

Тема 7. Нуклеофильные реакции в ряду карбонильных соединений
Реакции нуклеофильного замещения с участием sр2-гибридизованного атома углерода.
Механизм реакций этого типа рассмотрим на примере взаимодействия карбоновых кислот со спиртами (реакция этерификации). В карбоксильной группе кислоты реализуется р,- сопряжение, поскольку пара электронов атома кислорода гидроксильной группы ОН вступает в сопряжение с двойной углерод-кислородной связью (-связью):
Такое сопряжение является причиной, с одной стороны, повышенной кислотности карбоксильных соединений, а с другой уменьшения частичного положительного заряда (+) на атом углерода карбоксильной группы (sр2-гибридизованном атоме) что значительно затрудняет непосредственную атаку нуклеофила.
С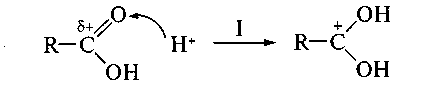
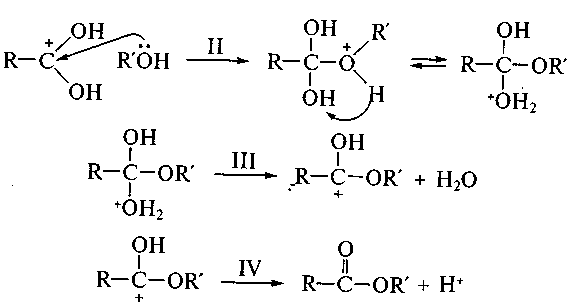 целью увеличения заряда на атоме углерода
используют дополнительное протонирование
— кислотный катализ (стадия I):
целью увеличения заряда на атоме углерода
используют дополнительное протонирование
— кислотный катализ (стадия I):
На стадии II происходит атака нуклеофила (молекулы спирта R'OH), протонирование гидроксильной группы с образованием хорошо уходящей группы Н2О, на стадии III — ее отщепление и на стадии IV — регенерация протона — возврат катализатора с образованием конечного продукта — сложного эфира.
Реакции нуклеофильного присоединения. Наиболее характерны реакции нуклеофильного присоединения (AN) для оксосоединений — альдегидов и кетонов. Механизм этих реакций имеет общие черты, это двухстадийный ионный процесс. Первая стадия (лимитирующая) представляет собой обратимую атаку нуклеофилом (Nu) с образованием так называемого тетраэдрического интермедиата. Вторая стадия — быстрая атака электрофилом:
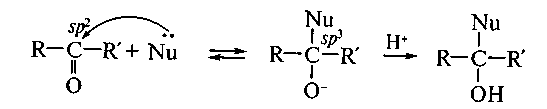
На реакционную способность оксосоединения оказывает влияние природа групп R и R'. Так, введение электронодонорных заместителей снижает реакционную способность, а электроноакцепторных — усиливает. Поэтому альдегиды более активны в реакциях AN, чем кетоны. Кроме того, реакционная способность зависит от природы нуклеофила. Например, тиолы RSH, являясь более сильными нуклеофилами, чем спирты ROH, вступают в реакцию AN как с альдегидами, так и с кетонами, образуя устойчивые к гидролизу тиоацетали, тогда как ацетали — продукты присоединения спиртов к альдегидам — к гидролизу не устойчивы
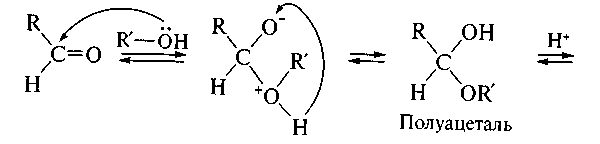
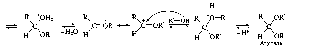
Обратите внимание, что последние стадии процесса представляют собой атаку нуклеофила (молекулы спирта R'OH) на электрофильный реакционный центр (карбкатион) и идут по механизму нуклеофильного замещения SN. Образующиеся промежуточные соединения — полуацетали — являются неустойчивыми. Стабилизация их возможна только в циклической форме при образовании циклических полуацеталей, например 5-гидроксипентаналя:
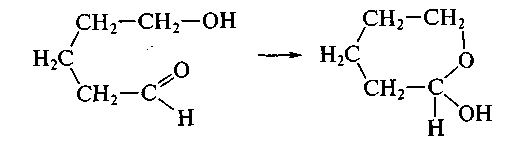
Д ругой
пример биологически важной реакции
этого типа - присоединение аминов и
некоторых других азотсодержащих
соединений к карбонильным соединениям
– альдегидам и кетонам. Реакция идет
по механизму нуклеофильного присоединения
– эли минирования (AN—E):
ругой
пример биологически важной реакции
этого типа - присоединение аминов и
некоторых других азотсодержащих
соединений к карбонильным соединениям
– альдегидам и кетонам. Реакция идет
по механизму нуклеофильного присоединения
– эли минирования (AN—E):
Другие азотсодержащие соединения, выступающие в этих реакциях в роли нуклеофила: гидразин NH2–NH2, фенилгидразин, С6Н5–NH–NH2,гидроксиламин NH2–ОН.
