

Билет 1.
Предмет, задачи и методы общей и неорганической химии, ее место в системе естественных наук, значение для развития кибернетики.
Химия изучает состав, свойства и превращения веществ, а также явления, которые сопровождают эти превращения, и законы, которым они подчиняются.
Предмет химии — химические элементы и их соединения, а также закономерности, которым подчиняются различные химические реакции.
Классические задачи Х. — установление состава и строения веществ — всё успешнее решаются с использованием новейших физических методов. Неотъемлемой чертой теоретической и экспериментальной Х. стало применение новейшей быстродействующей вычислительной техники для квантовохимических расчётов, выявления кинетических закономерностей, обработки спектроскопических данных, расчёта структуры и свойств сложных молекул.
Методы. Изучение химических объектов и явлений физическими методами, установление закономерностей химических превращений, исходя из общих принципов физики, лежит в основе физической химии. (рентгеновский структурный анализ, масс-спектроскопия, радиоспектроскопия и др) Люминесцентный анализ, метод меченых атомов, рентгеновский структурный анализ, электронография, полярография и др. физико-химические методы анализа находят широкое применение в аналитической химии.
Место в системе естественных наук. Без химических реакций сегодня невозможно представить научную картину мира, ведь окружающий мир - это прежде всего мир веществ неорганических и органических, постоянно взаимодействуют и принимают участие в различных типах преобразований, которые являются основой многих явлений природы. Она так же, как физика, ботаника, зоология, геология, изучает природу, весь окружающий мир - различные вещества и явления.
Кибернетика медицинская, научное направление, связанное с проникновением идей, методов и технических средств кибернетики в медицину. Развитие идей и методов кибернетики в медицине осуществляется в основном в направлениях создания диагностических систем для различных классов заболеваний с использованием универсальных или специализированных ЭВМ.Успехи химии, внедрение ее продуктов в медицину открывают безграничные возможности для преодоления ряда заболеваний, в первую очередь вирусных и сердечно – сосудистых.
Билет 2.
Массовая доля – амега=m вещ/m р-ра * 100%
Объемная доля – фи=Vвещ/Vр-ра * 100%
Молярная конц-я – С= nвещ/Vр-ра
Моляльная конц-я – Сm=nвещ/mр-ля
Эквивалент – реаль или услов частица вещ-ва, кот в данной кислот-основ реакции может отдавать, присоединять или зам-ть 1 атом или катион H, или в ОВР быть эквивалентной 1му электору.(Э)
Фактор эквивалентности – число, кот показ какая доля реаль или услов частиц вещ-ва Х эквивалентна 1 Н в дан реакции или 1 электрону в ОВР.(fэкв)
Молярная конц-я эквивалента – показ, какое кол-во вещ-ва наход-ся в единице Vр-ра.(Сэкв=nэкв/Vр-ра=m/Mэкв=m/VMfэкв)
Закон эквивалентов – все вещества реагируют и образуются в эквивалентных отношениях.
формула, выражающая Закон эквивалентов: m1Э2=m2Э1.
Билет 3.
Кинетическое уравнение, выражает зависимость скорости хим. реакции от концентраций компонентов реакционной смеси. Для простой (одностадийной) гомогенной реакции скорость v пропорциональна произведению концентраций реагирующих веществ и кинетическое уравнение записывается в виде: где [Ai] (i=1,2,...,l) - концентрация i-го вещества, ni-порядок реакции по i-му веществу, k-константа скорости р-ции.
Завис скорости реак-и от конц-и - скорость прямопропорц произвед конц-й реагир вещ-в, взятых в степенях их стехеометрич коэф-ов.
![]()
Порядок хим реакций
Порядок реакции
Порядок реакции, понятие кинетики химической. П. р. определяется как сумма показателей степеней n1 и n2 в уравнении
![]()
выражающем зависимость скорости реакции r от концентраций [A1] и [А2] исходных веществ (k — константа скорости). Реакции с n1+ n2 = 1, 2 и т.д. называются реакциями 1-го, 2-го и т.д. порядков. Отдельный показатель степени в уравнении (1) называется порядком реакции по соответствующему веществу.
У простых реакций скорость в одном направлении, согласно действующих масс закону, подчиняется уравнению (1), а n1и n2 совпадают с числом молекул веществ A1 и А2, участвующих в элементарном акте реакции. Скорости сложных реакций иногда также выражаются уравнениями вида (1), но при этом П. р. может не совпадать со стехиометрическим коэффициентом вещества в уравнении реакции (записанном с наименьшими целочисленными стехиометрическими коэффициентами), может быть дробным числом. В гетерогенно-каталитических реакциях обычны наряду с целыми дробные и нулевые порядки; встречаются также отрицательные порядки.
Константа скорости реакции.
Константа скорости реакции (удельная скорость реакции) — коэффициент пропорциональности в кинетическом уравнении.
Физический смысл константы скорости реакции k следует из уравнения закона действующих масс: k численно равна скорости реакции при концентрации каждого из реагирующих веществ равной 1 моль/л.
Константа скорости реакции зависит от температуры, от природы реагирующих веществ, но не зависит от их концентрации.
Закон действующих масс.
Закон действующих масс в кинетической форме (основное уравнение кинетики) гласит, что скорость элементарной химической реакции пропорциональна произведению концентраций реагентов в степенях, равных стехиометрическим коэффициентам в уравнении реакции
Химическое равновесие — состояние химической системы, в котором обратимо протекает одна или несколько химических реакций, причём скорости в каждой паре прямая-обратная реакция равны между собой. Для системы, находящейся в химическом равновесии, концентрации реагентов, температура и другие параметры системы не изменяются со временем.
Конста́нта равнове́сия — величина, определяющая для данной химической реакции соотношение между термодинамическими активностями (либо, в зависимости от условий протекания реакции, парциальными давлениями, концентрациями или фугитивностями) исходных веществ и продуктов в состоянии химического равновесия (в соответствии с законом действующих масс).К=К1/К2=Спродукта/Сисход веществ.
Билет 4.
Раствор - гомогенная (однородная) смесь, состоящая из частиц растворённого вещества, растворителя и продуктов их взаимодействия.
Растворитель - компонент, агрегатное состояние которого не изменяется при образовании раствора. В случае же растворов, образующихся при смешении газа с газом, жидкости с жидкостью, твёрдого вещества с твёрдым, растворителем считается компонент, количество которого в растворе преобладает.
Растворенное вещество – остальные компоненты
Растворимость - способность вещества образовывать с другим веществом однородную, термодинамически устойчивую систему переменного состава, состоящую из двух или большего числа компонентов.
Твёрдые, жидкие, газообразные растворы
Чаще под раствором подразумевается жидкое вещество, например раствор соли или спирта в воде (или даже раствор золота в ртути — амальгама).
Существуют также растворы газов в жидкостях, газов в газах и жидкостей в жидкостях, в последнем случае растворителем считается вода, или же компонент, которого больше.
В химической практике обычно под растворами понимают гомогенные системы, растворитель может быть жидким, твёрдым (твёрдый раствор), газообразным. Однако нередко допускается и микрогетерогенность — см. «Золи».
«Раствором» именуют и смесь цемента с водой, песком и так далее. Хотя это и не является раствором в химическом смысле этого слова.
Идеальный раствор - раствор, для которого выполняется первый закон Рауля.
Идеальными при любых концентрациях являются растворы, компоненты которых близки по физическим и химическим свойствам и образование которых не сопровождается объёмными и тепловыми эффектами. В этом случае силы межмолекулярного взаимодействия между однородными и разнородными частицами примерно одинаковы, и образование раствора обусловлено лишь энтропийным фактором.
Билет 5.
Коллигативные св-ва разбав р-ов – свойства, зависящие только от кинетически активных частиц, не зависящие от природы вещества.
