Gleichstromspannunsquelle von 10V angeschlossen. Nach Einschaltung des Rührers wird die Temperatur alle 30 Sekunden abgelesen. Beträgt die Temperaturdifferenz zwischen zwei Ablesungen nur 0,002 Grad (Beendingung der Vorperiode), schließt man den Stromkreis. Unter dauerndem Rühren beobachtet man die Hauptperiode und liest zugleich das Amperemeter genau ab. Ist ein Temperaturanstieg von etwa 0,5 Grad erfolgt, dann wird •der Stromkreis unterbrochen (Ende der Hauptperiode). Die Temperaturbeobachtung wird fortgesetzt, bis ein konstanter Temperaturgang festgestellt wird (Nachperiode). Es werden zwei Meßreihen aufgenommen.
b) Bestimmung der Neutralisationsent- h а 1 p i e AH für die Neutralisation von Salzsäure mit Natronlauge. Das Dewargefäß wird mit 280 ml Wasser und 10ml 1„ Natronlauge gefüllt. In die Einlaufpipette werden genau 10 ml
Salzsäure pipettiert. Die Einlaufpipette wird zusammen mit der Heizung, dem Thermometer und dem Rührer in das Kalorimetergefäß eingesetzt. Dabei ist darauf zu achten, daß die Auslauföffnung oberhalb des Flüssigkeitsspiegels des Kalorimeters verbleibt. Nach dem Zusammenbau der Apparatur wartet man eine gewisse Zeit, bis sich die Temperatur im Kalorimeter ausgeglichen hat. Erst dann beginnt man mit der Ablesung des Temperaturganges. Die Reaktion wird ausgelöst, indem man die Säure quantitativ durch Einblasen in das Kalorimetergefäß befördert. Die Temperatur wird alle 30 Sekunden abgelesen. Es werden zwei Meßreihen aufgenommen.
4. Auswertung.
Gang der Berechnung, a) Ermittlung von AT. Bei der graphischen Auswertung wird der gesamte protokollierte Temperaturgang gegen die Zeit t abgetragen (Ordina- tenachse umfaßt etwa 2 Grad). Eine durch die Meßpunkte der Vorperiode zu zeichnende Gerade wird in Richtung auf wachs ende Zeitwere verlängert. Analog wird der Gang der Meßpunkte der Nachperiode linear nach rückwärts extrapoliert, und man erhält zwei leicht gegen die Horizontale geneigte Hilfsgeraden. Zwischen beiden wird die Senkrechte in dem Zeitpunkt t errichtet, dem die Hälfte der gesamten Temperaturänderung entspricht. Die Strecke zwischen beiden Schnittpunkten ist AT.
Ermittlung der Wärmekapazität. Die Wärmekapazität С erhält man, indem man die zugeführte Wärmemenge nach der Gleichung 9=0,239 R • I2 • t berechnet und zusammen mit der aus der graphischen Darstellung ermittelten Temperaturdifferenz AT in die Gleichung q=C-AT einsetzt.
Ermittlung der Neutralisationsenthalpie. Die bei der Neutralisation von 10 ml In HCl freiwerdende Wärmemenge qp bei konstantem Druck ergibt sich nach der Gleichung aus der berechneten Wärmekapazität und der beobachteten Temperaturdifferenz AT. Es gilt
![]()
wobei АдЬ die auf eine beliebige Masse bezogene Reaktionsenthalpie ist und n die Zahl der umgesetzten Mole angibt. ArH ist mit negativem Vorzeichen zu verwenden, da die Wärmemenge nun nicht mehr als die vom Kalorimeter aufgenommene, sondern als die vom "System Reaktionsgemisch" abgegebene betrachtet wird. Von diesem Wert ist noch die Verdünnungsenthalpie von etwa —200 cal/mol abzuziehen.
TEXT 6. KONDUKTOMETRIE
Aufgaben, a) Bestimmen Sie experimentell:
den Leitwert G einer HCl-Lösung in Abhängigkeit von der Menge der zugesetzten 0,1 n NaOH.
Zeichnen Sie:
das konduktometrische Titrationsdiagramm G—f(V) für die durchgeführte Leitfähigkeitstitration.
Ermitteln Sie:
den Äquivalenzpunkt für die Titration.
Grundlagen des Versuches.
Die konduktometrische Titration beruht auf der Änderung der Leitfähigkeit der zu titrierenden Lösung, wenn infolge des Zusatzes der Titerlösung ein schnell wanderndes Ion durch ein langsamer wanderndes ersetzt wird. Besonders gut lassen sich starke Säuren und Basen konduktometrisch bestimmen, weil in diesem Falle durch den Zusatz von Titerlösung das schnell wandernde Wasserstoffion oder Hydroxylion durch das viel langsamer wandernde Kation oder Anion des sich bildenden Salzes ersetzt wird.
Wird eine starke Säure mit einer starken Lauge titriert, so nimmt die Leitfähigheit bis zum Äquivalenzpunkt proportional der zugesetzten Lauge ab; ton hier ab läßt die Vergrößerung der Zahl der OH-Ionen die Leitfähigkeit proportional der Laugenmenge wieder zunehmen.
Diese Verhältnisse werden durch Abb. 9 veranschaulicht, in der als Abszisse die Anzahl cm3 der zugefügten Base, als Ordinate die gemessene Leitfähigkeit aufgetragen sind. Die erste Gerade AB setzt sich zusammen aus der Leitfähigkeit der noch vorhandenen Säure (AF) und der des gebildeten Salzes (OB).
