Моногенні (молекулярні) спадкові захворювання
Причина генних захворювань — мутація гена. Генні мутації можуть з'являтися в структурних (основна маса хвороб) і регуляторних генах.
Більшість генних захворювань — це порушення обміну речовин. Послідовність патогенезу цих захворювань відбувається за схемою: ген — фермент — біохімічна реакція — ознака хвороби. Мутація гена спричинює такі наслідки: змінюється або зникає фермент, унаслідок цього не відбувається певна метаболічна реакція, що призводить до зміни або порушення розвитку окремих ознак організму.
Кожна генна мутація зумовлює зміну активності або відсутність білка. Від первинного аномального продукту починається порушений ланцюг біохімічних реакцій, який і призводить до клінічних проявів хвороби. На тлі порушеного обміну речовин розгортаються всі патологічні реакції, симптоми, синдроми. Крім того, на відміну від набутих захворювань, під час спадкових хвороб етіологічний чинник (функціонування мутантного гена) діє постійно. І через це спадкові захворювання тривають переважно безперервно і прогресують.
Для всіх моногенних захворювань характерні генетична гетерозиготність і клінічний поліморфізм.
За характером успадковування моногенні хвороби поділяють на три групи:
аутосомно-домінантні;
аутосомно-рецесивні;
зчеплені зі статтю.
Найпоширеніша і найбільш вивчена група моногенних захворювань — це ензимопатії (ферментопатії), тобто порушення структури білків-ферментів, які беруть участь в обміні речовин. У разі ензимопатії в організмі наявний дефіцит кінцевого продукту обміну внаслідок накопичення проміжних речовин.
Для ензимопатії характерний сімейний характер захворювання і хронічний, прогредієнтний, рецидивний перебіг.
Молекулярні хвороби вуглеводного, амінокислотного, ліпідного та мінерального обміну
Молекулярні хвороби вуглеводного обміну
Галактоземія — досить рідкісне захворювання (1 : 50 000 — 1 : ЗО 000), пов'язане з непереносністю новонародженими грудного молока. Під час вигодовування в дитини порушується обмі молочного цукру галактози. У нормі галактоза за допомогою фе] менту галактозо-1-фосфат-уридинтрансферази перетворюється організмі на галактозо-1-фосфат, який потім перетворюється на глюкозо-6-фосфат, що входить до метаболічного циклу глюкози.
У разі галактоземії, внаслідок мутації гена, у дитини спостерігається недостатність цього ферменту, що зумовлює накопичення надмірної кількості галактозо-1-фосфату та інших продуктів неповного розпаду лактози, які є токсичними для тканин організму. У дитини розвиваються цироз печінки, рання катаракта, уражуються нирки. Симптоми виявляються одразу після першого годування грудним молоком. З'являються блювання, пронос. Зменшується маса тіла, розвивається жовтяниця, гепатомегалія, анемія, мікроцефалія, з'являються кровотечі й геморагії на шкірі, часто бувають судоми, у подальшому — ознаки цирозу печінки і відставання в психічному розвитку. Якщо не буде належного лікування, такі діти вмирають у перші місяці життя. їх життя залежить від ранньої діагностики захворювання.
Різні типи галактоземії й труднощі біохімічної діагностики ускладнюють своєчасне розпізнавання цієї патології. Найнадійпішими діагностичними методами є виявлення галактози в крові 9а допомогою хроматографії на папері, а також пряме визначення активності галактозо-1-фосфат-уридинтрансферази в еритроцитах та виявлення галактози в сечі.
Ранню діагностику проводять також на ауксотрофних мікробах.
Лікують галактоземію спеціальною дієтою (без галактози) або припиняють годувати грудним молоком, замість нього вигодовують коров'ячим, яке містить менше галактози.
Фруктоземія — це спадкове захворювання, що передається за аутосомно-рецесивним типом. Воно пов'язане з різко зниженою активністю ферменту фруктозо- 1-фосфатальдолази. Поширеність його — 1 : 20 000 населення. При недостатній активності цього ферменту асиміляція фруктози затримується до фруктозо-1-фосфату, що накопичується в тканинах і завдає токсичного впливу на клітини.
