

2.Сущность наиболее широко используемых методов
2.1.Электрохимические методы анализа
Электрохимические методы анализа основаны на использовании ионообменных или электронообменных процессов, протекающих на поверхности электрода или в приэлектродном пространстве. Аналитическим сигналом служит любой электрический параметр (потенциал, сила тока, сопротивление, количество электричества и др.), функционально связанный с составом и концентрацией раствора. Электрохимические методы характеризуются высокой чувствительностью, низкими пределами обнаружения, широким интервалом определяемых содержаний, простотой и невысокой стоимостью аппаратуры.
2.1.1. Потенциометрия
Потенциометрический метод анализа основан на измерении электродвижущей силы (ЭДС) обратимых гальванических элементов. Гальванический элемент состоит из двух электродов: индикаторного и электрода сравнения, погруженных в один раствор (цепь без переноса) либо в два различающихся по составу раствора, связанных жидкостным контактом (цепь с переносом). В потенциометрии используют два класса индикаторных электродов.
1.Электроды, на межфазных границах которых протекают электронообменные процессы. Функционирование таких электродов основано на зависимости равновесного потенциала от состава и концентрации исследуемого раствора, описываемой уравнением Нернста:
![]()
где Е – равновесный потенциал; Е0 – стандартный потенциал, равный равновесному, если активность всех участвующих в электрохимической реакции компонентов равна единице; n – число электронов, участвующих в полуреакции; аОХ и аRed – активности окисленной и восстановленной форм.
В основном это активные металлические электроды (серебряный, медный, кадмиевый и др.) и инертные электроды (платиновый, золотой).
2. Электроды, на межфазных границах которых протекают ионообменные процессы – ионоселективные электроды (ИСЭ).
Потенциал системы, состоящей из внешнего электрода сравнения и ИЭС, погруженных в исследуемый раствор, описывается модифицированным уравнением Нернста (уравнение Никольского - Эйзенмана):
![]()
где const – константа, зависящая от значений стандартных потенциалов внутреннего и внешнего электродов сравнения и от природы мембраны ИСЭ; ai и zi, ak и zk – активности и заряды основного (потенциалопределяющего) и постороннего ионов соответственно; Ki/kпот – потенциометрический коэффициент селективности ИСЭ по отношению к потенциалопределяющему иону (i) в присутствии постороннего иона (к).
В качестве электрода сравнения в практической работе используют хлорид-серебряный или каломельный электроды. Их потенциалы постоянны и не зависят от состава раствора.
Различают прямую потенциометрию (ионометрию) – непосредственное измерение равновесного потенциала и нахождение активности ионов в растворе – и косвенную – потенциометрическое титрование – регистрация изменения потенциала в процессе химической реакции между определяемым веществом и титрантом. В потенциометрическом титровании используют кислотно-основные, окислительно-восстановительные реакции, а также осаждение и комплексообразование. Индикаторный электрод выбирают в зависимости от типа химической реакции и природы потенциалопределяющих ионов.
Прямая потенциометрия (ионометрия). При работе с ионоселективным электродом необходима его предварительная калибровка – установление зависимости между потенциалом электрода и активностью или концентрацией определяемых ионов.
Для измерения ЭДС гальванических элементов с ИСЭ наиболее часто используют электронные вольтметры с высоким входным сопротивлением (иономеры, рН - метры) различных марок.
Используя стандартные растворы определяемых соединений, получают градуировочный (калибровочный) график в координатах Е – lg ai (E – lg Ci), при этом считается, что коэффициент активности изучаемого иона либо известен, либо может быть легко вычислен.
Распространен и универсален метод постоянной ионной силы, в соответствии с которым к растворам потенциалопределяющего компонента добавляют избыток индеферентного электролита, что создает постоянную ионную силу как в стандартных, так и в исследуемых растворах. В этом случае можно использовать графическую зависимость Е – lg Ci.
По данным калибровки определяют следующие электрохимические характеристики:
1. Нернстовскую область электродной функции – интервал прямолинейной зависимости потенциала от активности (концентрации) потенциалопределяющих ионов.
2. Крутизну электродной функции – угловой коэффициент наклона градуировочного графика Е – lg Ci.
