

Принцип действия triple quadrupole систем
Ионы, образовавшиеся в источнике ионизации, отправляются на первый квадруполь, где отсеиваются все, кроме интересующих Вас ионов, которые попадают в ячейку соударений, заполненные аргоном или азотом. Традиционно используется аргон, поскольку азот может давать аддукты (согласно утверждениям некоторых представителей аналитического оборудования, кроме того, откачивать азот сложнее, чем аргон), что усложняет интерпретацию MS/MS спектра. В ячейке соударений происходит столкновение Ваших ионов с газом-мишенью и, под действием напряжения (которое варьируется пользователем) образуется следующее поколение ионов.
Системы типа QqTof не рассматриваются по причине их аналогичности описанным, за исключением того что окончание не квадруполь, а времяпролетник.
DESI (desorption electrospray ionization) – на рисунке ниже представлен принципиальный способ ионизации. Нет никакой необходимости в использовании хроматографического разделения. Только напряжение и электроспрей. Заряженные частицы растворителя попадают на поверхность образца, часть образца ионизируется и попадает через систему линз в масс-спектрометр.
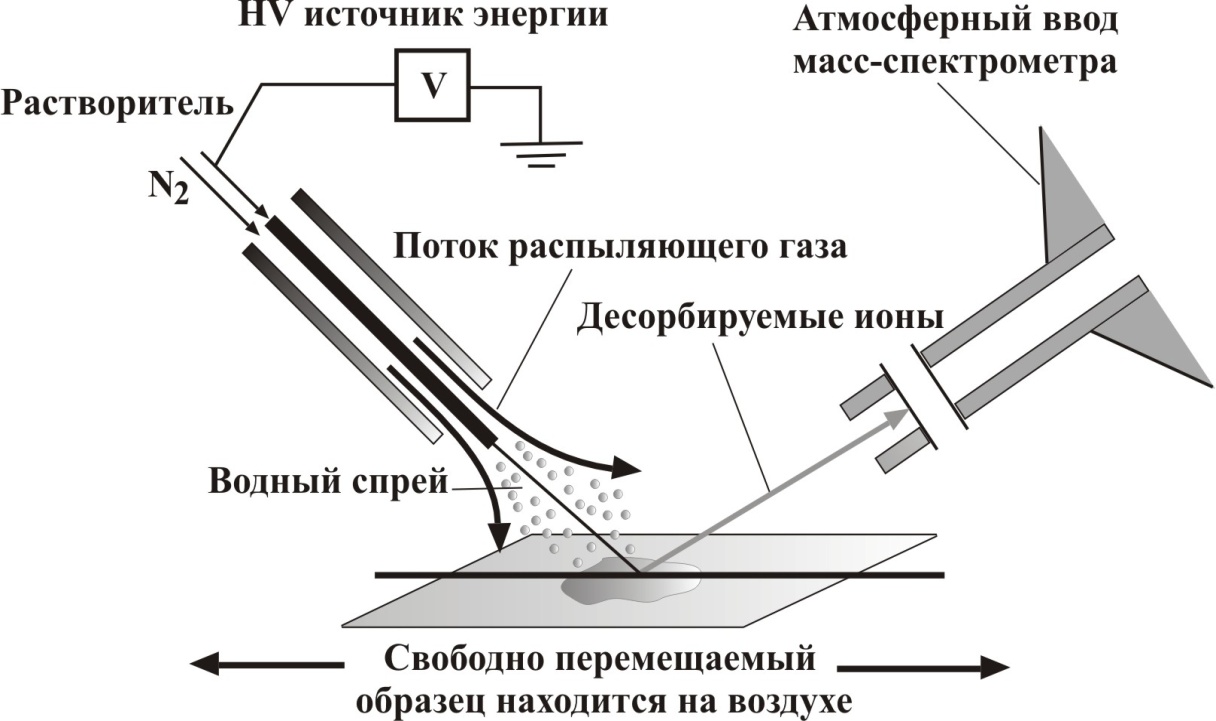
Spray ionization («Морковкин спрей и другие» (А. Т. Лебедев)) – принцип действия данного способа ионизации даже не буду объяснять. Все ясно из иллюстрации:
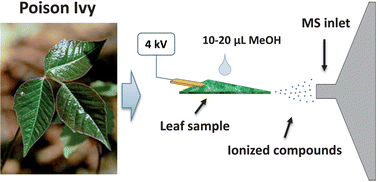
На треугольный кусочек образца (в данном случае листа) капают до 100 мкл метанола, подают высокое напряжение и все. Этого достаточно, чтобы ионизировать часть компонентов. И никакой пробоподготовки!
