

Тема 4. Кислотность и основность органических веществ.
Общая характеристика реакций органических соединений.
Кислотность и основность органических соединений.
Для оценки кислотности и основности органических соединений наибольшее значение имеют две теории – теория Бренстеда и теория Льюиса.
По теории Льюиса кислотные и основные свойства соединений определяются их способностью принимать или отдавать пару электронов с образованием связи. В соответствии с принципом ЖМКО кислоты и основания Льюиса делятся на жесткие и мягкие.
Кислотами Льюиса могут быть атомы, молекулы или катионы, обладающие вакантной орбиталью и способные принимать пару электронов с образованием ковалентной связи.
Кислоты Льюиса – акцепторы пары электронов; основания Льюиса – доноры пары электронов. Основания Льюиса (атом, молекула или анион) должны обладать по крайней мере одной парой валентных электронов, которую они способны предоставить партнеру для образования ковалентной связи. Все основания Льюиса представляют собой нуклеофильные реагенты.
По теории Бренстеда (протолитической теории) кислотность и основность соединений связывается с переносом протона Н+. Кислота и основание образуют сопряженную кислотно-основную пару, в которой чем сильнее кислота, тем слабее сопряженное ей основание, и напротив, чем сильнее основание, тем слабее сопряженная ему кислота.
Кислоты Бренстеда (протонные кислоты) – нейтральные молекулы или ионы, способные отдавать протон (доноры протонов).
Основания Бренстеда – нейтральные молекулы или ионы, способные присоединять протон (акцепторы протонов).
Кислотность и основность являются не абсолютными, а относительными свойствами соединений: кислотные свойства обнаруживаются лишь в присутствии основания; основные свойства – только в присутствии кислоты. В качестве растворителя при изучении кислотно-основных равновесий обычно используется вода.
В зависимости от природы элемента, с которым связан протон, различают ОН- кислоты (карбоновые кислоты, фенолы, спирты), SH-кислоты (тиолы), NН-кислоты (амины, амиды, имиды), СН-кислоты (углеводороды и их производные). Элемент и связанный с ним атом водорода называют кислотным центром. Во всех случаях присутствует сдвиг электронной плотности от атома водорода к более электроотрицательному атому, протону более или менее легко отщепиться. Чем выше электроотрицательность элемента, с которым связан протон, тем больше кислотность соединения (так, карбоновые кислоты являются более сильными кислотами, чем тиолы или амины).
Наличие в молекуле электроноакцепторных групп, обладающих отрицательными электронными эффектами, увеличивает положительный заряд на протоне, что приводит к усилению кислотных свойств.
Для образования ковалентной связи с протоном основания Бренстеда должны предоставлять либо неподелениую пару электронов, либо электроны -связи. В соответствии с этим основания Бренстеда делятся на п-основания и -основания.
n-основания могут быть нейтральными или отрицательно заряженными. Как правило, анионы обладают более сильно выраженным основным характером, чем нейтральные вещества. То есть амид-ион NН2– или гидроксид-ион НО– по основности превосходят аммиак NН3 и воду Н2О.
В -основаниях, к которым относятся алкены, алкадиены, арены, центром основности, т.е. местом присоединения протона, являются электроны -связи. Это очень слабые основания, так как протонируемые электронные пары несвободны.
Наличие электронодонорных заместителей увеличивает основность органических соединений.
Значения рН некоторых жидких систем организма.
Нормальное функционирование живых организмов возможно только в условиях определенного постоянства рН и других параметров их внутренней среды. Это постоянство поддерживается соответствующими буферными системами.
Общая характеристика реакций органических соединений
Большинство органических реакций включает несколько последовательных (элементарных) стадий. Детальное описание совокупности этих стадий называется механизмом. Механизм реакции — гипотеза, предлагаемая для объяснения экспериментальных данных. Он может уточняться и даже меняться с появлением новых фактов и углублением знаний.
Общая скорость сложной химической реакции определяется (лимитируется) скоростью ее наиболее медленной стадии, а скорость составляющих элементарных реакций — их энергией активации Еа. Последняя необходима для осуществления эффективного столкновения молекул, приводящего к взаимодействию. Ее можно определить также как энергию, необходимую для достижения системой переходного состояния, иначе называемого активированным комплексом, превращение которого в продукты реакции происходит уже самопроизвольно. Чем меньше величина энергии активации реакции, тем выше ее скорость.