Осмос – самопроизвольная диффузия молекул растворителя сквозь мембрану с избирательно проницаемостью.
Обратный осмос и роль в очистке воды. Обратный осмос является натуральным процессом, суть которого заключается в отделении мембраной молекул воды от растворенных в ней субстанций. Благодаря такой очистке из воды удаляются органические и неорганические соединения, а также тяжелые металлы, бактерии и вирусы.
Осмотическое давление – избыточное гидростатическое давление, возникающее в результате осмоса и приводящее к выравниванию скоростей взаимного проникновения молекул растворителя сквозь мембрану с избирательной проницаемостью.
Правило
Вант-Гоффа – при
увеличении температуры на каждые 10
градусов скорость хим реакции возрастает
в 2 раза![]()
Полупроницаемые мембраны – ограничена для веществ ( зависит от размера молекул) не все вещества могут пройти.
Осмолярность - сумма концентраций катионов анионов и неэлектролитов, т.е. всех кинетически активных частиц в 1л. раствора. Она выражается в миллиосмолях на литр (мосм/л).
Осмоляльность - концентрация тех же частиц, растворенных в килограмме воды, выражающаяся в миллиосмолях на килограмм (мосм/кг).
Осмотическое давление плазмы крови - Осмотическое давление плазмы в основном создается неорганическими солями, поскольку концентрация сахара, белков, мочевины и других органических веществ, растворенных в плазме, невелика. Благодаря осмотическому давлению происходит проникновение жидкости через клеточные оболочки, что обеспечивает обмен воды между кровью и тканями.
Билет 6.
Плазмолиз – сморщивание протопласта, от-хождение его от клеточной целлюлезной оболочки, наблюдающееся при погружении растительной клетки, окруженной твердой стенкой, в гипертонический раствор какого-либо вещества, при экзоосмосе.
Гемолиз – осмотический шок, лопание клетки при эндоосмосе.
Гипоизотонический раствор – раствор с меньшим осмотическим давлением.
Гиперизотонический раствор – с большим осмотическим давлением.
Изотонический раствор – одинаковые осмотические давления.
Коллоидно-осмотическое(онкотическое) давление плазмы крови – осмотическое давление, создаваемое за счет белков в плазме крови 2,5 – 4 кПа
Распределение воды в организме между сосудистым руслом и межклеточным пространством Немногим более половины воды организма находится внутри клеток. Из общего количества внеклеточной воды примерно 15— 20% приходится на плазму. Все остальное составляет внесосудпстая внеклеточная интерстициальная жидкость.
Проникновение воды через стенки клеток определяется существующей in vivo разницей осмотического давления между внутрии внеклеточными жидкостями. Перемещение воды через стенки кровеносных сосудов зависит от соотношения in vivo эффективного осмотического (или онкотического) давления плазмы крови п общего наружного гидростатического давления. Понимание этих факторов важно для правильной интерпретации результатов исследований электролитов плазмы крови.
Билет 7.
Давление насыщенного пара над раствором – давление, при котором при данной температуре в системе жидкость-пар наступает динамическое равновесие, харак-ся равенством испарения и конденсации.
Закон Рауля – давление пара раствора, содержащее нелетучее вещество прямо пропорционально доле растворителя. При постоянной температуре относительное понижение давления насыщенного пара растворителя над идеальным раствором нелетучего вещества равно молярной доле растворенного вещества. (это сумма результатов умножения значений давления насыщенного пара каждого компонента на мольную долю этого компонента в растворе.)
Повышение температуры кипения или
Понижение температуры замерзания идеального раствора нелетучего вещества прямопропорционально моляльной концентрации раствора.
Криоскопия
- метод
исследования растворов, в основе которого
лежит измерение понижения температуры
замерзания раствора по сравнению с
температурой замерзания чистого
растворителя. Был предложен Ф. Раулем
в 1882 году.Давление пара над раствором
нелетучего вещества практически
полностью определяется давлением пара
растворителя и может быть выражено
уравнением (согласно закону Рауля):![]() ,
х – мольная доля растворителя.
,
х – мольная доля растворителя.
Эбулиоскопия - метод исследования растворов, основанный на измерении повышения их температуры кипения по сравнению с чистым растворителем. Используется для определения молекулярной массы растворенного вещества, активности растворителя, степени диссоциации (или изотонического коэффициента).Температура кипения жидкости — такая температура, при которой давление пара над жидкостью равно внешнему давлению. В то же время давление пара над раствором нелетучего вещества практически полностью определяется давлением пара растворителя и, в соответствии с законом Рауля, может быть выражено уравнением:
Билет 8.
Коллигативными называются свойства растворов, которые не зависят от природы растворенного вещества, а только от его концентрации. Такие свойства проявляются в полной мере в идеальных растворах.
Идеальными называются растворы, при образовании которых не происходит изменения энтальпии и объема системы, не идут химические реакции между компонентами, а силы межмолекулярного взаимодействия между всеми компонентами одинакова. Наиболее близки к идеальным – разбавленные растворы неэлектролитов.
Для бесконечно разбавленных растворов, состояние которых близко к состоянию идеальных, такими свойствами являются:
– осмотическое давление;
– понижение давления насыщенного пара над раствором;
– повышение температуры кипения;
– понижение температуры замерзания раствора.
Изучение коллигативных свойств разбавленных растворов используется для определения молярной массы растворенного вещества, а также его степени диссоциации или показателя ассоциации.
Изотонический коэффициент (также фактор Вант-Гоффа; обозначается i) — безразмерный параметр, характеризующий поведение вещества в растворе. Он численно равен отношению значения некоторого коллигативного свойства раствора данного вещества и значения того же коллигативного свойства неэлектролита той же концентрации при неизменных прочих параметрах системы.
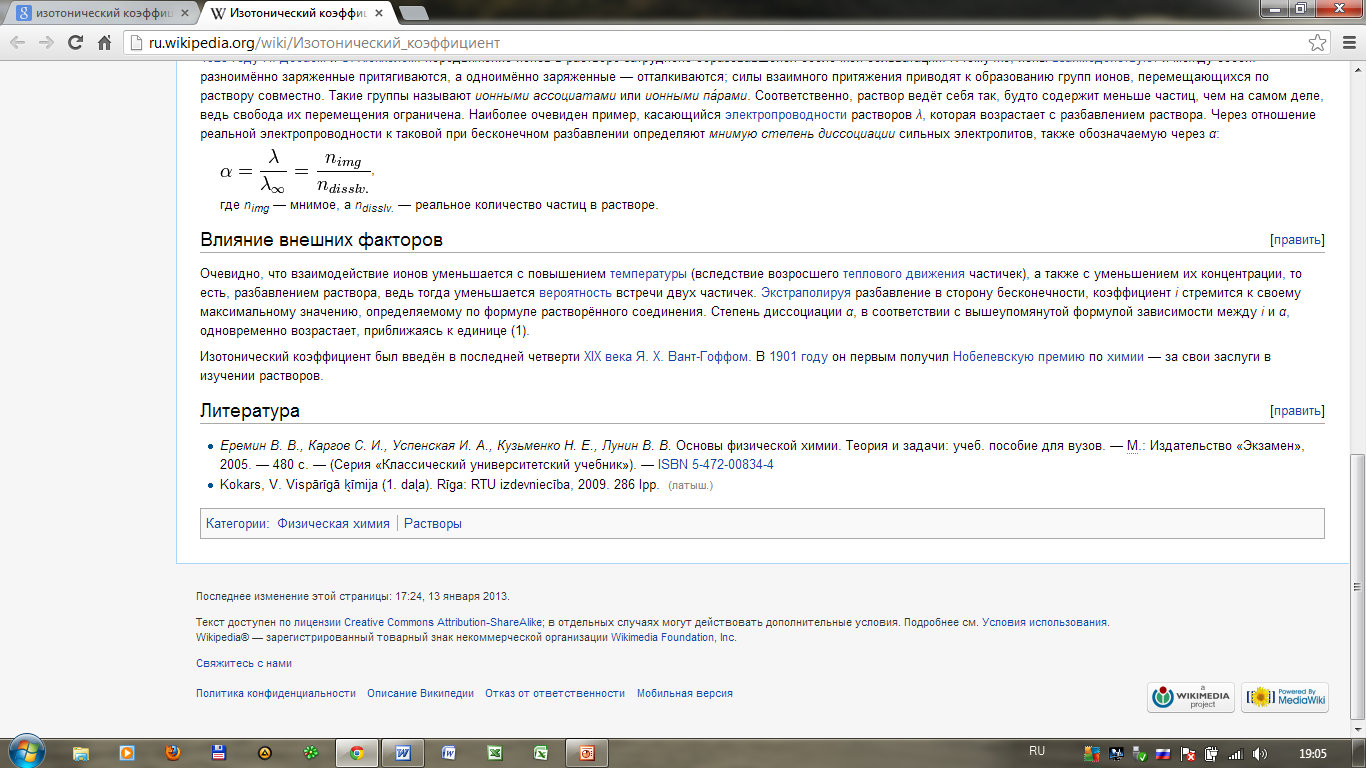
изотонический коэффициент показывает, насколько в растворе электролита больше частиц по сравнению с раствором неэлектролита аналогичной концентрации, и связан со способностью вещества распадаться в растворе на ионы, то есть, со степенью диссоциации.