Качественный анализ в гх-мс
Зачастую для качественного анализа используются коммерческие библиотеки масс-спектров, такие как Wiley или NIST. Ежегодно эти библиотеки пополняются, однако стоит ли им верить на 100%? Конечно, нет. Скажем больше – библиотеки позволяют предположить круг веществ-кандидатов. И Вы, как оператор прибора, определяете, какая степень совпадения для Вас приемлема. Лично для меня нижней планкой является Match factor = 90%. Немного об алгоритмах поиска и работы библиотек: первостепенным для них является совпадение значений m/z, а не относительных интенсивностей. И это верно, поскольку мы уже знаем, что относительная интенсивность будет меняться от прибора к прибору. Более того, относительные интенсивности каждого из ионов могут меняться в зависимости от концентраций аналитов, поскольку свой отпечаток может наложить и матрица. О матричном влиянии мы поговорим немного позже. Стоит отметить, что относительная интенсивность пика величина не постоянная, она вполне может колебаться в достаточно широких пределах. По этому поводу позволю себе привести небольшую вставку нормативного документа ВАДА:
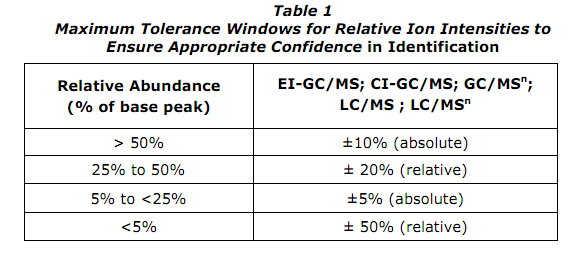
Итак, что же мы имеем? Колебания по абсолютным процентам – это значит колебания на 10% (например) по шкале общей. Колебания относительные – это относительно интенсивности иона в полученном спектре, а не по отношению к общей шкале.
Но что можно считать необходимым и достаточным условием надежной идентификации? Достаточно ли для этого одного лишь масс-спектра? Ответ однозначен: нет. Если исследуется новое соединение, то достаточным является снятие спектров ГХ-МС (ЭУ), ВЭЖХ-МС/МС со стандартом. В качестве стандарта используется вещество, полученное встречным синтезом. Однако, если есть возможность выделить соединение, то предпочтителен другой вариант: ГХ-МС (ЭУ), ВЭЖХ-МС/МС, ЯМР. По желанию добавляется ИК. Этот подход чаще применяют в органике, даже опуская ВЭЖХ-МС/МС, поскольку ЯМР предоставляет куда более полную информацию.
Итак, разобравшись со спектральными методами, перейдем к хроматографическим. Предположим, Вы синтезировали соединение, которое, предположительно, содержится в исследуемом образце, и планируете его использовать как стандарт. Тут же становится вопрос – а какие критерии идентификации по хроматографическим параметрам можно использовать? Вопрос отнюдь не праздный, поскольку разные вещества способны давать одинаковые спектры и иметь незначительные отличия во временах удерживания. Ключевое слово – незначительные. Каков же критерий существенной и несущественной разницы? И что можно использовать? Время удерживания или же индекс? Вопрос открытый до сих пор. На мой взгляд желательно использование индексов удерживания. По одной очень простой причине – это относительная величина, которая легко проверяется и является достаточно гибкой. Даже если в системе произошли какие-то изменения, Вы легко сможете продолжить работу, внеся лишь одну поправку, в отличии от использования времен удерживания (исключение – исправленное время удерживания). Кроме того, хорошим тоном в хроматографии считается указать индексы удерживания аналитов. Более того, в библиотеках НИСТ они указываются, если они были установлены.
