

7.3. Энергетический обмен мозга при болезни Альцгеймера
Нарушения энергетического обмена играют существенную роль в развитии болезни Альцгеймера (БА) - атрофического заболевания головного мозга, возникающего в позднем возрасте и приводящего к тотальному слабоумию. Болезни свойственно постепенное мало заметное начало в возрасте 40-90, но чаще после 65 лет, относительно медленное, но неуклонно прогредиентное течение с постепенным снижением памяти, распадом высших психических (когнитивных) функций и психической деятельности в целом (G. McKhann et al., 1984; С.И. Гаврилова с соавт., 1992). Патоморфологически БА характеризуется атрофией и аномально высокой плотностью сенильных бляшек и нейрофибриллярных клубков (Z.S. Khatchaturian, 1985).
БА является гетерогенным заболеванием, различные формы которого отличаются по клинической картине, генетике и особенностям патогенеза. Выделяют БА с ранним (в возрасте до 65 лет) и поздним началом (в возрасте 65 лет и старше), которым по ранее существовавшей отечественной классификации соответствовали болезнь Альцгеймера и сенильная деменция альцгеймеровского типа (СДАТ) (С.И Гаврилова с соавт., 1992). БА с ранним началом называют также БА пресенильного типа.
Этиопатогенез БА сложен и до конца не выяснен, однако не вызывает сомнения, что в развитии заболевания ведущими являются генетический и возрастной факторы. Их влияние на патогенез БА в значительной мере опосредуется изменениями церебрального энергетического обмена.
7.3.1. Роль изменений церебрального энергетического обмена в патогенезе болезни Альцгеймера
Нормальное старение мозга сопровождается легким, но постоянным снижением образования энергии в мозге вследствие из
140
менения работы различных систем, участвующих в церебральном энергообеспечении. Возникает уменьшение мозгового кровотока, повреждается ГЭБ, снижается метаболизм глюкозы и О2, наблюдаются нарушения работы митохондрий в нервных и глиальных клетках. В изменении метаболизма глюкозы важную роль играет десенситизация нейрональных инсулиновых рецепторов, которая особенно усиливается при стрессе под влиянием кортизола с последующей дисфункцией этих рецепторов в следствие изменения структуры и функции мембран. Снижение аэробного метаболизма глюкозы и относительное преобладание гликолиза вызывают внутриклеточный нейрональный ацидоз. Закисление способствует интенсификации свободно-радикального окисления, повреждающего мембраны, ядро и другие структуры нейронов (J. Bralet et al., 1991; S. Rehncrona et al., 1989; цит по: E. Roberts, T. Sick, 1996). Значительное снижение рН запускает механизмы апоптоза – запрограммированной клеточной смерти (D. Ding et al., 2000). Подробнее эти процессы описаны в главе 5.
Особенно значительные нарушения энергетического метаболизма у пожилых людей наблюдаются при стрессе. При этом активируются запасные пути энергетического метаболизма, связанные с использованием кетоновых тел в качестве энергетического субстрата, дополнительно усиливается гликолиз, что приводит к ацидозу и усилению свободно-радикальных процессов. Повышение выброса возбуждающего медиатора глютамата при стрессе ведет к накоплению внутриклеточного кальция, под влиянием которого активируются Са-зависимые протеазы, эндонуклеазы, фосфолипазы, вызывающие деградацию важнейших структур нейронов. Цепь перечисленных процессов приводит к нейродегенерации и способствует развитию БА.
Установлено, что болезнь Альцгеймера с ранним началом может быть обусловлена изменением ряда генов - гена белка-предшественника амилоида, расположенного в 21-й хромосоме (A. Goate et al., 1991), гена пресенилин 1, локализованного на 14-й хромосоме (E. Rogaev et al., 1995; R. Sherrington et al., 1995) и гена пресенилин 2, расположенного в 1-й хромосоме (E. Levy-Lahad et al., 1995). Аллель 4 аполипопротеин-Е гена (АРО-Е4), локализованного на 19 хромосоме, фактором риска для БА с поздним началом (A. Saunders et al., 1993).
