

4.2.2.6. Сцепленная с х-хромосомой недостаточность гипоксантин-гуанин—фосфорибозилтрансферазы (30800) [7055]
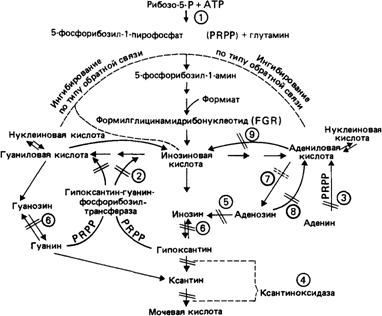
Дефекты ферментов и их роль в изучении механизмов действия генов. Некоторые дефекты ферментов сыграли заметную роль в изучении самых общих вопросов, касающихся действия генов и возникновения мутаций. Особенно важные результаты были получены при исследовании нарушений метаболизма пуринов, обусловленных недостаточностью гипоксантин-гуанин—фосфорибозилтрансферазы (HPRT) (рис. 4.21) [1294].
Синдром Леша-Найхана [1293]. В 1964 г. Леш и Найхан описали специфический синдром [1198], характеризующийся
|
Рис. 4.21. Известные дефекты метаболизма пуринов у человека. (1) Повышенная активность фосфорибозилпирофосфат —синтетазы у больных с избыточным синтезом мочевой кислоты и подагрой. (2) Почти полная недостаточность гипоксантин-гуанин — фосфорибозилтрансферазы (HPRT) у детей с синдромом Леша—Найхана и частичная недостаточность этого фермента у больных с избыточным синтезом мочевой кислоты при подагре. (3) Недостаточность аденинфосфорибозилтрансферазы (APRT) у больных с камнями в почках. В этом случае камни состоят из 2,8-диоксиаденина (не путать с мочекислыми камнями). (4) Недостаточность ксантиноксидазы у больных ксантонурией, для которых повышен риск образования камней в мочевыводящих путях и, в некоторых случаях, риск миалгии, обусловленной присутствием кристаллов ксантина в мышцах. (5) Недостаточность аденозиндезаминазы, связанная с общей тяжелой иммунной недостаточностью. (6) Недостаточность пуриннуклеозидфосфорилазы, связанная с дефектом Т-лимфоцитов. (7) Активность пурин-5-нуклеозидазы снижена в лимфоцитах больных агаммаглобулинемией, которая может быть вторичной при утрате В-клеток. (8) Недостаточность аденозинкиназы до сих пор наблюдалась только в культурах лимфобластов человека. Соответствующее заболевание еще предстоит идентифицировать. (9) Недостаточность миоаденилатдезаминазы у некоторых больных связана с явлениями слабости и судорогами в мышцах после сильных нагрузок, а также с отсутствием повышения концентрации ионов аммония в венозной крови в ответ на мышечные нагрузки [1294]. Эти дефекты ферментов служат замечательными примерами более, а часто менее характерных фенотипических последствий различных генетических блоков в единой цепи реакций. |
46 4. Действие генов
атетозом, гиперрефлексией и непременным самодеструктивным поведением, которое может выражаться даже в откусывании губ и пальцев [1164]. У всех пациентов (болеют только мальчики) наблюдается гиперурикемия и повышенная концентрация мочевой кислоты в моче, что приводит к образованию камней в почках и закупорке мочевых путей. Признак сцеплен с полом (разд. 3.1.4). Гетерозиготы клинически здоровы, однако поддаются выявлению.
В 1967 г. Сигмиллер и др. [1295] при обследовании больных с синдромом Леша - Найхана обнаружили практически полную недостаточность одного из ферментов метаболизма пуринов - гипоксантин-гуанин—фосфорибозилтрансферазы (HPRT) (у трех больных - в лизатах эритроцитов, у одного больного - в культуре фибробластов). Впоследствии дефект фермента был выявлен во многих тканях: печени, лейкоцитах и в мозге. Инозин-5'монофосфат образуется в нескольких реакциях, принадлежащих к разным метаболическим путям. Однако клетки могут использовать также готовые пуриновые основания и нуклеозиды, образующиеся при расщеплении нуклеиновых кислот. В этом пути «использования вторичного сырья» свободные пуриновые основания превращаются в соответствующие 5'-мононуклеотиды. Имеются два фермента, один специфичен для гипоксантина, второй для аденина (рис. 4.21). Если первый фермент дефектен, то вторичного использования гипоксантина и гуанина не происходит, вместо этого они превращаются в мочевую кислоту. В результате развивается гиперурикемия с образованием камней в почках; однако остается неясным, чем обусловлены симптомы со стороны ЦНС. Дефекты HPRT удобно изучать в культуре фибробластов, вот почему этот фермент использовали в качестве модели при решении ряда проблем.