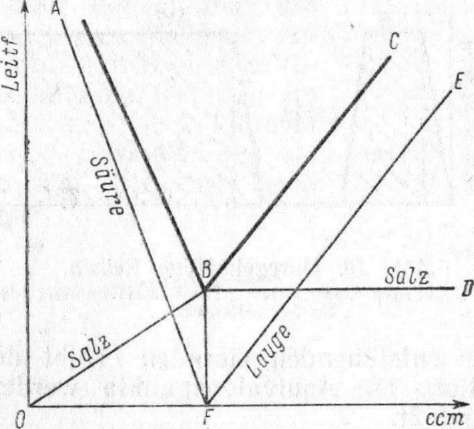
Nach dem Erreichen des Neutralitätspunktes erhält man die gemessene Leitfähigkeit FB des Salzes, vermehrt um die Leitfähigkeit FE des Luagenüberschusses. Der Schnittpunkt der Geraden AB und ВС ergibt also den Neutralitätspunkt. Er ist um so ausgeprägter, je stärker die verwendete Säure und Base sind, und wird besonders deutlich erhalten, wenn man zur Titration eine ca. 10 — bis 100 mal so konzentrierte Basenlösung benutzt wie die zu titrierende Säurelösung, da sich dann die Konzentrationsverhältnisse durch den Zusatz der Titerlösung am wenigsten ändern. Es empfiehlt sich daher, kleine Mengen einer möglichst konzentrierten Zugabelösung und dementsprechend in Vjoo cm3 geteilte Büretten zu verwenden.
3. Durchführung des Versuches.
Zubehör. a) Geräte: Leitfähigkeitsmeßgerät, zwei Elektroden, 25-ml-Mikrobürette, 100-ml-MeßzyIinder, 5-ml-Pipette, 250-ml-Becherglas, Glasstab, Spritzflasche.
b) Reagenzien: 0,1 n Salzsäure, 0,1 n Natronlauge.
Arbeitsablauf. 5 ml 0,1 n Salzsäure werden in die Leitfähigkeitsmeßzelle (250-ml-Becherglas mit Tauchelektrodenpaar) gegeben, mit etwa 100 ml destilliertem Wasser verdünnt, und der Leitwert G der Lösung wird gemessen. Nun wird aus einer Mikrobürette 1 ml 0,1 n Natronlauge hinzugegeben, die Lösung umgerührt und der Leitwert G erneut gemessen. Dieses wird wiederholt, bis man 10 ml Maßlösung zugesetzt hat.
Auswertung.
7.SW 2.5
W
Abb.
10.
Unregelmäßige
Reihen.
с —
Konzentration des flockenden Elektrolyten:
I. SW-erster Schwellenwert; 2-SW
— zweiter
Schwellenwert.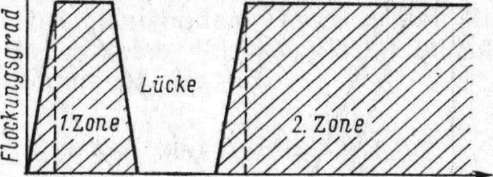
Gang der Berechnung. Den Leitwert G trägt man als Ordinate, die zugegebene Maßlösung als Abszisse auf. Der Schnittpunkt der entstehenden Geraden ergibt den Äquivalenzpunkt der Titration. Die Äquivalenzpunkte werden in einer Tabelle zusammengefaßt.
TEXT 7. UNREGELMÄßIGE REIHEN
Die unregelmäßige Reihen beobachtet man bei der Koagulation von verdünnten Solen mit mehrwertigen Ionen. Sie bedeuten, daß bei allmählich ansteigender Koagulatorkonzentration с die Flockung statt zunehmen an einer Stelle abnimmt. Dies ergibt zwei Flockungszonen (Abb. 10).
Hierzu kann ein verdünntes 0,05%iges Mastixsol dienen oder Eisenoxydsol. Das erste flockt man mit Aluminiumchlorid, das zweite mit Na2HP04. Bei zunehmender Elektrolytkonzentration nimmt die Flockung zunächst zu. Von einer bestimmten Konzentration an aber tritt keine Flockung mehr ein. Bei weiterer Zunahme der Elektrolytmenge erfolgt dann wieder Flockhung. Diese unregelmäßige Reihe der Flockung kann außer an Eisenoxydsol mit Na2HP04 oder an Mastixsol mit A1C13 auch an anderen Solen demonstriert werden.
Das Mastixsol wird erhalten, indem man 10 ccm 5%iger alkoholischer Mastixlösung in 90 ccm Wasser gießt, dann auf das zehnfache verdünnt und filtriert. Aus einem 10 ccm Meßglas gibt man von 10 ccm molarer Aluminiumchloridlösung 5 ccm in da$ erste Glas mit dem Sol, füllt das Meßglas mit Wasser auf 10 ccm auf, entnimmt wieder 5 ccm und so fort.
TEXT 8. QUELLUNGSGESCHWINDIGKEIT
Um die Geschwindigkeit der Quellung zu messen, gibt man in ein Meßglas 5 g körnige Gelatine und füllt mit Wasser auf 100 ccm auf. Nun notiert man die Zeit und wendet das Meßglas von Hand häufig um. Man liest das Volumen der gequollenen Gelatine im Meßglas anfangs nach je 2 Minuten und später nach je 10 Minuten ab. Die Endwerte bestimmt man nach einigen Stunden.

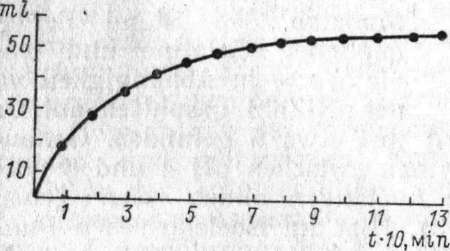
Abb. 11. Geschwindigkeit der Quellung. Abb. 12. Graduiertes.
У—Volumen des quellenden Stoffes; Zeit in dünnwandiges Rohr zur
Minutenxio. volumetriscnen Messung