Различные мутации, с которыми связано развитие БА, в конечном итоге вызывают накопление в мозге бета-амилоидного протеина (А), состоящего из 39-43 аминокислот и являющегося важнейшей составной частью сенильных бляшек. Мутации увеличивают продукцию А или приводят к образованию А c двумя дополнительных аминокислотными остатками. Удлинение А запускает ускоренную агрегацию этого протеина. A представляет собой нейротоксин, вызывающий нейродегенеративные изменения, причем его нейротоксическое действие может со
141
провождаться эпилептической активностью (F. LaFerla, 1995; G. Cole, S. Frautschy, 1996). У клинически здоровых родственников больных БА за много лет до развития заболевания в крови обнаружено повышение уровня A (N.R. Graff-Radford et al., 1998). Предполагается, что клиническая симптоматика манифестирует, когда A начинает откладываться в мозге. А значительно усиливает нейротоксическое действие возбуждающего медиатора глутамата (M. Mattson, 1990). В результате эксайтотоксических, т. е. возбуждающих нейротоксических, влияний задолго до развития болезни у людей, генетически предрасположенных к БА, увеличивается возбудимость мозговых структур и значительно снижается их резистентность к стрессу, что сопровождается характерными для стресса нарушениями энергетического обмена. Повышение возбудимости структур мозга у людей, генетически предрасположенных к БА, проявляется в виде нарастания амплитуды поздних компонентов зрительных вызванных потенциалов, появления заостренных по форме билатерально-синхронных высокоамплитудных тета- и дельта-волн при гипервентиляции, повышения УПП мозга (Н.В. Пономарева с соавт., 1998, 1999). Обнаружено также, что у родственников больных БА при функциональных нагрузках мозговой кровоток повышается значительно больше, чем в норме (S.Y. Bookheimer, 2000). Следствием повышенной чувствительности к стрессу является описанные ранее нарушения энергетического обмена, хроническое закисление мозга, нарастание интенсивности свободно-радикальных процессов, накопление внеклеточного кальция и в конечном итоге гибель нейронов.
Церебральный ацидоз влияет на специфические механизмы развития БА. В экспериментальных работах показано, что повышенная продукция лактата в мозге нарушает процессинг белка-предшественника амилоида и способствует образованию отложений бета-амилоидного протеина (G.J. Brewer, 1997; G.J. Bosman et al., 1997).
Возникающая в результате гибели нейронов церебральная атрофия является доминирующим изменением у больных БА (А.И. Ойфа, 1987). В наибольшей степени поражаются холинергические нейроны ассоциативных теменных, височных и лобных областей коры, базального ядра Мейнерта, гиппокампа и миндалины, а также норадренергические нейроны голубого пятна (E. Masliah, L. Hansn, 1994). Амилоидная ангиопатия выражена в мелких и средних сосудах коры (D.D. Orlovskaya, A.I. Oifa, 1984).
Большинство авторов, исследовавших энергетический обмен у больных БА, указывают на снижение по сравнению с нормой
142
интенсивности метаболизма глюкозы в теменных, височных и лобных ассоциативных корковых областях при относительной сохранности его в первичной сенсомоторной и зрительной коре, а также в мозжечке, базальных ганглиях и таламусе. Эти изменения выражены даже на ранних стадиях БА (F. Fazekas et al., 1989). У больных БА относительный метаболизм глюкозы в ассоциативных областях по отношению к проекционным намного меньше, чем в норме. У здоровых людей этот показатель и зависит от возраста. Метаболические различия менее заметны между больными БА и старыми людьми, чем между больными БА и молодыми испытуемыми. Локальный мозговой кровоток в ассоциативных областях у больных БА снижен, но значимых корреляций между скоростью кровотока и тяжестью БА или возрастом не отмечается. В связи со снижением аэробного окисления и относительным увеличением гликолиза при БА продукция лактата повышена(F.Fazekas et al, 1989; W. Heiss, 1991; N. Azari et al 1994).
У больных БА выявлены нарушения в работе митохондрий. Активность цитохромоксидазы – фермента дыхательной цепи снижена, что приводит к «ускользанию» электронов из дыхательной цепи и усилению продукции свободных радикалов кислорода (S. Chosh et al., 1997). Последние, являясь индукторами апоптоза, усиливают атрофию (C.W. Cotman, C.J. Pike, 1997).
По данным МРТ, при БА снижено образование АТФ. Энергии становится недостаточно для поддержания мембранных потенциалов нейронов и происходит частичная деполяризация мембраны. Вследствие этого даже обычные концентрации возбуждающего медиатора глютамата вызывают избыточную активацию NMDA-рецепторов, что приводит к накоплению Са2+ в нейроне, активации Са-зависимых эндонуклеаз, липаз и протеаз и деградации клеточных структур (М. Beal, 1992; J. Pettegrew et al., 1997).).