Но чаще всего используют времена удерживания. Почему? Это проще. Не надо ничего считать. В этом случае надо следить за стабильностью системы. Итак, какие критерии значимы, а какие – нет? Для индексов удерживания значимым является отличие больше, чем на 5 единиц, а для времен удерживания отличие времени удерживания аналита от времени удерживания стандарта на 2% от общего времени хроматограммы или же отличие на 0,1 мин, в зависмости от того, какая величина меньше. Именно такие требования выдвигает ВАДА, они являются достаточно строгими, но их соблюдение значительно повышает надежность вашего анализа.
Последний важный аспект, о котором мы еще не поговорили в рамках качественного анализа – это совмещение качественного и количественного анализа. В этом случае съемка проводится в режиме SIM (почему – см. в количественном анализе). Но для надежной идентификации выдвигается требование – соединение является идентифицированным, если имеются хотя бы 3 характеристичных иона: 1 количественный и как минимум 2 диагностических. Для чего это нужно? Посмотрите на рис. 1. Здесь Вы видите, что вершины пиков совпадают. Можно говорить о том, что все ионы принадлежат одному и тому же соединению.
Рис.1
А теперь обратите внимание на рис. 2. Вершина одного из ионов не совпадает с остальными. В этом случае можно утверждать, что с большой вероятностью мы имеем 2 соединения в одном пике.

Рис. 2
Именно поэтому стоит уделять большое внимание получаемым результатам.
Рассмотрим еще один пример. Вот хроматограмма по полному ионному току:
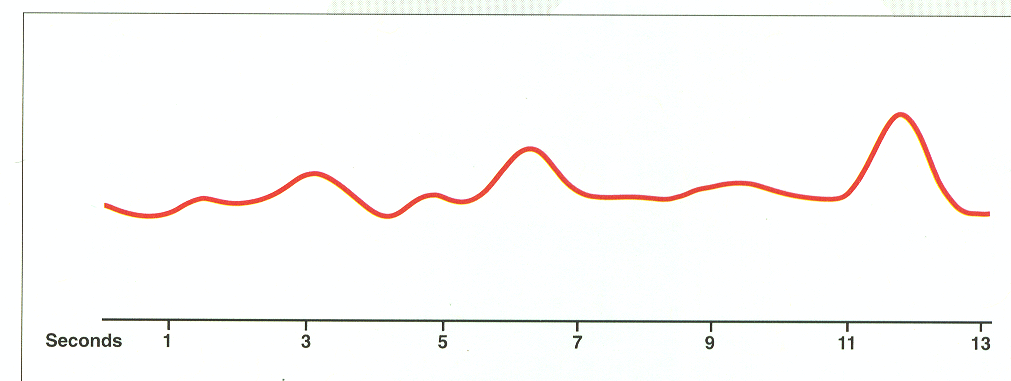
Ну нифига ж непонятно, правда? Да и казалось бы, сколько здесь может быть соединений? Ну, 7-8. А теперь выделим характеристичные ионы:
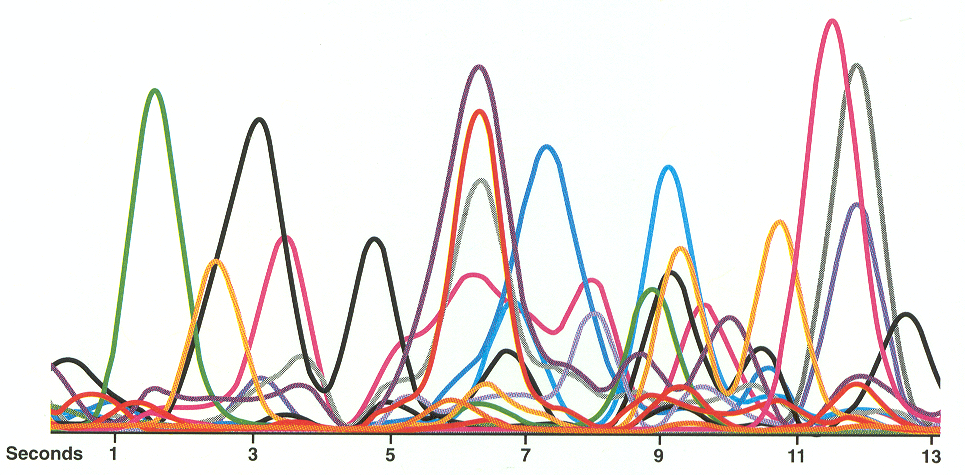
Что же мы видим? Да их здесь больше 30.