Клініка фруктоземії нагаДує галактоземію, але вона зазвичай виникає, коли дитина починає одержувати соки, плоди й овочі або споживати цукор. Організм дитини не сприймає фрукти. У неї з'являється блювання, характерний розвиток гіпотрофії, ге-иатоспленомегалії, асциту. Споживання великої кількості фруктози може призвести до гострої гіпоглікемії, що супроводжується судомами, тремором і розвитком коматозного стану.
Діагностика фруктоземії полягає у виявленні фруктозурії (реакція Селіванова) при навантаженні фруктозою, гіпоглікемії, гі-пофосфатемії, ознак ураження печінки.
Лікування фруктоземії полягає у виключенні з раціону соків, ягід, фруктів і овочів. При своєчасному виключенні фруктози дитина розвивається нормально, і прояви захворювання набувають зворотного розвитку.
Молекулярні хвороби амінокислотного обміну
Алкаптонурія. Алкаптонурію описав К. Бедекер у 1859 р., але біохімічно дослідив і вивчив Н. Геррод у 1908 р. Н. Геррод дійшов висновку, що:
алкаптонурія зумовлена недостатністю оксидази гомоген-тизинової кислоти, яка в нормі сприяє перетворенню цієї кислоти на метилоцтову кислоту;
індивід має виражену форму алкаптонурії або перебуває в нормальному стані; тобто або з організму людини виділяється кілька грамів гомогентизинової кислоти на день, або кислота зовсім не виділяється. Проміжних станів немає;
захворювання природжене;
захворювання трапляється в сибсів, а не в батьків;
у випадку алкаптонурії батьки зазвичай кузени. Алкаптонурія успадковується тільки за АР-типом, тому вона
трапляється в окремих родинах. Захворювання виявляється хронічним артритом унаслідок відкладання в суглобах, хрящах ті сполучних тканинах меланіноїдних речовин. Потім ці речовин; забарвлюються в колір вохри (охроноз). Забарвлення набуваютз також ендокард, великі судини, нирки, легені. Алкаптонурі, може супроводжуватися хворобами серця і судин, атероскле] зом, нирковокам'яною хворобою. У хворих на алкаптонурію внаслідок мутації гена відсутній фермент оксидаза гомогентизинової кислоти. Вона накопичується в організмі й частково виводитьс: із сечею. Сеча таких хворих на повітрі темнішає, тому що відбувається окиснення гомогентизинової кислоти. Це є важливою ознакою і дає змогу розпізнати хворобу в ранньому дитинстві й розпочати своєчасне лікування.
Лікують алкаптонурію великими дозами аскорбінової кислоти. Це значно пом'якшує симптоми хвороби.
Фенілкетонурія (ФКУ). Це захворювання зумовлене дефектої фені лаланінгідрокси лази, унаслідок чого амінокислота фенілаланін не може пройти процесу перетворення тирозин — тироксин меланін і адреналін. Фенілаланін накопичується в рідинах організму, в яких утворюються кетокислоти. Вони накопичуються організмі й стають токсичними продуктами для нервової системи, особливо для клітин мозку та його кори. Усе це спричинює затримку розвитку головного мозку, а також руйнування його клітин. У дитини розвивається тяжкий ступінь розумової відсталості. Недостатність тирозину призводить до зменшення утворення адреналіну і меланіну, тому в разі такого захворювання спостерігається депігментація волосся, райдужної оболонки. У людей з таким захворюванням світла шкіра, світле волосся й блакитні о1 (фенотипово — блакитноокі блондини).
Хвороба розвивається повільно й підступно. Новонароджений з ФКУ — клінічно здоровий. Це пояснюється тим, що відбувається вирівнювання ензимопатичного дефіциту в період внутріш ньоутробного життя через ензими матері, які проникають крізь плаценту. Але з перших тижнів життя в новонародженого розииваються клінічні ознаки неврологічної патології: підвищена ибудливість, посилені сухожилкові рефлекси, гіпертонія м'язів, тремтіння; судомні напади у вигляді кивків, диспепсія.