Предполагается, что снижение энергетического метаболизма играет ключевую роль в нарушении синтеза ацетилхолина, так как предшественник этого медиатора, ацетил-СоА, образуется в мозге из глюкозы главным образом гликолитическим путем. Нарушение нормального процессинга белка-предшественника амилоида, являющегося репаративным белком клеточных мембран, также связано со снижением энергетического метаболизма. В норме этот белок интегрирован в клеточные мембраны и разрезается протеазами в бета-области. Этот процесс зависит от энергии АТФ. При критическом снижении синтеза АТФ у больных БА белок-предшественник амилоида не может быть встроен в клеточные мембраны и соответственно нарушается его расщепление в бета-области. Вследствие этого происходит аномальный процессинг белка, и из его бета-фрагментов синтезируется бета-амилоид, который прогрессивно накапливается в сенильных бляшках и сосудистой стенке (W. Meiruge et al., 1994).
143
Хотя результаты экспериментальных исследований свидетельствуют о важном патогенетическом значении изменений энергетического метаболизма и КЩР мозга для развития БА, до появления методов компьютерной визуализации церебральных биохимических процессов и анализа УПП исследование изменений кислотно-щелочного баланса в мозге у больных БА было затруднено. В следующих разделах рассмотрены изменения УПП мозга у больных деменциями альцгеймеровского типа, а также связь этих изменений с биохимическими и электрофизиологическими показателями.
7.3.2. УПП головного мозга у больных деменциями альцгеймеровского типа
Нами обследовано 120 больных с разными формами БА. Пациенты проходили психиатрическое, неврологическое, ЭЭГ-и КТГ-обследования. Диагноcтика и определение стадий заболевания проводились сотрудниками отдела психической патологии позднего возраста Н.Д. Селезневой, И.В. Колыхаловым, Я.Б. Калыном (руководитель проф. С.И. Гаврилова). Все больные соответствовали NINCDS-ADRDA критериям для вероятной болезни Альцгеймера (J. McKhann et al., 1984). Основные результаты представленного ниже исследования опубликованы ранее (Н.В. Пономарева с соавт., 1991; С.И. Гаврилова с соавт., 1992).
У больных, страдающих деменциями альцгеймеровского типа, распределение УПП изменено по сравнению с контрольной группой здоровых испытуемых. Отличия определяются стадией и особенностями течения болезни. В общем случае на поздних стадиях заболевания наблюдаются более высокие усредненные УПП, что особенно характерно для БА с ранним началом. Нормальные или даже низкие значения могут иметь место в том случае, когда удается замедлить распад мозговой ткани или когда к атрофическим процессам в головном мозге присоединяется атрофия мышечной ткани. При этом в периферической крови также накапливается много кислых продуктов, благодаря этому соотношение концентрации водородных ионов в оттекающей от мозга и периферической крови может не отличаться от нормального или в некоторых случаях быть ниже его. Как правило, при БА нарушение церебрального гомеостаза рН выражено в большей мере, чем нарушение рН в периферической крови, поэтому значения УПП в подавляющем большинстве случаев повышены.
У больных БА пресенильного типа стадии мягкой деменции УПП в монополярных отведениях имеет тенденцию к повышению, однако отличия от нормы недостоверны. На стадиях умеренной (2а) и выраженной (2б) деменции, а также конечной стадии заболевания (3) регистрируются более высокие, чем в конт
144
рольной группе здоровых, значения усредненного УПП и УПП во всех монополярных отведениях (рис. 7.5). Достоверных отличий локальных потенциалов от нормы не выявлено. Сопоставление УПП с клиническими данными показывает, что более тяжелых нарушениях интеллекта и эмоциональной сферы наблюдаются более высокие значения УПП. Это понятно, если иметь в виду, что накопление кислых метаболитов в мозговой ткани связано с развитием атрофического процесса.
