

2.4.3. Визначення нуклеотидної послідовності днк
Секвенування −визначення нуклеотидної послідовності. Методи, що надали можливість ідентифікувати генетично важливі ділянки ДНК, мають велике значення. До основних методів секвенування належать хімічний і ферментативний.
Хімічне секвенування засновано на вибірковій хімічній деградації нуклеотидів. Метод розроблений у 1976 році А. Максамом і В. Гілбертом. Для цього методу необхідно отримати одноланцюгову молекулу ДНК, на одному з кінців якої роблять радіоактивну позначку за допомогою ізотопа фосфору 32Р, препарат позначеної ДНК поділяють на чотири порції і кожну обробляють реагентом, який специфічно руйнує одну або дві з чотирьох основ. Необхіднопідібрати умови реакції таким чином, щоб на одну молекулу було лише декілька пошкоджень. За обробки піперидином в ДНК утворюється розрив у тому місці, де була зруйнована основа.
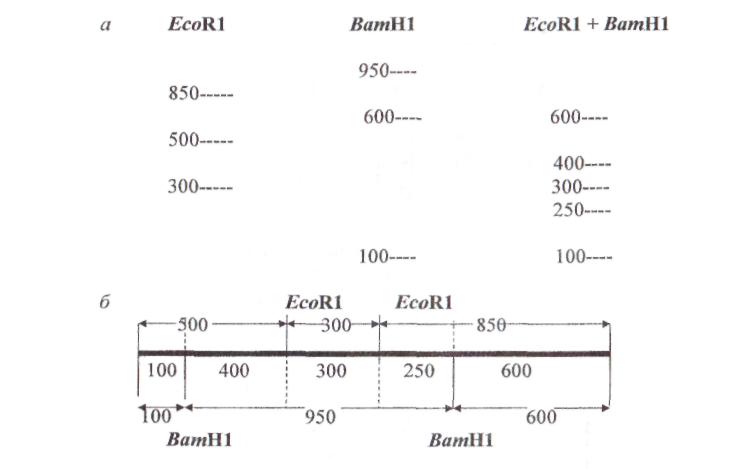
Рис. 15. Картирування сайтів рестрикції:
а - результати гель-електрофореза фрагментів ДНК;
б - рестрикційна карта,що побудована на підставі даних гель-електрофорезу
В результаті створюється набір позначених фрагментів, довжина яких визначається відстанню від зруйнованої основи до кінця молекули. Фрагменти, що створилися вусіх чотирьох реакціях, піддають електрофорезу на чотирьох доріжках в акриламідному гелі, потім роблять радіографію, і фрагменти, що містять радіоактивну позначку, залишають «відбитки» на рентгенівській плівці. За їх положенням можна визначити, на якій відстані від позначеного кінця була зруйнована основа. Таким чином, на підставі набору смуг на рентгенівській плівці визначають нуклеотидну послідовність фрагменту, що аналізується (рис. 16).

Нуклеотидна послідовність
ланцюга ДНК, що аналізується
Рис. 16. Схема методу секвенування за Максамом-Гілбертом
За допомогою цього методу вдалося встановити багато послідовностей різноманітних ДНК, але він має ряд недоліків, що пов'язані з тривалістю і трудомісткістю процедур.
У наш час найбільш широко використовується метод ферментативного секвенування або метод секвенування шляхом термінації (зупинки синтезу) ланцюга, запропонований Ф. Сенгером у 1977р. В основі методу Сенгера лежить принцип реплікації комплементарного ланцюга ДНК на одноланцюговій матриці, при тому що під час реплікації відбувається термінація − припинення синтезу дочірнього ланцюга у різних місцях, а наступний ланцюг знову починає синтезуватися від початку матриці до наступної термінації. Основним моментом ферментативного секвенування є термінація синтезу ланцюга, що будується. Агентами термінації, які викликають зупинку реплікації, є дідезоксинуклеотиди (ддАТФ, ддГТФ, ддТТФ, ддЦТФ) − це нуклеотиди позбавлені 2'- і 3'-гидроксильних груп при вуглеводних атомах цукрового кільця і тому подовження ланцюга, що відбувається за рахунок приєднання чергового нуклеозидтрифосфату до гідроксильної групи, припиняється.
Оскільки метод секвенування заснований на процесі реплікації ДНК, то основним ферментом є ДНК-полімераза. За наявності одноланцюгової ДНК-матриці, короткого полінуклеотидного праймера і нуклеотидів за принципом компліментарності відбувається синтез іншого ланцюга ДНК. При цьому подовження ланцюга здійснюється до того часу, коли замість дезоксинуклеотиду не приєднається дидезоксинуклеотид. Приєднання останнього викликає зупинку синтезу (рис. 17).