Ранні симптоми цього захворювання: блювання, яке імітує спазм чи вегетативні розлади, що проявляються пітливістю, акро-ціанозом. З'являється характерний для ФКУ мишачий запах сечі пі поту, зумовлений наявністю ортогідроксифенілоцтової кислоти. Пізніше, на 4—5-му місяці, спостерігаються затримка в розумовому й фізичному розвитку, мікроцефалія, збліднення шкірного покриву, волосся стає світлішим. Діти стають млявими, сонливими, не фіксують погляду на предметах, не здатні спілкуватися а батьками, у них ослаблена увага. У таких дітей пізно прорізуються зуби, затримується розвиток мовлення. Спостерігаються часті випадки нападів епілепсії. Психічні порушення прогресують до 4-річного віку. З віком у дитини знижується проникність гоматоенцефалічного бар'єра і зменшується загроза токсичного ураження мозку. Таким чигіом, особливо важливе значення має рання діагностика ФКУ і відповідне лікування суворою дієтою (усі продукти не повинні містити фенілаланіну).
Діагностика ФКУ: проведення тесту Гатрі в пологовому будинку дає змогу перевірити майже всіх новонароджених. Але деяких дітей не встигають обстежити, оскільки вони раніше 4-го дня життя потрапляють до інших відділень, а іноді їх з різних причин виписують без обстеження.
Щоб не пропустити виявлення хвороби, медичні працівники мають попереджати всіх батьків новонароджених про можливі наслідки непроходження тесту на ФКУ. Для цього потрібно обов'язково перевірити, чи є на обмінній карті позначка про аналіз крові на ФКУ. Якщо її немає, необхідно терміново здати кров дитини на аналіз.
У разі класичної ФКУ з першого тижня життя різко підвищується рівень фенілаланіну в сироватці крові — до ЗО мг % і більше.
Нині для діагностики ФКУ застосовують сучасний, екологічно чистий, кількісний флюорометричний метод на приладі "Флюороскан-2". За допомогою цього методу проводять контроль за лікуванням ФКУ.
Єдиним патогенетичним і досить успішним методом лікування ФКУ є дієтотерапія. З харчового раціону хворих повністю виключається фенілаланін. Дитину переводять на вигодовування спеціальним білковим гідролізатом, багатого на тирозин, триптофан, мінеральні солі і вітаміни, жири і вуглеводи (нефемікі нофелан, берлафен, лофенолакс, кетоніл, цимогран, гіпофенаї тощо). Однак фенілаланін належить до незамінних, тобто таки; що не синтезуються організмом людини, і має надходити в організм у кількості, потрібній для порівняно нормального фізично] розвитку дитини.
Не допустити розумової і фізичної неповноцінності дитини основне завдання лікування захворювання. Тому найголовніши] у лікуванні ФКУ є принцип гомеостатичності дієтотерапії. Вміє фенілаланіну в їжі має бути не більше, ніж 21 % вікової фізіол< гічної норми. Ця норма дає змогу запобігти як патологічному п] яву хвороби, так і порушенням фізичного розвитку.
За допомогою сучасних харчових раціонів для хворих на ФК' можна контролювати надходження фенілаланіну в організм, щоі воно точно відповідало його концентрації в крові. Рання діагностика і незволікання з призначенням дієтотерапії (у перші 2 З міс. життя) забезпечують нормальний розвиток дитини.
Для лікування важливою умовою є підтримання оптимальної кількості фенілаланіну в крові. У дітей грудного віку, які прох< дять лікування, цей показник має бути не нижчий ніж 2—3 мг % і не вищий ніж 6—8 мг %, у старших — до 10 мг %.
Контрольні аналізи проводять один раз на тиждень до вік; 6 міс, а з 6 міс. до 1 року — 2 рази на місяць.
У дітей з раннім виявленням і вчасно розпочатим лікування] хвороби прогнози сприятливі. Розумовий і фізичний розвиток таких дітей упродовж усього періоду лікування залишається но] мальним. Без проведення дієтотерапії в 100 % випадків розвивається тяжка розумова відсталість, ідіотія. На думку одних вчених, припиняти лікування можна у віці 4—6 років. Це безпечно не викликає побоювань щодо подальшого нормального розвитк; дитини. Інші вважають, що припиняти лікування в дошкільному віці передчасно, оскільки це може спричинити психічну деградацію пацієнтів.