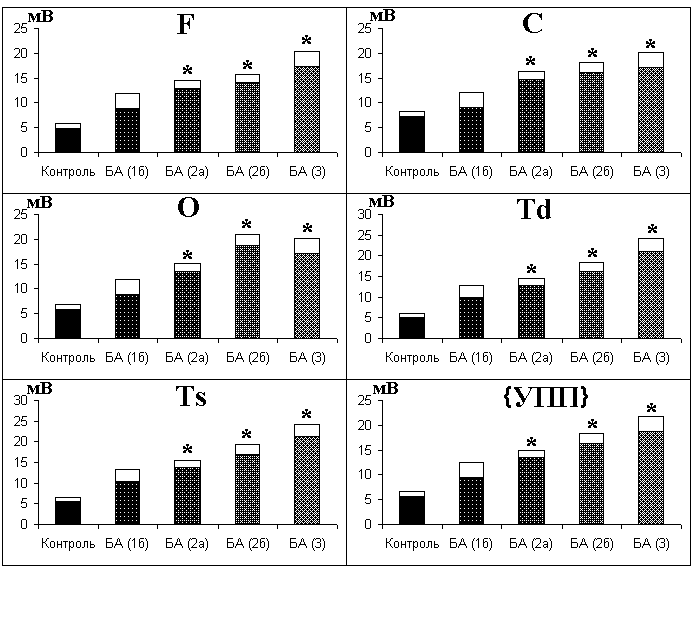
Рис. 7.5. Средние параметры УПП головного мозга у больных БА пресенильного типа и в контрольной группе здоровых испытуемых того же возраста.
По оси ординат - УПП, окрашенные столбики соответствуют средним значениям УПП, неокрашенные - стандартная ошибка. Контроль - группа здоровых испытуемых, соответствующих по возрасту больным БА. БА(1б), БА(2а), БА(2б), БА(3) - больные БА на стадиях 1б, 2а, 2б и 3 соответственно. * отмечены значения УПП у больных, достоверно (p<0,05) отличающиеся от нормы. F, C, O, Td, Ts, {УПП} - параметры УПП (см. раздел 4.5)
У пациентов, страдающих СДАТ и БА пресенильного типа, имеются определенные различия в распределении УПП. У больных СДАТ на стадии 2б наблюдается повышение УПП в левой височной области, снижение локального УПП в центральном отведении и повышение локального УПП в левом височном отведении (рис. 7.6). Снижение локального потенциала в центральном отведении характерно для нарушения корково-подкорковых отношений, связанных с поражением базальных ганглиев (раздел 7.4 «Энергети
145
ческий обмен при паркинсонизме»). Поэтому можно предположить, что у больных СДАТ атрофические изменения преобладают в височных областях и в подкорковых структурах мозга, в то время как при БА ранним началом – в коре. Это соответствует данным КТГ и особенностям клинической симптоматики. Для БА характерны выраженная амнезия и нарушение высших корковых функций (дисграфия, диспраксия и дисгнозия) при длительной сохранности основных свойств личности и эмоциональной сферы, в то время как при СДАТ доминируют эмоционально-личностные и амнестические нарушения со сдвигом ситуации в прошлое при менее значительном расстройстве высших корковых функций (С.И. Гаврилова с соавт., 1992).
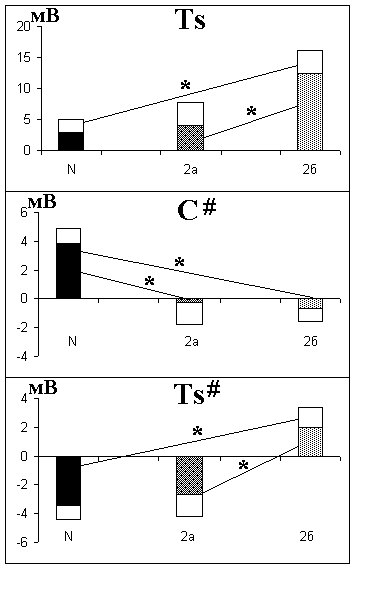
Рис. 7.6. Средние параметры УПП у больных СДАТ и в контрольной группе здоровых испытуемых того же возраста
Окрашенные столбики - среднее арифметическое УПП, неокрашенные - стандартная ошибка. N - контрольная группа здоровых испытуемых; 2а, 2б - больные СДАТ на стадиях 2а и 2б соответственно. * отмечены значения УПП у больных, достоверно (p<0,05) отличающиеся от нормы. Ts, C#, Ts# - параметры УПП (см. раздел 4.5)
Таким образом, существуют значимые отличия между УПП в норме и при деменциях альцгеймеровского типа. При БА пресенильного типа нарушения КЩР ацидотического характера имеют место в различных областях коры мозга, при СДАТ они в целом менее выражены и, судя по показателям УПП, наиболее заметны в левой височной области. Кроме того, снижение локального потенциала в центральной области при СДАТ указывает на изменение нейродинамики в результате подкорковых нарушений. Ацидоз способствует прогрессированию БА, усиливая аномальный процессинг белка-предшественника амилоида и повреждающий нейроны окислительный стресс.