

4.6. Механизм аутосомной доминантности
Аутосомно-рецессивные заболевания, как правило, обусловлены дефектами ферментов, которые возникают вследствие мутаций в соответствующих генах. Часто удается показать, что фермент имеет аномальную структуру или нестабилен (разд. 4.4.2) [1069]. У гетерозигот ферментативная активность составляет обычно 50% нормы, но это не приводит к заболеванию, поскольку такая активность фермента является достаточной для нормальной жизнедеятельности. Напротив, при аутосомно-доминантном наследовании гетерозиготы имеют все клинические признаки заболевания, т. е. присутствие лишь одной копии мутантного гена вызывает нарушение нормальной функции.
Механизмы аутосомно-доминантных наследственных болезней значительно более разнообразны, чем механизмы аутосомно-рецессивных заболеваний. Высказывалось предположение, что аутосомно-доминантные заболевания вызываются мутациями в генах структурных белков [132]. Это предположение выглядит еще более правдоподобным, если к числу структурных белков относить мембранные белки и рецепторы. Однако во многих случаях механизмы аутосомно-доминантных заболеваний остаются невыясненными. Мы не знаем, например, каким образом аномалия одного-единственного гена способна приводить к столь многочисленным проявлениям, как при нейрофиброматозе и туберозном скле-
120 4. Действие генов
розе. Нет никаких сведений и о механизмах патогенеза при таких заболеваниях, как хорея Гентингтона и полицистоз почек. Чтобы выяснить механизмы этих болезней, необходимы более глубокие знания генетики развития человека.
4.6.1. Аномальная агрегация субъединиц
Дисфибриногенемии (13480) [1112]. Для этой группы заболеваний характерно, что все симптомы проявляются у гетерозигот. Если такие гетерозиготы несут мутацию в гене, кодирующем белок с субъединичной структурой, то в организме будет присутствовать смесь мутантных и немутантных молекул. Вследствие этого нарушается формирование белкового агрегата (рис. 4.70). При некоторых формах дисфибриногенемии мутации, изменяющие молекулы фибриногена, приводят к кровотечениям. У определенных мутантных форм фибриноге-
|
Рис. 4.70. Схема агрегации полипептидных цепей у здоровых людей и у мутантов -гетерозигот. А. Индивид гомозиготен, синтезируются только нормальные полипептиды. Б. Индивид гетерозиготен, нормальные и аномальные полипептиды синтезируются в равных количествах; сборка белка нарушена. |
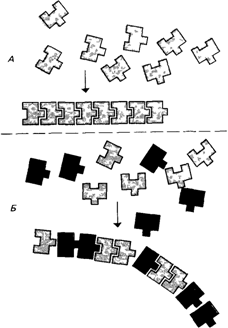
на измененным оказывается участок, ответственный за агрегацию с образованием фибрина. Например, при мутации Detroit имеется аминокислотная замена в сайте, ответственном за превращение фибриногена в фибрин, она обусловливает сильные кровотечения у больных с таким генотипом [1007]. Хотя при большинстве аномалий фибриногена количество этого белка не отличается от нормы, известен случай, когда уровень фибриногена был снижен из-за уменьшения времени жизни молекулы, что, вероятно, связано с ее нестабильностью [1210]. Некоторые генетические аномалии фибриногена считаются причиной тромбозов, однако не известно, какие именно мутации повышают свертываемость крови. Большая часть вариантов фибриногена не связана с клиническими осложнениями. Очевидно, что для выяснения взаимосвязи структуры и функции молекулы фибриногена необходимы дальнейшие исследования аминокислотных замен, характерных для фибриногенемий.