Билет 9.
Слабые и сильные электролиты.
Вещества, растворы которых являются электролитами (т.е. проводят электрический ток), при растворении распадаются на частицы (ионы), которые образуются в результате диссоциации растворенного вещества. Число частиц при этом увеличивается. Ионы, заряженные положительно получили название катионы, т.к. под действием электрического поля движутся к катоду. Ионы заряженные отрицательно – анионы, т.к. под действием электрического поля движутся к аноду. К электролитам относятся соли, кислоты и основания.
Al(NO3)3 Al ³+ + NO3ֿ
Электролиты диссоциируют не полностью. Способность вещества к диссоциации характеризуется значением степени электролитической диссоциации - . Степенью электролитической диссоциации называется отношение количества вещества электролита, распавшегося на ионы, к общему количеству растворенного электролита. = nионизированное / Nрастворенное
Степень диссоциации показывает, какая часть растворенного количества электролита при данных условиях находится в растворе в виде гидратированных ионов.
Для растворов с молярной концентрацией См = 0,1 моль/л в зависимости от величины степени диссоциации существуют следующие условные границы силы электролитов:
для сильных электролитов - > 30 %
для слабых электролитов - < 3 %
для электролитов средней силы – 3 % < < 30 %
Сильные электролиты в водных растворах полностью диссоциируют на ионы. Их диссоциация происходит необратимо:
HNO3 H+ + NO3-
Слабые электролиты в водном растворе диссоциируют частично, т.к. их диссоциация является обратимым равновесным процессом, что и отражается знаком обратимости в уравнениях диссоциации:
СНСООН ═ СНСОО- + Н+
Константа ионизации слабого электролита.
Н+•СН3СОО-
Кдисс = СН3СООН
Константа диссоциации электролита не зависит от концентрации раствора, но зависит от температуры, а также от природы растворенного вещества и растворителя и при данных условиях является постоянной величиной. Кдисс показывает отношение концентрации ионов в растворе слабого электролита к концентрации недиссоциированных молекул. У сильных электролитов константа диссоциации отсутствует.
Закон Оствальда:
С увеличением разведения (уменьшением концентрации электролита) степень диссоциации электролита возрастает.
Зная характеристики и Кдисс можно рассчитать концентрацию ионов в растворах электролитов:
См иона = См эл-та∙ ∙ n
для сильного электролита = 1, поэтому
См иона = См эл-та∙ n
для слабого электролита = √ Кдисс/См, поэтому
См иона = √ См эл-та∙ Кдисс ∙ n,
где n – число ионов данного типа, образовавшихся при диссоциации одной молекулы электролита.
Билет 10.
Положения теории сильных электролитов
Согласно этой теории растворы электролитов представляют собой идеальные растворы. Однако применение её к описанию сильных электролитов наталкивается на ряд трудностей. Даже в разбавленных растворах сильные электролиты не ведут себя как идеальные растворы. Это обстоятельство заставило пересмотреть классическую теорию электролитической диссоциации применительно к сильным электролитам. Поэтому была разработана электростатическая теория сильных электролитов, авторами которой были Дебай и Хюккель(1923). При выводе теории они исходили из следующих предположений:
1) В разбавленных растворах сильные электролиты нацело диссоциированы на ионы.
2) Между ионами имеет место только кулоновское электростатическое взаимодействие.
3) Каждый ион в растворе окружён сферой противоположно заряженных ионов.
4) В растворе имеет место статистическое распределение (распределение Больцмана) ионов.
5) Электростатическое взаимодействие рассматривается как взаимодействие центрального иона с его ионной атмосферой.
6) Ионы рассматриваются как бесструктурные шарики, что позволяет пренебречь их размером.
7) Диэлектрическая постоянная растворителя не изменяется с растворением в нём электролита.
Активность компонентов раствора — эффективная (кажущаяся) концентрация компонентов с учётом различных взаимодействий между ними в растворе, то есть с учётом отклонения поведения системы от модели идеального раствора.
Активность
отличается от общей концентрации на
некоторую величину. Отношение активности
(![]() )
к общей концентрации вещества в растворе
называется коэффициентом
активности:
)
к общей концентрации вещества в растворе
называется коэффициентом
активности:
![]()
Коэффициент активности служит мерой отклонения поведения раствора (или компонента раствора) от идеального. Отклонения от идеальности могут быть обусловлены различными химическими и физическими причинами — дипольные взаимодействия, поляризация, образование водородных связей, ассоциация, диссоциация, сольватация и др.[1]
Ионная сила раствора — мера интенсивности электрического поля, создаваемого ионами в растворе. Полусумма произведений из концентрации всех ионов в растворе на квадрат их заряда. Формула впервые была выведена Льюисом:
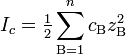 ,
,
где cB — молярные концентрации отдельных ионов (моль/л), zB заряды ионов
Суммирование проводится по всем типам ионов, присутствующих в растворе. Если в растворе присутствуют два или несколько электролитов, то вычисляется общая суммарная ионная сила раствора.Например, для раствора NaCl с концентрацией 0,001 моль/л, в котором присутствуют два вида однозарядных ионов Na+ и Cl− с концентрациями также равными 0,001 моль/л, ионная сила будет вычисляться следующим образом:
I(NaCl) = 0,5(z²(Na+)•c(Na+) + z²(Cl−)•c(Cl−)) = 0,5(1²•c(NaCl) + (-1)²•c(NaCl)) = c(NaCl)
Билет 11.
Диссоциация воды.Чистая вода, хоть и плохо (по сравнению с растворами электролитов), но может проводить электрический ток. Это вызвано способностью молекулы воды распадаться (диссоциировать) на два иона которые и являются проводниками электрического тока в чистой воде (ниже под диссоциацией подразумевается электролитическая диссоциация - распад на ионы):H2O ↔ H+ + OH
Ио́нное
произведе́ние воды́ —
произведение концентраций ионов водорода Н+ и
ионов гидроксила OH− в воде или
в водных растворах, константа
автопротолиза воды.![]()
Обозначим произведение K·[H2O] = Kв = 1,8·10−16 моль/л·55,56 моль/л = 10−14моль²/л² = [H+]·[OH−] (при 25 °C).Константа Kв, равная произведению концентраций протонов и гидроксид-ионов, называется ионным произведением воды. Она является постоянной не только для чистой воды, но также и для разбавленных водных растворов веществ. C повышением температуры диссоциация воды увеличивается, следовательно, растёт и Kв, при понижении температуры — наоборот.pH воды
Для удобства, концентрации [H+] и [HO-] выражают в виде водородного показателя pHи гидроксильного показателя pOH. pH и pOH - это отрицательные десятичные логарифмы концентраций [H+] и [HO-] (правильнее использовать не концентрацию, а активность) соответственно:pH = -lg[H+]
рН>7 среда щелочная
pH<7 среда кислая
pH=7 среда нейтральна
Кислотно-основные индикаторы — органические соединения, способные изменять цвет в растворе при изменении кислотности (pH).