Продуктами реакций AN—E этих случаях являются соединения, называемые гидразонами, фенил-гидразонами, оксимами.
Р еакции
конденсации.
Протекают в присутствии катализаторов,
чаще щелочной природы. Приводят к
усложнению углеродного скелета.
Характерным примером являются альдольная
и кротоновая конденсации:
еакции
конденсации.
Протекают в присутствии катализаторов,
чаще щелочной природы. Приводят к
усложнению углеродного скелета.
Характерным примером являются альдольная
и кротоновая конденсации:
Реакции нуклеофильного замещения в ряду карбоновых кислот.
Только с чисто формальных позиций можно рассматривать карбоксильную группу как комбинацию карбонильной и гидроксильной функций. Фактически их взаимное влияние друг на друга таково, что полностью изменяет их собственные свойства.
П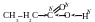 оляризация
двойной связи С=О сильно возрастает
за счет дополнительного сдвига свободной
электронной пары с соседнего атома
кислорода гидроксильной группы:
оляризация
двойной связи С=О сильно возрастает
за счет дополнительного сдвига свободной
электронной пары с соседнего атома
кислорода гидроксильной группы:
Следствием является значительное ослабление связи О–Н в гидроксиле и легкость отщепления атома водорода от него в виде протона (Н+). Появление пониженной электронной плотности (+) на центральном углеродном атоме карбоксила приводит также к оттягиванию -электронов соседней связи С – С к карбоксильной группе и появлению (как у альдегидов и кетонов) пониженной электронной плотности (+) на - углеродном атоме кислоты.
Самыми слабыми кислотными свойствами обладают предельные монокарбоновые кислоты. Кислотность двухосновных, непредельных или замещённых кислот (галогенкислоты, гидрокси- и оксокислоты и т.п.) заметно выще, что определяется электроноакцепторным действием заместителей и стабилизацией промежуточного аниона.
В общем производные карбоновых кислот по сравнению с альдегидами и кетонами труднее подвергаются нуклеофильной атаке, так как электрофильность карбонильного атома углерода обычно снижается за счет - М-эффекта функционального заместителя, связанного с атомом углерода карбонильной группы. По этой причине для проведения реакций оказывается необходимым кислотный катализ – протонирование aтома кислорода карбонильной группы, что ведет к появлению дополнительного заряда на атоме углерода, что облегчает атаку нуклеофилом.
В итоге механизм катализируемой кислотой реакции по сравнению с механизмом некатализируемой реакции включает предварительную стадию протонирования и заключительную стадию депротонирования.
Реакции конденсации, в основе которых лежит способность одного карбонильного соединения присоединяться к карбонильной группе этого же или другого карбонильного вбединения, характерны и для производных карбоновых кислот, в частности рюжных эфиров и тиоэфиров. Такие реакции имеют большое биологическое значение. С их помощью в организме происходит образование новых связей углерод—углерод. Непременными участниками реакций по типу альдольного присоединения in vivo являются тиоэфиры карбоновых кислот — производные кофермента А. При конденсации ацетилкофермента А по типу альдольного присоединения из двух молекул ацетилкофермента А образуются ацетоацетилкофермент А и кофермент А.
Контрольные задания
1. Примером биологически важной реакции является присоединение аминов и некоторых других азотсодержащих соединений к карбонильным соединениям – альдегидам и кетонам. Напишите уравнение присоединения гидразина к пентаналю. Назовите продукт реакции. Поясните, можно ли с помощью этой реакции провести выделение и идентификацию пентаналя?
2. Наличие какой функциональной группы в молекуле формальдегида подтверждает реакция получения его динитрофенилгидразона:
а) карбоксильной; б) гидроксильной; в) карбонильной; г) метильной; д) атома водорода.
3. Для чего можно использовать реакцию альдегида с динитрофенилгидразином:
1) идентификации альдегида; 2) окисления альдегида; 3) восстановления альдегида; 4) выделения альдегида из смеси с карбоновой кислотой; 5) выделения альдегида из смеси с кетоном.
4. Приведите формулу изомера пропаналя. Будут ли для него характерны те реакции нуклеофильного присоединения, которые характерны для пропаналя?
5. Одним из феромонов является этиловый эфир 4-фенил-2-бутеновой кислоты (этилциннамат). Получите это соединение по реакции этерификации.
6. В реакции этерификации группа –ОН отщепляется (для образования воды) от молекулы: а) спирта; б) альдегида; в) кетона; г) кислоты? Приведите уравнение реакции этерификации.
7. Почему реакцию этерификации ведут, как правило, при температуре 100-105 С:
1) чтобы сдвинуть равновесие в сторону образования сложного эфира;
2) чтобы сдвинуть равновесие в сторону образования кислоты;
3) чтобы сдвинуть равновесие в сторону образования спирта.