У пробірках збирають чотири реакційні суміші, що мають однаковий склад: фрагмент ДНК-матриці, в якій визначають нуклеотидну послідовність, чотири дезоксинуклеотиди достатньої кількості для здійснення росту комплементарного ланцюга, радіоактивно позначений короткий праймер, комплементарний фрагменту ДНК-матриці, з якого ДНК-полімераза подовжує синтез. У кожну пробірку потім додають лише один з дидезоксинуклеотидів. У всіх пробірках буде відбуватися термінація ланцюга, що синтезується, лише в тому місці, де ДНК-полімераза приєднуватиме дідезоксинуклеотид, який був доданий у реакційну суміш, тобто в одній пробірці синтез припиняється за приєднання до комплементарного ланцюга ддАТФ, в іншій − ддГТФ і т.д. Оскількиобрив ланцюга відбувається у випадкових місцях, то створюється набір фрагментів усіх можливих довжин, починаючих з праймера до кінця фрагменту, що досліджується.

Рис. 17. Схема методу секвенування за Сенгером:
а - припинення росту ланцюга ДНК; б - радіоавтографія гелю
Фрагменти ДНК, що були отримані, розподіляють у поліакриламідному гелі (з точністю до одного нуклеотиду), проводять радіографію і на підставі побаченого розподілу фрагментів у чотирьох пробірках установлюють нуклеотидну послідовність.
Використання флуоресцентних барвників, пов'язаних з нуклеотидами термінації, дозволяє проводити усі реакції в одній пробірці. Крім того, розроблено принципово нові підходи до секвенування, наприклад, секвенування шляхом гібридизації з фіксованими олігонуклеотидними чипами, секвенування з використанням екзонуклеаз та інші. Усі ці методи дозволяють досить швидко вирішувати проблеми секвенування як великих ділянок ДЇЖ, так і будь-яких геномів. Отримані результати вносять у бази даних – банки генів.
Необхідність отримання інформації про послідовність нуклеотидів ДНК усе зростаючих масштабів викликала потребу щодо розгляду питання про автоматизацію процесу секвенування ДНК, оскільки існуючі ручні методи вже не дозволяли справляться з такими великими обсягами роботи.
Автоматичне
секвенування – це, в першу чергу,
електрофоретичне розподілення позначених
продуктів реакції
Для
позначення знов синтезованих ланцюгів
ДНК в умовах
Автоматичні секвенатори ДНК керуються спеціально створеними комп’ютерними програмами. Так, наприклад, прибори фірми AppliedBiosystemsкомплектуються програмами збору і аналізу даних.
Сучасні автоматичні секвенатори набагато перевершують людину за продуктивністю, більш того, кожен рік такі автомати вдосконалюють. Усе це веде до того, що секвенування фрагментів ДНК стає простою процедурою, доступною для будь-якої молекулярно-біологічної лабораторії.
Розшифрування
великих геномів прокаріотичних
і еукаріотичних
клітин потребувало поєднання зусиль
різних лабораторій і формування
еукаріотичних
клітин потребувало поєднання зусиль
різних лабораторій і формування
У липні
1995 р. була опублікована перша
Першим еукаріотичним організмом, повна послідовність якого (12680000 п.н.) визначена у 1996 p., стали дріжджі Saccharomycescerevisiae. Це був результат зусиль значного числа дослідників, що працюють у лабораторіях і великих дослідних центрах країн Західної Європи, США і Японії.
Значні успіхи у розшифруванні послідовності молекул ДНК спричинили те, що у 1990 р. у США була прийнята офіційна програма щодо розшифрування генома людини (HumanGenomeProject, HGP), в межах якої планувалося за вкладення 3 млрд. доларів США завершити секвенування повного генома людини протягом 15 років. Основним виконавцем HGPстала компанія CeleraGenomics. Аналогічні проекти щодо генома людини почали реалізовуватися у Західній Європі, Японії, Росії.
У 2001 р. учасники американської програми HGPі Міжнародного консорціуму з секвенування генома людини, що поєднує численні наукові організації США, Великої Британії, Німеччини, Франції, Японії і Китаю, незалежно оголосили про завершення секвенування більшої частини (понад 95%) генома людини. На підставі отриманих даних стало можливим зробити ряд висновків про організацію генома людини.