Лакмус, метилоранж, фенолфталеин….
Широко применяются смеси индикаторов, позволяющие определить значение рН растворов в большом диапазоне концентраций (1-10; 0-12). Растворами таких смесей - «универсальных индикаторов» обычно пропитывают полоски «индикаторной бумаги», с помощью которых можно быстро (с точностью до единиц рН, или даже десятых долей рН) определить кислотность исследуемых водных растворов. Для более точного определения полученный при нанесении капли раствора цвет индикаторной бумаги немедленно сравнивают с эталонной цветовой шкалой.
Билет 12.
Протолитическая теория - кислотные или основные свойства частиц обусловлены их способностью отдавать или присоединять катион водорода (протон Н+)
NH4+ + S2– NH3 + HS–
Кт Ос Ос Кт
Пары "сопряженная кислота / сопряженное основание":
NH4+/NH3 и HS –/ S2–
Кислоты отдают катион водорода – доноры. Основания присоединяют катион водорода – акцепторы.
Совокупность кислоты и ее сопряженного основания – сопряженная кислотно-основная пара.
Сильные кислоты при диссоциации превращаются в слабые сопряженные основания.
Амфолиты – молекулы или ионы, которые способны отдавать или присоединять протоны, способны вступать в реакции как килоты или основания.
Протолитические процессы – переход протона от кислоты к основанию – протолиз.
Протолитическое равновеие – в результате конкуренции за протон между основаниями взаимодействующих кислотно-основных пар, равновесие смещается в сторону образования более слабой кислоты..
Чем больше показатель кислотности кислоты, тем меньше константа кислотности, тем слабее кислота.
Чем больше показатель кислотности основания, тем меньше константа кислотности основания, тем основание сильее.
Вода – как амфипротонный растворитель. (не нашла)
Нейтрализа́ция (от лат. neuter — ни тот, ни другой) — взаимодействие кислот с основаниями, в результате которого образуются соли и вода.
Часто реакции нейтрализации экзотермичны. К примеру, реакция гидроксида натрия и соляной кислоты:
НСl + NaOH = NaCl + Н2О
Гидро́лиз (от др.-греч. ὕδωρ — вода и λύσις — разложение) — один из видов химических реакций сольволиза, где при взаимодействии веществ с водой происходит разложение исходного вещества с образованием новых соединений. Механизм гидролиза соединений различных классов: соли, углеводы, белки, сложные эфиры, жиры и др. имеет существенные различия.
Слаб к-та + слаб основания – гидролиз по аниону и катиону, среда нейтральная
Слаб. К-та + сильн основание – гидролиз по катиону, среда щелочная
Сильн к-та + слаб основание – гидролиз по аниону, среда кислая
Сильн к-та + сильн основание – гидролизу не подвергается
Под степенью гидролиза подразумевается отношение части соли, подвергающейся гидролизу, к общей концентрации её ионов в растворе. Обозначается α (или hгидр); α = (cгидр/cобщ)·100 % где cгидр — число молей гидролизованной соли, cобщ — общее число молей растворённой соли. Степень гидролиза соли тем выше, чем слабее кислота или основание, её образующие.
Константа гидролиза — константа равновесия гидролитической реакции. Так константа гидролиза соли равна отношению произведения равновесных концентраций продуктов реакции гидролиза к равновесной концентрации соли с учетом стехиометрических коэффициентов.
Ионизация – диссоциация – распад на ионы.
I стадия: H3BO3 ↔ H+ + H2BO3−,
II стадия: H2BO3− ↔ H+ + HBO32−,
III стадия: HBO32− ↔ H+ + BO33−,
Билет 13.
Буферными называют растворы, рН которых практически на изменяется от добавления к ним небольших количеств сильной кислоты или щелочи, а также при разведении. Простейший буферный раствор - это смесь слабой кислоты и соли, имеющей с этой кислотой общий анион (например, смесь уксусной кислоты СН3СООН и ацетата натрия СН3СООNa), либо смесь слабого основания и соли, имеющей с этим основанием общий катион (например, смесь гидроксида аммония NH4OH с хлоридом аммония NH4Cl).
Сопряженные кислотно-основные пары В /ВН+ и А- /НА называют буферными системами.
Классификация кислотно-основных буферных систем
Кислотные – слаб к-та(донор протона) и соль к-ты (акцептор протона)
Основные – слаб основание (акцептор протона) и соль основания (донор протона)
Механизм действия – совмещенные протолитические процессы, связанные с диссоциацией протолита(обратимый процесс) и диссоциацией соли(необратимый процесс).
Рассчет рН для
Буферные системы
К буферным системам относятся смеси :
а)слабой кислоты и ее соли , например CH3COO H + CH3COO Na
б) слабого основания и его соли, например NH4OH + NH4Cl
в) смесь кислых солей разной кислотности , например
NaH2PO4 + Na2HPO4
г) смесь кислой и средней солей , например NaНCO3 + Na2 CO3
д) смесь основных солей разной основности , например
Al(OH)2Cl + Al(OH)Cl2 и т.д.
Расчет рН в буферных системах ведут по формулам (10-11)
рН = рКк – lg Cк /Сс (10)
рН = 14 – рКо+ lg Cо /Сс (11)
Механизм защитного действия буферных систем по поддержанию постоянства рН среды сводится к связыванию добавляемых в раствор ионов и компонентами буферной системы в малодиссоциирующие соединения.
Каждая буферная система характеризуется определенной концентрацией ионов водорода (рН), которую стремится сохранить при добавлении кислоты, основания или при разбавлении. Определяется рН буферных растворов по уравнению Гендерсона-Гассельбаха.
![]()
Величина, характеризующая способность буферного раствора противодействовать смещению реакции среды при добавлении кислот и щелочей, называется буферной емкостью.
В=C·V/∆pH·Vбуф,
где В - буферная емкость,С - концентрация кислоты или основания,V - объем данного электролита,Vбуф –объем буферного раствора ,∆pH – изменение рН.
Буферная емкость зависит от ряда факторов:
1. Чем выше концентрации компонентов буферного раствора, тем больше его буферная емкость.
2![]() . Буферная
емкость зависит от отношения концентраций
компонентов , а следовательно , и от рН
буфера. При рН=рКа буферная
емкость максимальна.
. Буферная
емкость зависит от отношения концентраций
компонентов , а следовательно , и от рН
буфера. При рН=рКа буферная
емкость максимальна.
3. Установлено, что достаточное буферное действие наблюдается, если концентрация одного из компонентов превышает концентрацию другого не более, чем в 10 раз.
Интервал рН=рКа±1 называется зоной буферного действия.
4. При разбавлении буферного раствора величина буферной емкости уменьшается вследствие снижения концентрации компонентов раствора.
Билет 14.



Б
 илет
15.
илет
15.
Гетерогенные равновесия
Рассмотрим
гетерогенную систему, состоящую из
малорастворимого осадка сильного
электролита и насыщенного раствора над
ним, между которыми устанавливается
динамическое химическое равновесие.