Молекулярні хвороби ліпідного обміну
Хвороба Тея—Сакса. Це одна з форм амавротичної ідіоті (рання, дитяча). Характеризується клінічно прогресивним зниженням гостроти зору, прогресивною деградацією інтелекту, судомами та іншими неврологічними симптомами. У віці 4—6 міс. раніше активна дитина втрачає рухливість, цікавість до навю і и цінного середовища, не впізнає батьків, припиняє гратися, сміятися, не фіксує погляду, сліпне. Відзначається гіперреакція на :шук (аж до тонічних судом). На очному дні визначається симптом вишневої кісточки; наявна атрофія зорового нерва. Хвороба швидко прогресує, і через 1,5—2 роки від початку захворювання дитина помирає.
Первинний біохімічний дефект — зниження активності фруктозо- 1-фосфатальдолазигексозамінідази А. Хвороба частіше трапляється серед євреїв-ашкеназі, а також у разі кровноспорід-нених шлюбів. Ефективної терапії немає. Практикують симптоматичне лікування.
З метою діагностики проводять біохімічне дослідження крові й очного дна. Можлива пренатальна діагностика. Особливу увагу слід приділяти профілактиці цього захворювання та виявленню гетерозиготних носіїв патологічного гена.
Хвороба Гоше (глюкоцереброзний ліпідоз). Це генне захворювання, пов'язане з порушенйям ліпідного обміну. Перетворення одного ліпіду на інший відбувається за допомогою відповідного ферменту внаслідок мутації гена. Це призводить до накопичення в клітинах певних ліпідів.
У разі хвороби Гоше в клітинах головного мозку і внутрішніх органів накопичується глюкоцереброзид (сполука цераміду з глюкозою).
Розрізняють три форми хвороби: гостру, підгостру і хронічну. Розвиток її починається в певному віці, і в кожної переважає симптоматика, характерна тільки для певної з них.
У разі гострої форми симптоми захворювання з'являються в перші 2—3 міс. життя. Причому перше місце посідають неврологічні порушення. У дітей спостерігаються м'язова ригідність, порушення зору, косоокість, утруднення ковтання, спазм гортані, психічна деградація, остеопороз кистей, деформація стегон за типом колб Ерленгастера. Часто трапляються переломи кісток унаслідок розростання клітин Гоше в кістковому мозку. Зменшення кісткового мозку спричинює гіпохромну анемію, тромбо-пенію, які супроводжуються носовими та іншими кровотечами. Кардинальними ознаками є помітне збільшення печінки й селезінки, що виявляється у віці 3—6 міс, а також збільшення живота. Можливі бронхопневмонії внаслідок аспірації. На очному дні з'являється вишнево-червона пляма. Смерть настає від дихальних розладів на першому році життя.
У разі підгострої форми також переважають неврологічні симптоми: змінюється поведінка, спостерігаються судоми, відбуваються мозочкові зміни. Недоумство, збільшується живіт унаслідок гепатоспленомегалії, з'являється біль у кістках. Патологічні переломи. Асептичний некроз головки стегнової кістки, анемія, тромбоцитопенія. Також спостерігаються аномальна пігментація обличчя, шиї, гомілок, остеопороз. Клітини Гоше виявляються в кістковому мозку, печінці, лімфатичних вузлах. У тканинах мозку, печінки, селезінки, кістковому мозку наявна велика кількість глюкоцереброзидів. У лейкоцитах знижується активність В-глюкозидази. Усі ці симптоми в разі підгострої форми м'якші, ніж під час гострої.
У разі хронічної форми ушкоджуються лише внутрішні органи, головний мозок не залучається до процесу.
Молекулярні хвороби мінерального обміну
Хвороба Вільсона—Коновалова. Це спадкове захворювання, що характеризується поєднанням цирозу печінки з дистрофічним процесом у головному мозку. Успадковується за аутосомно-рецесивним типом.
Для прояву захворювання мають значення екзогенні впливи, які вражають печінку (інтоксикації та інфекції). Основну роль у патогенезі гепатоцеребральної дистрофії відіграють генетично зумовлені порушення обміну білків і міді. Порушення синтезу білків призводить до гіпераміноацидурії і гіпопротеїнемії, страждає й обмін нуклеотидів. Особливо велике значення має зменшення вмісту церулоплазміну — білка, який містить мідь і має ферментативні властивості оксидази. У результаті мідь виявляється слабко зв'язаною з альбуміном і амінокислотами крові, легко відщеплюється від них, у великій кількості виділяється із сечею і відкладається в тканинах — головним чином у печінці, головному мозку і рогівці.