Гетерогенность на молекулярном уровне. В разных семьях обычно встречаются различные мутации. Об этом свидетельствуют результаты изучения таких характеристик фермента, как остаточная активность, константа Михаэлиса—Ментен, термолабильность, ингибирование конечными продуктами - GMP и IMP и других. Иногда выраженная недостаточность HPRT имела место и в отсутствие признаков синдрома Леша-Найхана. Дефектная гипоксантингуанин—фосфорибозилтрансфераза была выявлена у некоторых взрослых больных подагрой [1162; 1294]. Однако подавляющее большинство людей, страдающих этим заболеванием, обладают нормальной HPRT. У тех немногих больных подагрой, которые имеют дефектный фермент, этот признак наследуется сцепленно с Х-хромосомой. Это еще раз указывает, что мутации действительно затрагивают один и тот же локус.
Доказательства инактивации Х-хромосомы. Одно из наиболее убедительных доказательств гипотезы Лайон (разд. 2.2.3.3) было получено при изучении активности фермента на уровне отдельных клеток у гетерозигот с мутациями по HPRT [1164]. Более того, эти исследования позволили по-новому взглянуть на метаболические взаимоотношения между клетками.
Метаболическая кооперация. Изучение культуры фибробластов кожи дает возможность идентифицировать гетерозиготных носителей мутации Если получить клоны из отдельных фибробластов и определить активность фермента, измерив с помощью радиоавтографии количество поглощаемого клетками гипоксантина, меченного тритием, то оказывается, что примерно у половины клонов активность HPRT нормальная, а у половины фермент дефектен. Однако в культурах фибробластов, которые получены без клонирования, подавляющее большинство гетерозиготных клеток обладали активностью. Очевидно, при контакте клеток, дефектных по HPRT, с нормальными клетками их метаболический дефект подвергается коррекции [1090]. Этот вывод подтверждается опытами, в которых нормальные и дефектные клетки смешивали в одной культуре. Феномен назвали «метаболической кооперацией».
Рассмотрим три возможных механизма такой кооперации (рис. 4.22).
4 Действие генов 47
|
Рис. 4.22. Возможные механизмы метаболической кооперации между клетками с активной HPRT и с неактивным ферментом в культуре гетерозиготных клеток |
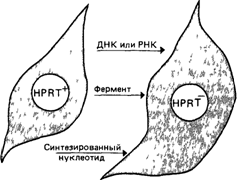
1 Нормальные клетки снабжают дефектные клетки ДНК или мРНК, в результате последние приобретают способность синтезировать функционально активный фермент
2 Дефектные клетки получают от нормальных готовый фермент. В основе этого предположения аналогия с коррекцией дефектов при мукополисахаридозах (разд. 4 2 2 3). Инкубация культуры фибробластов, дефектных по HPRT, с нормальными клетками, разрушенными ультразвуком, приводила к частичному восстановлению функции фермента.
3. Нормальные клетки синтезируют нуклеотид (конечный продукт), который переносится в дефектные клетки Большинство экспериментальных данных свидетельствует в пользу этого механизма. Если дефектные фибробласты отделить от нормальных клеток, их мутантный фенотип быстро восстанавливается, несмотря на то что нормальная гипоксантин-гуанин—фосфорибозилтрансфераза стабильна в этих условиях в течение многих часов. В другом эксперименте лимфоциты нормальной женщины инкубировали в среде, содержащей меченный тритием гипоксантин. Затем такие клетки смешивали в обычной среде с лимфоцитами мужчины, страдающего синдромом Леша—Найхана. Спустя некоторое время Y-хромосомы мужских клеток с дефектным ферментом, окрашивали акрихинипритом, после чего проводили радиоавтографию. В мужских клетках обнаруживалась метка, что указывало на перенос нуклеотидов из нормальных клеток в дефектные В подобных опытах метаболическая кооперация была продемонстрирована также между преинкубированными нормальными эритроцитами и мутантными лимфоцитами или фибробластами. Оказалось, что в дефектные клетки переносится инозимонофосфат или одно из его производных; по-видимому, активную роль в этом процессе играет мембрана клетки [1053].
Другие проблемы, связанные с недостаточностью HPRT. Дефектная HPRT стала важной модельной системой для изучения мутационного процесса. 1. Эта система дает возможность идентифицировать гемизигот и гетерозигот, измеряя активность фермента в фибробластах, и таким образом сравнивать частоты спонтанных мутаций у мужчин и женщин (разд. 5.15). 2 Ген HPRT экспрессируется в клетках амниотической жидкости, поэтому недостаточность гипоксантин-гуанин—фосфорибозилтрансферазы удается диагностировать с помощью амниоцентеза. Этим дефект HPRT в корне отличается от других патологических состояний, наследуемых сцепленно с Х-хромосомой, например гемофилии или мышечной дистрофии Дюшенна, при которых биохимический дефект не проявляется в клетках амниотической жидкости. 3. Разработана система отбора точковых мутаций в культурах фибробластов, основанная на использовании необычного субстрата, 8-азагуанина. Она позволяет изучать на клеточном уровне возникновение спонтанных и индуцированных мутаций. В нормальных клетках 8-азагуанин утилизируется HPRT, что приводит к гибели клеток. Клетки, дефектные по HPRT, не способны метаболизировать это соединение и выживают.