Использование катализатора существенно снижает скорость реакции за счёт понижения энергии активации из-за образования активированного промежуточного комплекса. В живых организмах роль высокоспецифичных катализаторов выполняют ферменты.
Фермент карбоангидраза катализирует биохимические реакции гидратации альдегидов, сложных эфиров, а также диоксида углерода. Жизненная важность этого фермента определяется тем, что он регулирует кислотность крови, а посредством этого (конечно, наряду с другими факторами) – интенсивность дыхательного процесса. Конкретная реакция, которую катализирует карбоангидраза, представляет собой равновесное превращение воды и диоксида углерода в угольную кислоту.
Н2О + СО2 ↔ Н2СО3 ↔ HCО3– + Н+
Именно эта реакция используется организмом для удаления из клеток углекислого газа, образовавшегося в них в результате жизнедеятельности. Некатализируемая гидратация СО2 протекает слишком медленно, чтобы обеспечивать его эффективный транспорт от тканей к легким. Активность же карбоангидразы поражает воображение: одна молекула фермента катализирует каждую минуту гидратацию 3,6107 молекул диоксида углерода.
Принципиальная схема работы карбоангидразы заключается в следующем. Карбоангидраза представляет собой белок, состоящий из фрагментов 260 аминокислот. Молекула воды теряет протон на активном участке фермента, который выступает как основание. При этом образуется сопряженное основание – гидроксид-ион, который присоединяется к молекуле диоксида углерода точно так же, как это происходит в реакциях гидроксид-иона с другими карбонильными соединениями. По существу, это присоединение представляет собой кислотно-основную реакцию Льюиса.
Кислотность воды, однако, не столь высока, чтобы протон от нее легко было бы оторвать. Поэтому карбоангидраза нуждается в помощи. Эту помощь ей оказывает кофактор - один из микроэлементов, присутствующих в организме, а именно ион Zn2+. Как кислота Льюиса он координируется по атому кислорода молекулы воды и существенно облегчает тем самым отрыв протона активным участком карбоангидразы. На модельных реакциях было определено влияние иона цинка как кофактора. Этот ион увеличивает скорость реакции гидратации карбонильного соединения более чем в 6 млн. раз по сравнению с некатализируемой реакцией.
Реакционная способность всегда должна рассматриваться только по отношению к реакционному партнеру. Само вещество при этом называют субстратом, а действующее на него соединение (реакционную частицу) – реагентом. Субстратом, как правило, называют то вещество, в котором у атома углерода происходит разрыв старой и образование новой связи. В биохимических процессах реагентами считают ферменты, а вещества, подвергающиеся их действию, субстратами. В ходе химического превращения обычно затрагивается не вся молекула, а только ее часть – реакционный центр.
Типы реакций в органической химии
Многообразие органических реакций приводит к целесообразности их классификации по следующим признакам:
1. По электронной природе реагентов (нуклеофильные, электрофильные, свободнорадикальные реакции).
Нуклеофильные реагенты – это одно- или многоатомные анионы или молекулы, имеющие центры с повышенной электронной плотностью. К ним относятся такие анионы и молекулы, как HO-, RO-, Cl-, Br-, RCOO-, CN-, R-, NH3, C2H5OH и т.д.
Электрофильные реагенты – это катионы, простые или сложные молекулы, которые сами по себе или же в присутствии катализатора обладают повышенным сродством к электронной паре или отрицательно заряженным центрам молекул. К ним относятся катионы H+, Cl+, +NO2, +SO3H, R+ и молекулы со свободными орбиталями AlCl3, ZnCl2 и т.п.
Свободные радикалы – это электронейтральные частицы, имеющие неспаренный электрон, например: Cl, NO2.
2. По изменению числа частиц в ходе реакции (замещение, присоединение, отщепление, разложение, ОВР и др.).
В случае реакций замещения в молекуле один атом (или группа атомов) замещается другим атомом (или группой атомов), в результате чего образуются новые соединения:
СН3–СН3 + С12 СН3–СН2С1 + НC1
При протекании реакций присоединения из двух (или нескольких) молекул образуется одно новое вещество:
CH2 = CH2 + HBr → CH2Br–СH3
В результате реакции отщепления образуется новое органическое вещество, содержащее кратную связь:
СН3–СН2С1 + NaOH(спиртовой р-р) СН2 = СН2 + NaC1 + Н2О
Реакции разложения приводят к образованию из одного вещества двух или более веществ более простого строения:
НСООН → СО2 + Н2
3. По частным признакам (гидратация и дегидратация, гидрирование и дегидрирование, нитрование, сульфирование, галогенирование, ацилирование, алкилирование, карбоксилирование и декарбоксилирование, енолизация, замыкание и размыкание циклов, изомеризация, окислительная деструкция, пиролиз, полимеризация, конденсация и др.).