Надлишок вільної міді пригнічує активність окисних і деяких інших ферментів, що призводить до загибелі клітин. Ураження печінки з розпадом її тканини і зниженням бар'єрної функції веде до аутоінтоксикації продуктами гепатоаутолізу і чужорідними продуктами, що надходять з кишок.
Діагноз установлюють за даними анамнезу про спадковий характер захворювання, наявністю рогівкового кільця, симптомів патології печінки (діагностична пункція печінки виявляє цироз і
великий вміст міді), явищ геморагічного діабету й особливо гіпер-купрурії (виділення за добу понад 200 мкг міді) і гіпераміноаци-дурії (понад 350 мкг за добу).
У лікуванні ефект дають тіолові препарати, що зв'язують мідь і виводять її з організму.
Муковісцидоз. Муковісцидоз, або кістозний фіброз (підшлункової залози), — одне зі спадкових захворювань, яке трапляється найчастіше, має моногенну природу та аутосомно-рецесивний тип успадковування.
Частота хворих на муковісцидоз у Європі становить приблизно І : 2000 новонароджених. Серед негритянського населення частота випадків становить майже 1 : 7000 новонароджених. Хворих на муковісцидоз практично немає в Китаї і Японії. Серед монголоїдної раси муковісцидоз трапляється з частотою 1 : 30 000 новонароджених. В Україні частота захворювань — 1: 2500 новонароджених.
Найпоширенішою причиною муковісцидозу є мутація (деле-ція) гена.
Патогенез ґрунтується на порушенні провідності йонів хлору мембранами клітин епітелію. Це спричинює зміну фізико-хімічних властивостей слизу ендокринних залоз і призводить до утворення густого, в'язкого секрету, обтурації, застійних явищ, фіброзу і склерозу в різних органах і тканинах організму дитини. За цих умов різко порушується сольовий обмін унаслідок надмірного потовиділення ("солона дитина", "гіркі сльози"), через під-иищену концентрацію натрію та хлору в поті, нігтях, волоссі.
Згідно із сучасними даними, первинну ланку муковісцидозу можна представити так:
етап — порушення нуклеотидної послідовності ДНК-кодувальної послідовності гена трансмембранного регуляторного білка муковісцидозу (ТРБМ) (понад 150 різних мутацій).
етап — структурно-функціональні порушення продукту гена ТРБМ; білка хлорного каналу клітинних мембран.
етап — функціональні порушення проникності клітинних мембран апікального епітелію. Дефектний ген функціонує як постійно "закритий" канал.
Для хвороби характерний поступовий розвиток, на 2—3-му місяці життя з'являється бронхіт. У подальшому в нього тривалий перебіг, і він не піддається антибактеріальній терапії. Хворі скаржаться на кашель, задишку, підвищення температури тіла,
зниження апетиту, посилюється загальна слабкість. З прогр< ванням хвороби кашель стає постійним, виснажливим, напад* подібним, кашлюкоподібним. Емфізема легень розвивається 100 % випадків. Найтяжчі ускладнення — абсцедивна пневмонія, пневмоторакс, легенева кровотеча.
Симптоми муковісцидозу дуже схожі з клінічними проява] пневмонії, але мають характерні ознаки.
За наявності змішаної форми (кишкової і легеневої) симптоми можуть виявлятися майже одночасно. Часто захворюванню починається з кишкових симптомів, а легеневий симптом приєднується пізніше. Кишковий прояв муковісцидозу пов'язані з порушенням активності ферментів підшлункової залози, залі кишок й ураженням печінки.
У період загострення кишкової форми муковісцидозу хво] перебувають у тяжкому стані. У них виражений симптом недостатності всмоктування. Гнильні процеси в кишках спричинююті здуття живота, появу рясних, жирних, дьогтеподібних випорожнень світло-жовтого або сірого кольору. Випорожнення буваю' до 6—8 разів на добу, з різким гнильним запахом. У багатьох хворих спостерігаються випадання прямої кишки, біліарний ци] печінки, гіпотрофія.
Меконіальний ілеус трапляється в 10—20 % новонароджені з муковісцидозом. За цих умов розвивається картина кишкові непрохідності: блювання з домішкою жовчі, невідходження м< конію, збільшення живота. Найзагрозливіше ускладнення — м< конієвий перитоніт.