При контакте осадка (например, BaSO4)
с водой в системе протекают процессы:
1) растворения –– полярные молекулы
Н2О
часть ионов из кристаллической решетки
BaSO4
переводят в жидкую фазу; 2) осаждения ––
под влиянием электростатического поля
кристаллической решетки BaSO4
часть ионов Ва2+
и
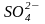 переходят из жидкой фазы в твердую,
достраивая кристаллическую решетку
соли.С течением времени скорость
растворения станет равной скорости
осаждения и установится динамические
равновесие между кристаллическим
осадком малорастворимой соли BaSO4
и его водным раствором, содержащим ионы
Ва2+(p)
и
переходят из жидкой фазы в твердую,
достраивая кристаллическую решетку
соли.С течением времени скорость
растворения станет равной скорости
осаждения и установится динамические
равновесие между кристаллическим
осадком малорастворимой соли BaSO4
и его водным раствором, содержащим ионы
Ва2+(p)
и
Таким образом, в насыщенном растворе малорастворимого сильного электролита произведение равновесных активностей его ионов есть величина постоянная при данной температуре.
Константа растворимости
![]()
Произведение растворимости (ПР, Ksp) — произведение концентраций ионов малорастворимого электролита в его насыщенном растворе при постоянной температуре и давлении. Произведение растворимости — величина постоянная.
При постоянной температуре в насыщенных водных растворах малорастворимых электролитов устанавливается равновесие между твердым веществом и ионами, образующими это вещество. Например, в случае для CaCO3 это равновесие можно записать в виде:
![]()
Константа этого равновесия рассчитывается по уравнению:
![]()
В приближении идеального раствора с учётом того, что активность чистого компонента равна единице, уравнение упрощается до выражения
![]()
Билет 16.
Условия образования и растворения осадков.
Образование осадка происходит в том случае, когда произведение концентраций ионов, входящих в его состав, превышает величину произведения растворимости ПР(KA) малорастворимого электролита К+ + Аˉ ↔ КА; [К+ ] [Аˉ] > ПР(КA),
т. е. когда возникает местное (относительное) пересыщение раствора, которое рассчитывают по формуле: (Q - S) /S, где Q - концентрация растворенного вещества в какой-либо момент времени, моль/см3; S - растворимость вещества в момент равновесия, моль/см3 В этом месте появляется зародыш будущего кристалла (процесс зародышеобра-зования). Для этого требуется определенное время, называемое индукционным периодом. При дальнейшем прибавлении осадителя более вероятным становится процесс роста кристаллов, а не дальнейшее образование центров кристаллизации, которые соединяются в более крупные агрегаты, состоящие из десятков и сотен молекул (процесс агрегации). Размер частиц при этом увеличивается, и более крупные агрегаты под действием силы тяжести выпадают в осадок. На этой стадии отдельные частицы, будучи диполями, ориентируются по отношению друг к другу так, что их противоположно заряженные стороны сближаются (процесс ориентации). Если скорость ориентации больше скорости агрегации, то образуется правильная кристаллическая решетка, если же наоборот, выпадает аморфный осадок. Чем меньше растворимость вещества, тем быстрее образуется осадок и мельче кристаллы. Одни и те же малорастворимые вещества могут быть выделены как в кристаллическом, так и в аморфном состоянии, что определяется условиями осаждения.
Билет
17.
Комплексные
соединения или координационные
соединения — частицы которые образуются
в результате присоединения к данному
иону (или атому), называемому
комплексообразователем, нейтральных
молекул или других ионов, называемых
лигандами. Теория комплексных соедин.
(координационная теория)была предложена
в1893 г.А.Вернером. Комплексные соединения
мало диссоциируют в растворе (в отличие
от двойных солей)., либо вообще не
диссоциировать на ионы (соединения
типа неэлектролитов, например карбонилы
металлов).
Комплексообразователь
— центральный
атом
комплексной частицы. Обычно — атом
элемента, образующего металл, но это
может быть и атом кислорода, азота,
серы, йода и других элементов, образующих
неметаллы. Комплексообразователь
обычно положительно заряжен и в таком
случае именуется в современной научной
литературе металлоцентром; заряд
комплексообразователя может быть также
отрицательным или =0
Лиганды
— атомы или изолированные группы
атомов, располагающиеся вокруг
комплексообразователя. Лигандами могут
быть частицы, до образования комплексного
соединения представлявшие собой
молекулы (H2O, CO, NH3 и др.), анионы (OH−, Cl−,
PO43− и др.), а также катион H+. Комплексная
частица —
сложная частица, способная к
самостоятельному существованию в
кристалле или растворе, образованная
из других, более простых частиц, также
способных к самостоятельному
существованию. Иногда комплексными
частицами называют сложные химические
частицы, все или часть связей в которых
образованы по донорно-акцепторному
механизму. Внутренняя
сфера
комплексного соединения — центральный
атом со связанными с ним лигандами, то
есть, собственно, комплексная частица.
Внешняя сфера
— остальные частицы, связанные с
комплексной частицей ионной или
межмолекулярными связями, включая
водородные. Координационное
число (КЧ) —
число связей, образуемых центральным
атомом с лигандами. Для комплексных
соединений с монодентантными лигандами
КЧ равно числу лигандов, а в случае
полидентантных лигандов — числу таких
лигандов, умноженному на дентатность.
Дентатность
лиганда
определяется числом координационных
мест, занимаемых лигандом в координационной
сфере комплексообразователя. Различают
монодентатные лиганды, связанные с
центральным атомом через один из своих
атомов, то есть одной ковалентной
связью, бидентатные (связанные с
центральным атомом через два своих
атома, то есть, двумя связями), три- ,
тетрадентатные и т. д. (монодентатными
(например, NH3), два — бидентатными
(оксалат-анион [O-C(=O)-C(=O)-O]2−))
Природа связи
между центральным атомом и лигандами
может быть двоякой. С одной стороны,
связь обусловлена силами электростатического
притяжения. С другой — между центральным
атомом и лигандами может образоваться
связь по донорно-акцепторному механизму
Во многих комплексных соединениях
связь между центральным атомом и
лигандами обусловлена как силами
электростатического притяжения, так
и связью, образующейся за счёт неподеленных
электронных пар комплексообразователя
и свободных орбиталей лигандов.
Существует несколько
классификаций
комплексных соединений
в основу которых положены различные
принципы.
- По заряду комплекса
1) Катионные комплексы образованы в
результате координации вокруг
положительного иона нейтральных молекул
(H2O, NH3 … [(Zn(NH3)4)]Cl2 — хлорид тетраамминцинка(II)
[Co(NH3)6]Cl2 — хлорид гексаамминкобальта(II)
2) Анионные комплексы:
в роли комплексообразователя выступает
атом с положительной степенью окисления,
а лигандами являются простые или сложные
анионы.K2[BeF4] — тетрафторобериллат(II)
калия Li[AlH4] — тетрагидридоалюминат(III)
лития
3) Нейтральные
комплексы образуются при координации
молекул вокруг нейтрального атома, а
также при одновременной координации
вокруг положительного иона —
комплексообразователя отрицательных
ионов и молекул.
[Ni(CO)4] —
тетракарбонилникель [Pt(NH3)2Cl2] —
дихлородиамминплатина(II)
- По числу мест,
занимаемых лигандами в координационной
сфере
1) Монодентатные
лиганды. Такие лиганды бывают нейтральными
(молекулы Н2О, NH3, CO, NO и др.) и заряженными
(ионы CN−, F−, Cl−, OH−, SCN−, S2O32− и др.).
2) Бидентатные
лиганды. Примерами служат лиганды: ион
аминоуксусной кислоты H2N — CH2 — COO−,
оксалатный ион −O — CO — CO — O−,
карбонат-ион СО32−, сульфат-ион SO42−.
3) Полидентатные
лиганды. Например, комплексоны —
органические лиганды, содержащие в
своём составе несколько групп −С≡N
или −COOH (этилендиаминтетрауксусная
кислота — ЭДТА). Циклические комплексы,
образуемые некоторыми полидентатными
лигандами, относят к хелатным (гемоглобин
и др.).