4. По механизмам элементарных стадий реакций (нуклеофильное замещение SN, электрофильное замещение SE, свободнорадикальное замещение SR, парное отщепление, или элиминирование Е, нуклеофильное или электрофильное присоединение AdE и AdN и т. д.).
Рассмотрим несколько примеров.
Пример 1. При действии на организм больших доз гидразина или его производных наблюдаются нервные расстройства. Какова химическая основа действия гидразина, если известно, что он реагирует с коферментом пиридоксальфосфатом?
Решение. Пиридоксальфосфат — гетероциклическое соединение, содержащее в цикле наряду с другими заместителями альдегидную группу. Гидразин NH2–NH2 как нуклеофильный реагент взаимодействует с карбонильным атомом углерода. Поляризованная -связь карбонильной группы легко разрывается, и между карбонильным атомом углерода и атомом азота возникает ковалентная связь донорно-акцепторного типа за счет пары электронов атома азота молекулы гидразина.
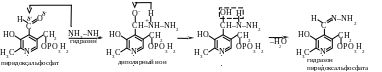
Образующийся диполярный ион в результате перехода протона от положительно заряженного атома азота (кислотный центр) к аниону (основный центр) превращается в нейтральное соединение. В этом соединении у атома углерода содержатся одновременно две электроноакцепторные группы, поэтому оно неустойчиво и переходит в более стабильное состояние путем отщепления молекулы воды. Конечным продуктом описанной реакции присоединения-отщепления является гидразон пиридоксальфосфата.
Образование гидразона приводит к блокированию альдегидной группы пиридоксальфосфата, что нарушает его взаимодействие как кофермента с аминогруппой глутаминовой кислоты. Эта реакция является одним из этапов превращения в организме глутаминовой кислоты в -аминомасляную. Блокирование же кофермента гидразином приводит к недостатку -аминомасляной кислоты, тормозящей проведение нервных импульсов.
Пример 2. В процессе метаболизма в живых организмах фумаровая кислота превращается в яблочную. Каким путем можно получить яблочную кислоту из фумаровой в условиях in vitro?
Р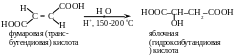 ешение.
Фумаровая кислота — ненасыщенная
двухосновная кислота, которую можно
рассматривать как замещенный алкен.
Яблочная кислота принадлежит к насыщенным
двухосновным гидроксикислотам.
ешение.
Фумаровая кислота — ненасыщенная
двухосновная кислота, которую можно
рассматривать как замещенный алкен.
Яблочная кислота принадлежит к насыщенным
двухосновным гидроксикислотам.
Переход от фумаровой кислоты к яблочной осуществляют путем присоединения воды по кратной связи, т. е. с помощью реакции гидратации. Гидратацию алкенов проводят в разбавленном водном растворе сильной кислоты, например серной. Кислота служит источником электрофильной частицы — протона Н+.
Электронная плотность углерод-углеродной -связи в молекуле фумаровой кислоты уменьшена вследствие электроноакцепторного действия двух карбоксильных групп. Поэтому гидратацию фумаровой кислоты осуществляют в сравнительно жестких условиях (нагревание с разбавленным водным раствором кислоты при температуре 150-200 °С).
Гидратация фумаровой кислоты протекает по обычному для алкенов механизму электрофильного присоединения АЕ. Протон взаимодействует с кратной связью в молекуле фумаровой кислоты. Образовавшийся карбокатион атакуется нуклеофильным реагентом – молекулой воды. Алкилоксониевый ион, являясь сильной кислотой, отщепляет протон (возврат катализатора). В результате образуется продукт реакции – яблочная кислота.
Гидратация фумаровой кислоты in vitro приводит к образованию рацемата – смеси равных количеств двух энантиомеров яблочной кислоты. В организме эта реакция катализируется ферментом фумаразой, для которого характерна строгая пространственная специфичность, что ведет к образованию только L-яблочной кислоты. Это пример селективного (избирательного) протекания реакции.
Направление химической реакции определяется совокупностью многих факторов.