3. Предел обнаружения потенциалопределяющего иона (Сmin), для этого можно использовать два приема:
а) экстраполируют прямолинейные участки зависимости Е – lg Ci; полученная точка пересечения соответствует на оси абсцисс величине Cmin;
б) на экстраполированном линейном участке электродной функции находят точку, отстоящую от экспериментальной кривой на 18/z мВ (Cmin в ионометрии соответствует концентрация, для которой отклонение от нернстовской зависимости составляет 59/z lg 2, т.е. 18z мВ, где z – заряд потенциалопределяющего иона). В случае отклонения крутизны электродной функции от теоретической величины для определения Сmin используют значение, экспериментально найденное из градуировочного графика.
4. Время отклика ИСЭ – время достижения стационарного потенциала.
5. Селективность электрода относительно определяемого иона в присутствии посторонних ионов.
Потенциометрическое титрование. Потенциометрическое титрование основано на регистрации изменения потенциала индикаторного электрода в процессе химической реакции между определяемым компонентом и титрантом. Конечную точку титрования находят по скачку потенциала, отвечающему моменту завершения реакции. Из экспериментальных данных, записанных в виде таблицы (Vтитранта – Е), можно построить кривую титрования в интегральной форме или в виде первой и второй производной и найти конечную точку титрования (к.т.т.) графически (рис 2.1.).
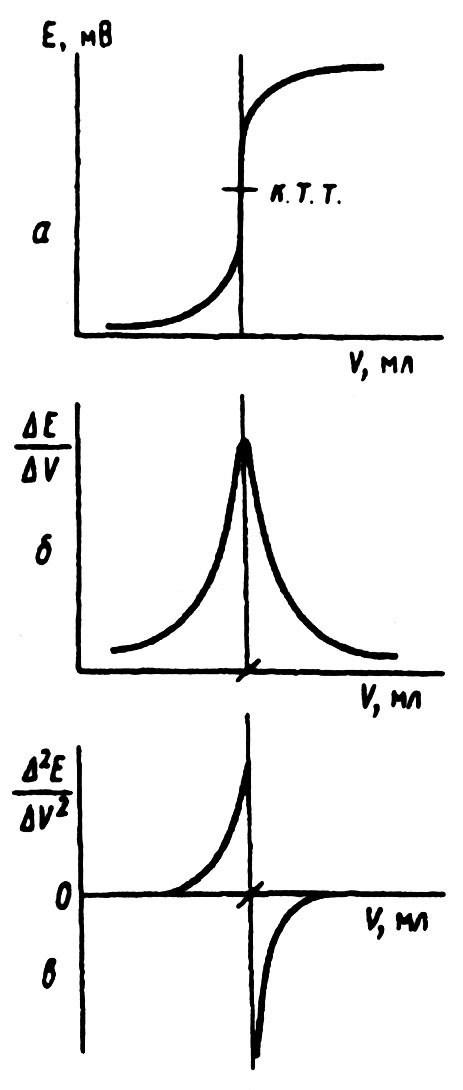
Рис.2.1. Графический способ нахождения конечной точки титрования:
а- интегральная кривая; б- первая производная; в- вторая производная.
Во всех этих случаях полагают, что кривая титрования симметрична относительно точки эквивалентности, поскольку за конечную точку принимают точку максимального наклона кривой. Если скачок титрования большой, то погрешность при выполнении этого допущения невелика. На практике проводят две параллельные касательные к пологим верхней и нижней частям кривой и соединяют их прямой таким образом, чтобы точка пересечения ее с кривой титрования делила эту прямую на две равные части (точка к.т.т. на рис 2.1, а). Опускают перпендикуляр из этой точки на ось абсцисс (ось V – объем титранта) и получают объем титранта, отвечающий к.т.т. Более точным способом нахождения к.т.т. является графическое изображение зависимости величин первой производной от объема (рис. 2.1, б); максимум на этой кривой соответствует к.т.т. В случае асимметричных кривых титрования или при малом скачке потенциала следует пользоваться второй производной (рис. 2.1, в).
Потенциометрическое титрование позволяет проводить количественное определение в темно-окрашенных и мутных растворах, а также в смесях.