- По природе лиганда
1) Аммиакаты — комплексы, в которых
лигандами служат молекулы аммиака,
например: [Cu(NH3)4]SO4, [Co(NH3)6]Cl3, [Pt(NH3)6]Cl4 и др.
2) Аквакомплексы
— в которых лигандом выступает вода:
[Co(H2O)6]Cl2, [Al(H2O)6]Cl3 и др.
3) Карбонилы —
комплексные соединения, в которых
лигандами являются молекулы оксида
углерода(II): [Fe(CO)5], [Ni(CO)4].
4) Ацидокомплексы
— комплексы, в которых лигандами
являются кислотные остатки. К ним
относятся комплексные соли: K2[PtCl4],
комплексные кислоты: H2[CoCl4], H2[SiF6].
5) Гидроксокомплексы
— комплексные соединения, в которых в
качестве лигандов выступают гидроксид-ионы:
Na2[Zn(OH)4], Na2[Sn(OH)6] и др.
Номенклатура 1)
В названии комплексного соединения
первым указывают отрицательно заряженную
часть — анион, затем положительную
часть — катион.
2) Название
комплексной части начинают с указания
состава внутренней сферы. Во внутренней
сфере прежде всего называют лиганды —
анионы, прибавляя к их латинскому
названию окончание «о». Например: Cl−
— хлоро, CN− — циано, SCN− — тиоцианато,
NO3− — нитрато, SO32− — сульфито, OH− —
гидроксо и т. д. При этом пользуются
терминами: для координированного
аммиака — аммин, для воды — аква, для
оксида углерода(II) — карбонил.
3) Число монодентатных
лигандов указывают греческими
числительными: 1 — моно (часто не
приводится), 2 — ди, 3 — три, 4 — тетра, 5
— пента, 6 — гекса. Для полидентатных
лигандов (например, этилендиамин,
оксалат) используют бис-, трис-, тетраки
4) Затем называют
комплексообразователь, используя
корень его латинского названия и
окончание -ат, после чего римскими
цифрами указывают (в скобках) степень
окисления комплексообразователя. 5)
После обозначения состава внутренней
сферы называют внешнюю сферу. 6) В
названии нейтральных комплексных
частиц комплексообразователь указывается
в именительном падеже, а степень его
не указывается, так как она однозначно
определяется, исходя из электронейтральности
комплекса.Пр: K3[Fe(CN)6] — гексацианоферрат(III)
калия (NH4)2[PtCl4(OH)2] — дигидроксотетрахлороплатинат(IV)
аммония
[Сr(H2O)3F3] —
трифторотриаквахром [Сo(NH3)3Cl(NO2)2] —
динитритохлоротриамминкобальт
Билет 18.
Первичная диссоциация комплексного соединения - это его распад на комплексный ион, образованный внутренней сферой и ионы внешней сферы. В водных растворах первичная диссоциация комплексных соединений обусловлена разрывом ионной связи между внутренней и внешней сферами, она практически необратима:
K3[Fe(CN)6] ® 3K+ + [Fe(CN)6]3–
Na[Al(OH)4] ® Na+ + [Al(OH)4]–
[Cu(NH3)4]SO4 ® [Cu(NH3)4]2+ + SO42–
Pt(NH3)4Cl2]Cl2 ® [Pt(NH3)4Cl2]2+ + 2Cl–
Образующийся комплексный ион ведет себя как целая самостоятельная частица с характерными для нее свойствами. Поэтому в водных растворах комплексных соединений, как правило, нельзя обнаружить присутствие ионов или молекул, входящих в состав внутренней сферы. В растворе красной кровяной соли нельзя обнаружить присутствие ионов Fe3+ и CN–, а в 1М растворе хлорида дихлоротетраамминплатины(IV) [Pt(NH3)4Cl2]Cl2 обнаруживается только присутствие 2 моль хлорид-ионов, а не 4 моль.
Но насколько устойчива внутренняя сфера комплекса? Может ли происходить отщепление лигандов от комплексообразователя? Действительно, это возможный процесс, он называется вторичной диссоциацией комплексного соединения.
Вторичная диссоциация комплексного соединения – это распад внутренней сферы комплекса на составляющие ее компоненты.
Так как при этом разрушаются не ионные, а ковалентные связи комплексообразователя с лигандами, этот процесс затруднен и обратим. Он происходит ступенчато:
[Ag(NH3)2]+ ? [Ag(NH3)]+ + NH3 1-ая ступень
Ag(NH3)]+ ? Ag+ + NH3 2-ая ступень
Вторичная диссоциация характеризуется константой равновесия, причем для каждой из стадий можно вычислить свою константу. Для количественной оценки устойчивости внутренней сферы комплексного соединения используют константу равновесия, описывающую полную ее диссоциацию. Эту константу называют константой нестойкости комплекса Кнест.
Константы образования и нестойкости комплексов.
Полная константа образования комплекса bn(обр) характеризует устойчивость комплексного соединения: чем больше значение bn(обр), тем более устойчив комплекс данного состава.
Если вместо равновесия в реакциях образования комплексов рассматривать обратный процесс – реакции диссоциации комплексов (или реакции обмена лигандов на молекулы растворителя), то соответствующие константы будут носить название ступенчатых констант нестойкости комплексов:
Таким образом, полная константа образования дает возможность судить об отсутствии склонности комплекса к полной диссоциации, а ступенчатая константа образования свидетельствует об устойчивости отдельных форм комплексных ионов или нейтральных комплексов.
Билет
19.
Существует
несколько классификаций
комплексных соединений
в основу которых положены различные
принципы.
- По заряду комплекса
1) Катионные
комплексы образованы в результате
координации вокруг положительного
иона нейтральных молекул (H2O, NH3 и др.).
[(Zn(NH3)4)]Cl2 — хлорид
тетраамминцинка(II) [Co(NH3)6]Cl2 — хлорид
гексаамминкобальта(II)
2) Анионные
комплексы: в роли комплексообразователя
выступает атом с положительной степенью
окисления, а лигандами являются простые
или сложные анионы.
K2[BeF4] —
тетрафторобериллат(II) калия Li[AlH4] —
тетрагидридоалюминат(III) лития
3) Нейтральные
комплексы образуются при координации
молекул вокруг нейтрального атома, а
также при одновременной координации
вокруг положительного иона —
комплексообразователя отрицательных
ионов и молекул.
[Ni(CO)4] —
тетракарбонилникель [Pt(NH3)2Cl2] —
дихлородиамминплатина(II)
- По числу мест,
занимаемых лигандами в координационной
сфере
1) Монодентатные
лиганды. Такие лиганды бывают нейтральными
(молекулы Н2О, NH3, CO, NO и др.) и заряженными
(ионы CN−, F−, Cl−, OH−, SCN−, S2O32− и др.).
2) Бидентатные
лиганды. Примерами служат лиганды: ион
аминоуксусной кислоты H2N — CH2 — COO−,
оксалатный ион −O — CO — CO — O−,
карбонат-ион СО32−, сульфат-ион SO42−.
3) Полидентатные
лиганды. Например, комплексоны —
органические лиганды, содержащие в
своём составе несколько групп −С≡N
или −COOH (этилендиаминтетрауксусная
кислота — ЭДТА). Циклические комплексы,
образуемые некоторыми полидентатными
лигандами, относят к хелатным (гемоглобин
и др.).
- По природе лиганда
1) Аммиакаты —
комплексы, в которых лигандами служат
молекулы аммиака, например: [Cu(NH3)4]SO4,
[Co(NH3)6]Cl3, [Pt(NH3)6]Cl4 и др.
2) Аквакомплексы
— в которых лигандом выступает вода:
[Co(H2O)6]Cl2, [Al(H2O)6]Cl3 и др.
3) Карбонилы —
комплексные соединения, в которых
лигандами являются молекулы оксида
углерода(II): [Fe(CO)5], [Ni(CO)4].