Серед багатьох методів діагностування муковісцидозу найчастіше вдаються до антенатальної (пренатальної) діагностики, яка дає можливість запобігти народженню хворої дитини (вчас-, но перервати вагітність). Антенатальна діагностика муковісцидозу ґрунтується на аналізі мутації гена ТРБМ у зразках ДНК (ДНК-діагностика), взятих із матеріалу плода. Цей матеріал різні терміни вагітності отримують шляхом біопсії хоріона (] триместр вагітності), амніоцентезу (II триместр), кордоцентез; (III триместр). Цей метод характеризується високою точністю інформативністю.
Під час біохімічної пренатальної діагностики муковісцидозу, яку проводять на матеріалі амніотичної рідини, найголовнішим є визначення активності деяких ферментів кишкових мікроворси-нок плода.
Ці методи, які застосовують у більшості країн світу і в Украї-ііі, є найефективнішим засобом подолання муковісцидозу.
Рання антенатальна і постнатальна діагностика муковісци-ДОЗу ґрунтується на типових клінічних симптомах, генетичному анамнезі, виявленні високої концентрації хлоридів у потовій рідині й відсутності панкреатичних ферментів у дуоденальному І місті.
"Потовий тест" фактично є патогномонічним для муковісцидозу.
Пренатальна діагностика спадкової патології
Пренатальна діагностика природжених і спадкових хвороб — це комплексна галузь медицини, яка швидко розвивається. Вона застосовує й ультразвукову діагностику (УЗД), й оперативну техніку (хоріонбіопсію, амніо- і кордоцентез, біопсію м'язів і шкіри плода), і лабораторні Методи (цитогенетичні, біохімічні, молекулярно-генетичні).
Пренатальна діагностика має винятково важливе значення під час медико-генетичного консультування, оскільки це дає можливість перейти від вірогідного до однозначного прогнозу-шшня здоров'я дитини в родинах з генетичним обтяженням. Пренатальну діагностику здійснюють у І і II триместрах вагітності, тобто в періоди, коли у випадку виявлення патології ще можна перервати вагітність. На сьогодні можлива діагностика практично всіх хромосомних синдромів і близько 100 спадкових хвороб, біохімічний дефект яких встановлено вірогідно.
Питання про проведення пренатального переривання вагітності має статися тільки після оцінювання таких критеріїв.
1. Хвороба має бути досить тяжкою, щоб було виправдане пе- реривання вагітності.
2. Лікування хвороби плода неможливе і незадовільне.
Родина, що консультується, повинна бути згодна на переривання вагітності.
Існує точний тест для встановлення пренатального діагнозу.
Досить високий генетичний ризик несприятливого результату вагітності.
При організації розвитку системи пренатальної діагностики мають виконуватися такі умови:
Діагностичні процедури мають бути безпечні для здоров'; матері і плода.
Частота ускладнень вагітності після пренатальної діагностики не повинна помітно підвищуватися зі спонтанним рівнем, тобто процедура не повинна підвищувати ймовірності втрати плі да відразу чи після її проведення у віддалений період.
Лікарі, що володіють технікою пренатальної діагностики, мають знати ймовірність установлення псевдопозитивних чі псевдонегативних діагнозів, іншими словами, повинні добре знати обмеження методу.
4. Пренатальна діагностика має включати два етапи:
виявлення жінок з підвищеним ризиком несприятливого в генетичному плані результату вагітності під час медико-генетичного консультування чи первинного обстеження всі: вагітних, у тому числі з використанням методів просіваючої діагностики;
власне пренатальна діагностика. Аналізи проводять тількз жінкам, що мають фактори ризику.
5. Група фахівців з пренатальної діагностики (акушер- гінеколог, лікар-генетик, лікар-лаборант-генетик) повинні знати діагностичні обмеження методу не взагалі, а в їхній власній лабо- раторії.
6. Група фахівців повинна суворо дотримуватися стандартів для процедур і лабораторних аналізів, здійснювати точний конт- роль якості роботи, а також мати статистику завершення вагіт- ностей і розбіжностей діагнозів (контроль після абортів чи післ; народження).