Иммунная недостаточность, связанная с дефектами аденозиндезаминазы и нуклеозидфосфорилазы (рис 4 21) Дефект другого фермента, участ-
48 4. Действие генов
вующего в метаболизме нуклеозидов, приводит к иному фенотипу. Этот случай тем более интересен, что дефектной является редкая форма полиморфного фермента. Дефекты одного или нескольких компонентов иммунной системы могут обусловливать повышенную восприимчивость к бактериальным инфекциям. Классическим примером такой патологии является гипогаммаглобулинемия, которая наследуется сцепленно с полом и обусловлена дефектом созревания В-лимфоцитов [1092; 1261]. В-лимфоциты служат местом образования гуморальных антител, и их отсутствие приводит к нарушению синтеза γ-глобулинов. Т-лимфоциты обеспечивают клеточный иммунитет и при этом заболевании остаются интактными.
Различают несколько форм иммунной недостаточности. Одна из них, называемая острой комбинированной обусловливает повышенную восприимчивость к заражению самыми разнообразными бактериями, вирусами и грибами. При этой форме нарушены функции как В-, так и Т-лимфоцитов. Иногда оказываются дефектными только Т-лимфоциты. Оказалось, что в основе таких дефектов лежит нарушение дифференцировки стволовых клеток в зрелые лимфоидные клетки [1274; 1267]. Эта группа заболеваний гетерогенна по этиологии, поскольку известны как случаи с аутосомно-рецессивным наследованием, так и варианты, которые наследуются сцепленно с Х-хромосомой. Среди вариантов с аутосомно-рецессивным наследованием обнаружена дополнительная гетерогенность. Так, комбинированная иммунная недостаточность может быть вызвана дефектом аденозиндезаминазы (24275) или нуклеозидфосфорилазы (16405) [1294].
Аденозиндезаминаза (ADA) катализирует необратимое дезаминирование и гидролиз аденозина до инозина и иона аммония. Нуклеозидфосфорилаза катализирует превращение инозина в гипоксантин и гуанозина в гуанин. Она обладает также небольшой активностью, обеспечивающей превращение аденозина в аденин. Эти ферменты играют ключевую роль в метаболизме РНК и ДНК.
Аденозиндезаминаза кодируется геном, локализованным в 20-й хромосоме (разд. 3.4). Электрофорез в крахмальном геле продемонстрировал полиморфизм этого фермента. Наиболее распространенный аллель обозначается ADA1 часто встречающийся полиморфный вариант ADA2. В западных популяциях аллель ADA2 встречается с частотой около 0,05 [1309]. Описаны и другие варианты ADA [1294]. Недостаточность ADA является аутосомно-рецессивным признаком. У больных детей в эритроцитах и других тканях активность аденозиндезаминазы полностью отсутствует [1098]. У их родителей, как правило, обнаруживаются промежуточные количества фермента, при этом клинически они вполне нормальны. У больных родителей выявляется остаточная активность ADA [1294]. Оказалось, что дефекты обусловлены структурными мутациями в гене ADA, которые приводят к почти полной потере функциональной активности фермента у больных гомозигот. Разработана пренатальная диагностика недостаточности аденозиндезаминазы [1126].
Нуклеозидфосфорилаза кодируется локусом в 14-й хромосоме [1096]. Обнаружено несколько редких вариантов фермента [1064]. У больных активность нуклеозидфосфорилазы полностью отсутствует, у родителей активность фермента промежуточная [1294]; заболевание наследуется по аутосомно-рецессивному типу. Иммунологическими методами показано, что по меньшей мере две мутации вызывают недостаточность нуклеозидфосфорилазы [1256]. При одной из них имеется перекрестно реагирующий материал (ПРМ), при другой его нет (см. разд. 4.2.2.2). Предполагается, что это мутации в структурном гене фермента. Вполне возможно, что больные являются не истинными гомозиготами, а составными гетерозиготами с двумя различными мутациями.
Обычно у больных с недостаточностью аденозиндезаминазы сильно нарушена функция В- и Т-лимфоцитов [1294], в то же время при нарушении нуклеозидфосфорилазы функция В-клеток интактна и синтез иммуноглобулинов происходит нормально. При дефекте обоих ферментов наблюдается поразительная дисфункция Т-лимфоцитов. Это проявляется в лимфопении, неспособности лимфоцитов реагировать на митогены и ненормальных кожных реакциях на различные антигены.
Точный биохимический механизм, который приводит к иммунологическим нарушениям в этих случаях, не установлен. Высказывались предположения о том, что недостаточность аденозиндезаминазы обусловливает накопление дезокси-АТР, который ингибирует образование пиримидиновых дезоксирибонуклеотидов, что влечет за собой нарушение синтеза ДНК, пролиферации лимфоцитов и, наконец, иммунного ответа. Недостаточность нуклеозидфосфорилазы может иметь сходный механизм [1294].
Наиболее эффективный метод исправления этих нарушений - пересадка костного мозга. В ряде случаев ослабления клинических симптомов, вызванных недостаточностью аденозиндезаминазы, удавалось добиться переливанием нормальных эритроцитов, которые служили источником недостающего фермента [1266].
4. Действие генов 49