4) Ацидокомплексы
— комплексы, в которых лигандами
являются кислотные остатки. К ним
относятся комплексные соли: K2[PtCl4],
комплексные кислоты: H2[CoCl4], H2[SiF6].
5) Гидроксокомплексы
— комплексные соединения, в которых в
качестве лигандов выступают гидроксид-ионы:
Na2[Zn(OH)4], Na2[Sn(OH)6] и др. Особое
место среди комплексных соединений
занимают хелаты.
Это комплексы,
в которых полидентатные лиганды образуют
с центральным атомом хелатные
(клешневид- ные) циклы (от
англ. chelate –
клешня). Хелатные комплексы образует,
например, биден- татный этилендиамин
(En):
В
этом комплексе молекулы этилендиамина
образуют два пятичленных хелатных
цикла, каждый из которых включает атом
платины и по два атома азота и углерода
лиганда. Поскольку в состав хелатных
циклов входит атом металла, то их также
называют металлоциклами.
Образование
хелатных циклов приводит к повышению
устойчивости комплексных соединений,
по сравнению с комплексами тех же
металлов
Для живых
организмов(животных, растений, бактерий)
очень важны комплексные соединения
металлов, в которых четыре координационных
места занимает одна и та же частица,
наз. ываемая порфином. В составе
гемоглобина, миоглобина, цитохромов,
каталазы и пероксидазы порфирины
выступают в виде комплексов с ионами
железа – гемов. В организме непрерывно
происходят образование и разрушение
биокомплексов из катионов биометаллов
(железо, медь, цинк, кобальт) и биолигандов
(порфиринов, аминокислот, полипептидов).
Обмен веществ с окружающей средой
поддерживает концентрации вещества
на определенном уровне, обеспечивая
состояние металло-лигандного гомеостаза.
Распределение
того или иного катиона металла между
биолигандами в биосредах определяется
как прочностью образующихся комплексов,
так и концентрациями этих лигандов.
Для каждого из катионов биометаллов
характерна своя совокупность реакций
металло-лигандного равновесия.
Поступление, метаболизм, накопление и
выделение катионов металлов (а в целом
любых микроэлементов) регулируются
специальной системой микроэлементозного
гомеостаза. В совокупности существуют
тысячи патологических явлений -
микроэлементозов, связанных с теми или
иными металлоизбыточными или
металлодефицитными состояниями.
Нарушение металло-лигандного гомеостаза
возможно по разным причинам: из-за
дефицита или избытка катионов биометаллов,
из-за поступления катионов токсичных
металлов, из-за поступления или
образования посторонних лигандов.
Хелатный эффект!!!! также влияет на устойчивость комплексов. Металлы в растворах преимущественно связываются с хелатными лигандами, а не с
монодентными – это и есть хелатный эффект.Конкурентное взаимодействие лежит в основе хелотерапии (лечение
отравлений металлами свинца, ртути, которые являются промышленными
ядами с помощью хелатных лигандов).
Билет 20.
Реакции, протекающие с изменением
степени окисления атомов, входящих в
состав реагирующих веществ, называются
окислительно – восстановительными.
Другими словами это реакции, связанные
с передачей электронов от одних атомов
к другим.
Окисление – процесс
отдачи электронов атомом, молекулой
или ионом, степень окисления при этом
повышается.
Ca – 2e- = Ca+2 H02 – 2e-
= 2H+
Восстановление –
процесс присоединения электронов
атомом, молекулой, ионом. Степень
окисления при этом понижается. Fe+3 + e-
= Fe2+ Cl02 + 2e- = 2Cl-
Восстановители –
молекулы или ионы отдающие электроны,
сами при этом восстановители окисляются.
Окислители –
молекулы или ионы, присоединяющие
электроны ,во время реакции окислители
восстанавливаются.
Окисление и
восстановление протекают одновременно.
В зависимости от
того между какими атомами и в каких
веществах происходит переход электронов
все окислительно-восстановительные
процессы можно разделить на 3 типа:
1) Межмолекулярные
2) Дисмутационные
( диспропорционирования)
3) Внутримолекулярные
4) Компропорционированния
1.Межмолекулярные
реакции окислителения-восстановления
– это реакции, в ходе которых переход
электронов происходит между частицами
различных веществ. В выше рассматриваемых
реакциях окислитель и восстановитель
находятся в разных веществах
Mn+4O2 +
4HCl-1 =t Cl02 ↑ + Mn+2Cl2 + 2H2O
Mn+4 + 2e =
Mn+2 1
2Cl- - 2e
= Cl2 1
2. Диспропорционирования
– когда атомы или ионы одного и того и
того же элемента , содержащиеся в одной
молекуле, являются и окислителем и
восстановителем.
4KCl+5O3 =t
KCl- + 3KCl+7O4
Cl+5 – 2e
= Cl7+ 3
Cl+5 + 6e =
Cl- 1
Диспропорционировать
могут вещества, один из элементов
которых находится в промежуточной
степени окисления, т.к. степень окисления
одной части атомов понижается за счет
другой части таких же атомов, степень
окисления которых повышается.
3.Внутримолекулярные
– когда окислитель и восстановитель
одно и тоже вещество, но изменяют степень
окисления в нем атомы различных
элементов.
1) (N-3H4)2
Cr2+6O7 = N02 + Cr+23 O3 + 4H2O
2N-3 -6e =
N02 1
2Cr+6 + 6e
=2Cr+3 1
2) 2Hg+2O-2
= Hg0 + O02
3) 2КClO3
= 2KCl- + 3O2
Cl+5 + 6e =
Cl- 1
2O3 – 6e = 3O2 1
4. Компропорционированния
– реакции в которых участвуют два
вещества, cодержащие атомы одного и
того же элемента в разных степенях
окисления
Cu0 +
Cu+2Cl2 = 2Cu+1Cl
Применяются два
вида составления уравнений
окислительно-восстановительных реакций:
1) Метод электронного
баланса.
2) Метод полуреакций.
По методу
электронного баланса сравнивают степени
окисления атомов в исходных и конечных
веществах, причем число электронов
отданных восстановителем, должно
ровняться числу электронов, присоединенных
окислителем.
Метод полуреакций
применяется для реакций между
газообразными, твердыми или жидкими
веществами, протекающих без
электролитической диссоциации.
Например: 1) ^ Метод
электронного баланса
+3 +7
+5 +2
H3AsO3 + KMnO4 + H2SO4
→H3AsO4 + MnSO4 + K2SO4 + H2O
Ортомышьяковистая
Ортомышьяковая
кислота кислота
Из схемы реакции
видно, что степень окисления атома
мышьяка до реакции +3 , после +5, степень
окисления марганца изменилась от +7 до
+2.
Отражаем это
изменение степени окисления в электронных
уравнениях.
Восстановитель
As+3 – 2e- = As+5 5 процесс окисления
Окислитель Mn+7
+5e- = Mn+2 2 процесс восстановления
или методом
полуреакций
MnO4- + 8H+ +5e = Mn+2 + 4H2O
2
H3AsO3 +
H2O – 2e = H3AsO4 + 2H+ 5
Общее число
электронов, отданных восстановителем,
должно быть равно общему числу электронов,
принятых окислителем. Найдя наименьшее
общее кратное определяем, что молекул
восстановителя должно быть 5, а молекул
окислителя 2, т.е. находим соответствующие
коэффициенты в уравнении. Уравнение
будет иметь вид:
5H3AsO3 + 2KMnO4 + 3H2SO4 =
5H3AsO4 + 2MnSO4 + K2SO4 + 3H2O
Билет 21. Реакции, протекающие с изменением степени окисления атомов, входящих в состав реагирующих веществ, называются окислительно – восстановительными. Другими словами это реакции, связанные с передачей электронов от одних атомов к другим.