Комп’ютерний
аналіз дозволив виявити 26588 транскрибуючих
і
Отримані дані про структуру генома дозволяють значно поширити наше розуміння генетики людини, природи різноманітних спадкових хвороб. Інформація, що накопичується, дозволяє з’ясовувати молекулярні механізми патогенного впливу мікроорганізмів, вивчати захисні механізми, що реалізуються людиною, твариною, рослиною у відповідь на певну інфекцію. Це створює базу для ефективної розробки сучасних засобів і методів лікування інфекційних захворювань.
Таблиця 4
Приклади секвенування повних геномів різних типів організмів
Організми |
Розмір генома, млн. п.н. |
Передбачена кількість генів |
Рік завершення секвенування |
Бактерія (Haemophilus influenzae) |
1,8 |
1743 |
1995 |
Дріжджі (Saccharomycescerevisiae) |
12,1 |
6102 |
1996 |
Кільчастий хробак (Caenorhabditis elegans) |
97,0 |
19099 |
1998 |
Рослина (Arabidopsis thaliana) |
100,0 |
25000 |
2000 |
Комаха (Drosophila melanogaster) |
180,0 |
13061 |
2000 |
Людина (Homo sapiens) |
3000,0 |
35000…45000 |
2004 |
Полімеразна ланцюгова реакція. Чисельні (ампліфіковані у мільйон разів) копії певних фрагментів ДНК отримують invitroза допомогою полімеразної ланцюгової реакції (ПЛР), яку поширено використовують у наш час. Вперш її запропонували у 1985 році Р.К. Сейки, С. Шарф, Ф. Фалуна, К.Б. Мулліс, Г.Е. Хорн, Х.А. Ерліх і Н. Арнхейм.ПЛР високо специфічна і чутлива, за її допомогою можна виявити, розмножити і дослідити навіть одиничну копію гена у вихідному матеріалі. Зазвичай 30 циклів ампліфікації протікає протягом трьох годин.
Для здійснення ПЛР необхідно:
два синтетичних олігонуклеотидних праймери (короткі олігонуклеотиди, що гібридизуються із матрицею і є запалом за її копіювання), завдовжки близько 20 нуклеотидів, комплементарні ділянкам ДНК з протилежних ланцюгів, що фланкирують (оточують) послідовність-мішень, яку розмножують; їх 3'-гідроксильні кінці після отжигу (процес утворення дволанцюгових молекул (ДНК-ДНК або ДНК-РНК) з одноланцюгових полінуклеотидних комплементарних ланцюгів) з ДНК повинні бути орієнтовані назустріч один одному;
ДНК-мішень завдовжки від 100 до ~ 35000 п.н.;
термостабільна ДНК-полімераза, що не втрачає активності за температури 95°С і вищої;
чотири дезоксирибонуклеотиди.
Типова ПЛР-ампліфікація складається з багаторазового повторення таких реакцій.
1.Денатурація. Перший етап ПЛР полягає в тепловій денатурації зразка ДНК витримуванням його за температури 95°С протягом, що найменше 1 хв. Крім ДНК, в реакційній суміші міститься надлишок двох* праймерів, термостабільна ДНК-полімераза Taq, що виділена з бактерій Thermusaquaticus, і чотири дезоксирибонуклеотида.
2.Ренатурація. Температуру суміші повільно знижують, приблизно до 55°С, при цьому праймери спаровуються із комплементарними послідовностями ДНК.
3.Синтез.Температуру підвищують приблизно до 75°С – величини, оптимальної для ДНК-полімерази Taq. Починається синтез комплементарного ланцюгу ДНК, що ініціюється 3'-гідроксильною групою праймера (рис. 18).