Показання для пренатальної діагностики:
вік матері визначений у 35 років;
наявність у родині попередньої дитини з хромосомною патологією, у тому числі із синдромом Дауна;
перебудова батьківських хромосом;
наявність у родині захворювань, успадковуваних зчеплено зі статтю;
природжені "помилки" метаболізму;
інші показання для цитогенетичної пренатальної діагностики.
Інвазивні методи дослідження в пренатальній діагностиці
Амніоцентез — прокол плодового міхура з метою одержання навколоплідної рідини і злущених клітин амніона плода. Діагностичне значення методу не викликає сумнівів. Ця процедура здійснюється на 15—18-му тижні вагітності, ризик ускладнень вагітності при амніоцентезі становить 0,2 %.
Кордоцентез, тобто взяття крові з пуповини, стали використовувати частіше після того, як цю процедуру почали здійснювати під контролем УЗД, тобто без фетоскопії. Процедуру проводять у термін з 18-го по 22-й тижні вагітності; зразки крові є об'єктом для цитогенетичних (культивуються лімфоцити), молекулярно-генетичних і біохімічних методів діагностики спадкових хвороб.
Кордоцентез застосовують для діагностики хромосомних та гематологічних спадкових хвороб (гемоглобінопатії, коагулопа-тії, тромбоцитопенії), імунодефіцитів, гематологічного статусу в разі резус-сенсибілізації, внутрішньоутробних інфекцій.
Біопсія тканин плода як'діагностична процедура здійснюється у II триместрі вагітності під контролем УЗД.
Неінвазивні методи дослідження в пренатальній діагностиці. Основним неінвазивним методом пренатальної діагностики є ультразвукове дослідження (УЗД), яке необхідно проводити усім вагітним. Ультразвукове сканування плода проводять не менше як два рази кожній жінці. Перший огляд не пізніше ніж через 15—16 тиж., другий — у 25—26 тиж.
РОЗДІЛ 8
Біологія індивідуального розвитку.
Молекулярно-генетичні механізми онтогенезу.
Патологічні порушення онтогенезу людини
Гаметогенез
Процес формування статевих клітин (гамет) відомий під за гальною назвою гаметогенез. Він характеризується важливим біологічними процесами і відбувається з деякими відмінностям під час дозрівання сперматозоїдів (сперматогенез) і яйцекліт (овогенез).
Сперматогенез. Сім'яник складається з безлічі канальців. поперечному перерізі крізь канадець можна спостерігати кіл ка шарів клітин. Це послідовні стадії розвитку сперматозоїд' (мал. 18).
Зовнішній шар (зона розмноження) утворений сперматогонія ми — клітинами кулястої форми, з відносно великим ядром і зн чною кількістю цитоплазми. У період ембріонального розвитку після народження до статевого дозрівання сперматогонії ділятьс шляхом мітозу, унаслідок чого збільшується кількість клітин розміри сім'яника.
Після настання статевої зрілості частина сперматогоній тако продовжує ділитися мітотично й утворює клітини, частина з яки переміщується в наступну зону — зону росту, яка розташован ближче до просвіту канальця. Тут відбувається значне збільшен ня розмірів клітин унаслідок підвищення кількості цитоплазм На цій стадії їх називають первинними сперматоцитами.
Третя зона розвитку чоловічих гамет називається зоною д зрівання. У цей період відбуваються два поділи мейозу, швидко проходять один за одним, у результаті зазнає перебудо ви хромосомний апарат. З кожного первинного сперматоцит після мейозу спочатку утворюються два вторинні сперматоци
4
т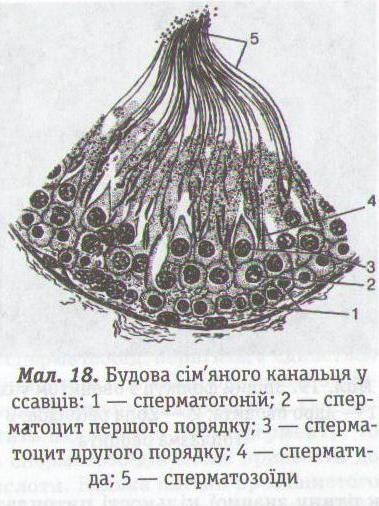 и,
а потім — чотири сперматиди, у яких
овальна форма і значно менші розміри.