Статические факторы. Реакционная способность соединений существенно зависит от распределения в их молекулах электронной плотности, которое в свою очередь определяется электронными эффектами заместителей и наличием сопряженных и ароматических фрагментов. Характерная для большинства соединений неравномерность в распределении электронной плотности является причиной появления в молекуле реакционных центров, предопределяющих направление атаки тем или иным реагентом (электронный фактор).
Пространственное строение молекулы определяет пространственный фактор, когда из-за относительно большого пространственного объема заместителей, окружающих реакционный центр, к нему может быть затруднен подход атакующей частицы. При этом реакция либо не будет осуществляться совсем, либо будет идти по иному направлению с участием другого, более доступного реакционного центра, если он имеется в молекуле.
Динамические факторы. Многостадийные процессы обычно включают стадии промежуточного образования нестабильных интермедиатов, обладающих высокой реакционной способностью. Часто можно предположить образование не одного, а нескольких интермедиатов. Реакция предпочтительно будет проходить через стадию образования относительно более устойчивого интермедиата. Относительная устойчивость интермедиатов, в частности часто выступающих в качестве высокореакционных промежуточных частиц карбокатионов, карбанионов и свободных радикалов, определяется возможностью делокализации в этих частицах электронной плотности.
Селективность реакций
Во многих случаях в органическом соединении присутствуют несколько неравноценных реакционных центров. В зависимости от строения продуктов реакции говорят о региоселективности, хемоселективности и стереоселективности реакции.
Региоселективность — предпочтительное протекание реакции по одному из нескольких реакционных центров молекулы.
Например, при взаимодействии пропана с бромом при УФ облучении в реакции преимущественно участвует один реакционный центр — связь С—Н вторичного атома углерода.
С Н3—СН2—СН3
+ Вr2
hv
120°С
СН3—СНВr—СН3
+ НВr
Н3—СН2—СН3
+ Вr2
hv
120°С
СН3—СНВr—СН3
+ НВr
Пропан 2-Бромпропан
Второй возможный изомер, 1-бромпропан, в этом процессе практически не образуется.
Хемоселективность — предпочтительное протекание реакции по одной из родственных функциональных групп.
Н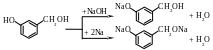 апример,
из двух нижеприведенных реакций с
участием п-гидроксибензилового спирта
только взаимодействие его с гидроксидом
натрия относится к хемоселективным
процессам.
апример,
из двух нижеприведенных реакций с
участием п-гидроксибензилового спирта
только взаимодействие его с гидроксидом
натрия относится к хемоселективным
процессам.
Стереоселективность — предпочтительное образование в реакции одного из нескольких возможных стереоизомеров.
Например, при катализируемом ферментом присоединении воды к фумаровой кислоте in vivo образуется только один из двух возможных здесь стереоизомеров — L-яблочная кислота
На способность соединений вступать в реакции также оказывают влияние индукционный и мезомерный эффекты заместителей.
Контрольные задания
1. Какая из кислот обладает самыми сильными кислотными свойствами:
а) уксусная; б) трибромуксусная; в) бромуксусная; г) стеариновая?
2. Какой электронный эффект атома галогена является причиной изменения кислотности у галогенсодержащих кислот: 1) положительный индукционный, 2) отрицательный индукционный, 3) положительный мезомерный, 4) отрицательный мезомерный?
3. Какое из соединений обладает более выраженными основными свойствами:
а) метиламин, б) диметиламин, в) анилин, г) 4-нитроанилин.
4. Укажите электронные эффекты (электронный эффект) метильного радикала:
1) положительный мезомерный; 2) отрицательный мезомерный; 3) нет эффекта,
4) положительный индуктивный; 5) отрицательный индуктивный.
5. К какому типу относится реакция гидролиза галогенпроизводных углеводородов:
СН3–СН2С1 + NaOH(водный р-р) СН3–СН2ОН + NaC1
а) электрофильное присоединение; б) нуклеофильное замещение;
в) элиминирование; г) электрофильное замещение; д) радикальное замещение.
6. По какому механизму протекает реакция синтеза кислоты из альдегида:
а) радикального замещения; б) нуклеофильного присоединения;
в) радикального присоединения; в) электрофильного присоединения;
г) электрофильного замещения; д) окисления-восстановления?
7. При обесцвечивании пропеном бромной воды образуется: 1,2-дибромпропан
К какому типу реакций относится взаимодействие пропена с водой в присутствии серной кислоты: 1) замещение; 2) элиминирование; 3) разложение; 4) присоединение