Рис. 18. Схема полімеразної ланцюгової реакції (ПЛР):
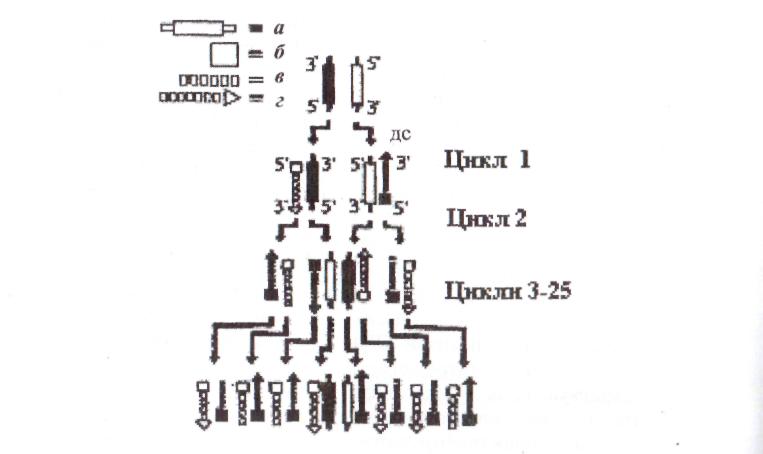
а - ДНК-мішень, б - праймер у ПЛР, в - нова ДНК, г - напрямок ампліфікації, де - денатурація і синтез
У першому циклі після денатурації ДНК і зв'язування з неюпраймерів ДНК-полімераза створює дволанцюгові структури, в яких батьківські ланцюги ДНК поєднані із знов синтезованими комплементарними ланцюгами різної довжини, але із фіксованим 5'-закінченям. Уже у другому циклі створюються два ланцюги специфічної послідовності, що мають задані розміри. У третьому циклі ці ланцюги дають начало дволанцюговим детермінованим за розміром фрагментам ДНК. У кожному наступному циклі їх кількість подвоюється і теоретично досягає за 22 цикли, а на практиці – за ЗО циклів більше мільйона копій. Вихідні молекули ДНК і структури, що створилися у першому циклі, присутні в реакційній суміші наприкінці ПЛР у незначній кількості.
Для здійснення ПЛР необхідно знати послідовність як найменш 17 п.н. з обох боків дослідного фрагменту ДНК. Зазвичай синтезують два дезоксинуклеотиди довжиною 20...30 основ, які являють собою кінцеві послідовності фрагменту ДНК, що нас цікавить. Використовуючи комплементарні до цих ділянок олігомери -праймери, запускають ампліфікацію (процес збільшення копій гену). Надлишкову кількість праймерів змішують з геномної ДНК, а потім послідовно здійснюють реакції денатурації, випалу (ренатурації) і нарощування ланцюга (подовження праймера). Теплова денатурація супроводжується розплітанням подвійної спіралі ДНК. За зниження температури має місце випал олігонуклеотидів, тобто здійснюється гібридизація олігонуклеотидних праймерів із своїми комплементарними послідовностями. Ріст ланцюга праймерів каталізує ДНК-полімерза в присутності дезоксирибонуклеозид-трифосфатів, і в результаті добудовується новий комплементарний ланцюг ДНК.
За повторних циклів теплової денатурації, випалу і синтезу нові ланцюги ДНК, що утворилися, є шаблонами (матрицями) для праймерів, що викликає експоненціальне нарощування ділянки ДНК, обмеженої праймерами, що входять до складу цих фрагментів.
У разі ампліфікації геномної ДНК з клітин (тканин) хребетних тварин інколи відбувається накопичення фрагментів нез'ясованого походження. Тоді здійснюють серію додаткових ампліфікацій, використовуючи інший набір олігонуклеотидних праймерів.
На перших етапах використовували в ПЛР так звані фрагменти Кленова ДНК-полімерази І Е. соli, які виявилися термолабільними, і після кожного циклу денатурації необхідно було додавати нову Порцію ферменту. Крім того, за пониженої температури випалузнижувалася специфічність ампліфікації.
Після виділення термостабільних ДНК-полімераз, зокрема, з бактерій Thermusaquaticus(Taq-полімераза) суттєво збільшилася ефективність ПЛР. Taq -полімераза зберігає активність після теплової денатурації ДНК і не виникає необхідності замінювати фермент після кожного циклу. Температурний оптимум реакції становить 70...75°С, що значно підвищує специфічність, кількісний вихід і можливу довжину копій фрагментів ДНК, що амплифікуються.
Успіх певної ПЛР суттєво залежить від правильного вибору праймерів. Для мінімізації можливості неправильного спарування, праймер повинен відповідати деяким вимогам. Його довжина має бути 17...30 нуклеотидів за вмісту GC-napблизько 50%. Чим менший вмістGC-nap у праймері, тім більшою повинна бути його довжина. Необхідно запобігти появі вторинних структур у праймері і більше трьох-чотирьох однакових нуклеотидів підряд. Праймери, що використовуються в одній реакції, не повинні мати комплементарні ділянки.
Область використання ПЛР велика і постійно розширюється: діагностика генетичних захворювань; виявлення генетичного матеріалу патогенних мікроорганізмів у клінічних зразках; синтез зондів для гібридизації; створення бібліотек кДНК (копійних ДНК) з малої кількості мРНК; мультиплікація ДНК з метою секвенування; спрямований мутагенез таін.