Четверта зона розвитку
— формування. Сперматиди переміщуються
ближчі
до
просвіту канальця, де з
них
формуються сперматозоїди.
и,
а потім — чотири сперматиди, у яких
овальна форма і значно менші розміри.
Четверта зона розвитку
— формування. Сперматиди переміщуються
ближчі
до
просвіту канальця, де з
них
формуються сперматозоїди.
3
У більшості тварин сперматогенез
відбувається в певні періоди року.
У проміжках між ними в каналі цих
сім'яників містяться
лише сперматогонії.
У людини і
більшості свійських
тварин сперматогенез
постійний.
Овогенез. Фази овогенезу
подібні до фаз сперматогенезу.
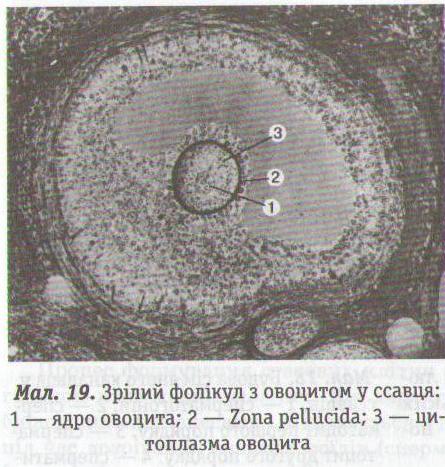
У цьому процесі також є період розмноження, коли інтенсивно діляться овогонії — дрібні клітини з відносно великим ядром і незначною кількістю цитоплазми. У ссавців і людини цей період закінчується ще до народження. Сформовані первинні овоцити ;осрі гаються бел ВМІВ тривалий час (місяці і роки). З настанням статевої зрілості окремі овоцити періодично вступають у період росту, клітини збільшуються, у них нагромаджуються жовток, жир, пігменти. У цитоплазмі клітини, в її органелах і мембранах нідбуваються складні морфологічні та біохімічні перетворення. Кожний овоцит оточений дрібними фолікулярними клітинами, І ксі забезпечують його живлення (мал. 19).
Потім настає період дозрівання, коли відбуваються два послідовні поділи мейозу з перебудовою хромосомного апарату. Крім того, ці поділи супроводжуються нерівномірним розподілом цитоплазми між дочірніми клітинами. При поділі первинного ово-цита утворюється одна велика клітина — вторинний овоцит, яка вбирає майже всю цитоплазму, і маленька клітина — первинний полоцит. Під час другого мейотичного поділу цитоплазма знову розподіляється нерівномірно. Утворюються один великий вторинний овоцит і вторинний полоцит. У цей час первинний поло
цит також може поді литися на дві кліти ни. Таким чином, і одного первинног овоцита утворюютьс один вторинний ово цит і три полоцит (редукційні тільця) Потім із вторинно овоцита формуєтьс яйцеклітина, а поло цити розсмоктуют ся або зберігаютьс на поверхні яйця але не беруть учас в подальшому розви тку. Нерівномірни розподіл забезпечу надходження в яйце
клітину значної кількості цитоплазми і поживних речовин, як будуть потрібні в майбутньому для розвитку зародка.
У ссавців і людини періоди розмноження і росту яйцекліти відбуваються у фолікулах. Зрілий фолікул заповнений рідиною усередині його — яйцеклітина. Під час овуляції стінка фолікула тріскається, яйцеклітина потрапляє в черевну порожнину, а потім, як правило, — у труби матки. Період дозрівання яйцеклітин відбувається в маткових трубах, де вони й запліднюються.
У багатьох тварин овогенез і дозрівання яйцеклітин здійсню ються тільки в певні сезони року. У жінок зазвичай щомісячно дозріває одна яйцеклітина, а за весь період статевої зрілості — близько 400. Для людини має суттєве значення те, що первинні овоцити формуються ще до-народження, зберігаються все життя і тільки поступово деякі з них починають дозрівати і дають яйцеклітини. Це означає, що різні несприятливі чинники, які діють упродовж життя на жіночий організм, можуть вплинути на їхній подальший розвиток. Так, отруйні речовини (зокрема нікотин і алкоголь), які потрапляють в організм, можуть проникнути в овоцит і потім спричинити порушення нормального розвитку майбутнього потомства.