Окисление – процесс отдачи электронов атомом, молекулой или ионом, степень окисления при этом повышается.
Ca – 2e- = Ca+2 H02 – 2e- = 2H+
Восстановление – процесс присоединения электронов атомом, молекулой, ионом. Степень окисления при этом понижается.
Fe+3 + e- = Fe2+ Cl02 + 2e- = 2Cl-
Восстановители – молекулы или ионы отдающие электроны, сами при этом восстановители окисляются.
Окислители – молекулы или ионы, присоединяющие электроны ,во время реакции окислители восстанавливаются.
Вещества, являющиеся окислителями во многих реакциях, представляют собой типичные (сильные) окислители. К ним относятся F2, Cl2, O2, KClO3, H2SO4, HNO3, KMnO4, MnO2, K2Cr2O7, PbO2 и др.
Типичными (сильными) восстановителями являются H2, C (графит), Zn, Al, Ca, KI, HCl (конц.), H2S, CO и др.
Многие вещества могут проявлять как окислительные, так и восстановительные свойства. К таким веществам принадлежат KNO2, H2O2, SO2, Nа2SO3 и др.
Окислительно-восстановительные свойства веществ связаны с положением элементов в Периодической системе Д.И. Менделеева. Простые вещества - неметаллы обладают большими окислительными свойствами, а металлы - большими восстановительными свойствами (O2, Cl2 - окислители, Na, Ba, Al, Zn - восстановители).
В каждой группе Периодической системы элемент с более высоким порядковым номером обладает более ярко выраженными восстановительными свойствами в своей группе, а элемент с меньшим порядковым номером - более сильными окислительными свойствами. Так, кальций Ca - более сильный восстановитель, чем магний Mg, а молекулярный хлор Cl2 - более сильный окислитель, чем иод I2.
Соединения, содержащие атомы элементов в низшей степени окисления, будут восстановителями за счет этих атомов (например, NH3 - восстановитель за счет атома N−III, H2S - за счет атома S−II, KI за счет атома I−I). Соединения, включающие атомы элементов в высшей степени окисления, будут окислителями (например, HNO3 за счет атома N+V, K2Cr2O7 - за счет атома Cr+VI, KMnO4 - за счет атома Mn+VII).
Химические свойства веществ, в отличие от физических, не зависят от агрегатного состояния. Так, сера в любом агрегатном состоянии при сгорании образует сернистый газ (т.е. проявляет одно и то же химическое свойство), но физические свойства серы в разных агрегатных состояниях весьма различны (например, плотность твердой серы равна 2,1 г/см3, жидкой серы 1,8 г/см3 и газообразной серы 0,004 г/см3).
Все вещества состоят из частиц и характеризуются определённым набором химических свойств - способностью веществ участвовать в химических реакциях.
Степень окисления - это условный заряд атома в молекуле, вычисленный в предположении, что молекула состоит из ионов и в целом электронейтральна.
Наиболее электроотрицательные элементы в соединении имеют отрицательные степени окисления, а атомы элементов с меньшей электроотрицательностью - положительные.
Степень окисления - формальное понятие; в ряде случаев степень окисления не совпадает с валентностью.
Для вычисления степени окисления элемента следует учитывать следующие положения:
1. Степени окисления атомов в простых веществах равны нулю (Na0; H20).
2. Алгебраическая сумма степеней окисления всех атомов, входящих в состав молекулы, всегда равна нулю, а в сложном ионе эта сумма равна заряду иона.
3. Постоянную степень окисления имеют атомы: щелочных металлов (+1), щелочноземельных металлов (+2), водорода (+1) (кроме гидридов NaH, CaH2 и др., где степень окисления водорода -1), кислорода (-2) (кроме F2-1O+2 и пероксидов, содержащих группу –O–O–, в которой степень окисления кислорода -1).
4. Для элементов положительная степень окисления не может превышать величину, равную номеру группы периодической системы.
Примеры:
V2+5O5-2; Na2+1B4+3O7-2; K+1Cl+7O4-2; N-3H3+1; K2+1H+1P+5O4-2; Na2+1Cr2+6O7-2
Билет 22. Окислительно-восстановительную реакцию можно представить в виде сочетания двух сопряженных пар.
В ходе реакций окислитель превращается в сопряженный восстановитель (продукт восстановления), а восстановитель в сопряженный окислитель (продукт окисления). Их рассматривают как окислительно-восстановительные пары:
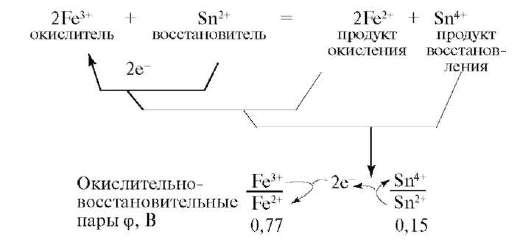
Поэтому окислительно-восстановительные реакции представляют единство двух противоположных процессов окисления и восстановления, которые в системах не могут существовать один без другого. В этом мы видим проявление универсального закона единства и борьбы противоположностей. Реакция произойдет, если сродство к электрону окислителя больше, чем потенциал ионизации восстановителя. Для этого введено понятие электроотрицательности - величины, характеризующей способность атомов отдавать или принимать электроны.
Составление уравнений окислительно-восстановительных реакций проводится методом электронного баланса и методом полуреакций. Методу полуреакций необходимо отдать предпочтение. Применение его связано с применением ионов, реально существующих, видна роль среды. При составлении уравнений необходимо выяснить, какие из веществ, вступающих в реакцию, выполняют роль окислителя, а какие - восстановителя, влияние на ход реакции pH среды и каковы возможные продукты реакции. Окислительно-восстановительные свойства проявляют соединения, которые содержат атомы, имеющие большое число валентных электронов, обладающих различной энергией. Такими свойствами обладают соединения d-элементов (IB, VIIB, VIIIB групп) и р-элементов (VIIA, VIA, VA групп). Соединения, которые содержат элемент в высшей степени окисления, проявляют только окислительные свойства (КМnО4, H2SO4), в низшей - только восстановительные свойства (H2S), в промежуточной - могут вести себя двояко (Na2SO3). После составления уравнений полуреакций, ионного уравнения составляют уравнение реакции в молекулярной форме:
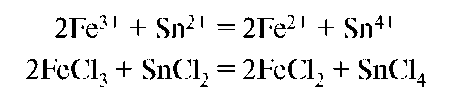
Проверка правильности составления уравнения: число атомов и зарядов левой части уравнения должно быть равно числу атомов и зарядов правой части уравнения для каждого элемента.
Окислительно-востановительная
двойственность – способность одного и того же вещества, в зависимости от
реагентов и от условий проведения реакции, выступать как в роли окислителя так
и в роли восстановителя. Окислительно-востановительная двойственность
характерна для простых веществ – неметаллов. Например, фосфор по отношению к
металлам выступает в роли окислителя: 3P + 2Ca = Ca2P3
в то же время фосфор выступает в роли восстановителя по отношению к фтору,
кислороду или хлору. Например: 4P
+ 5O2 = 2P2O4
Билет 23 – 28.
Билет 29.
Теория строения органических соединений Бутлерова.Теория химического строения органических веществ была сформулирована А. М. Бутлеровым в 1861 году.У этой теории четыре положения:Атомы в молекуле соединены в определённой последовательности в соответствии с их валентностью. Эта последовательность называется химическим строением.Свойства вещества зависят не только от качественного и количественного состава молекулы, но и от её химического строения. Вещества, имеющие один и тот же состав, но разное строение, называются изомерами, а само их существование изомерией.Атомы и группы атомов в молекуле взаимно влияют друг на друга непосредственно или посредством других атомов.Строение вещества познаваемо, возможен синтез веществ с заданным строением.
Билет 30.
Пространственное строение органических соединений. Стереохимические формулы.
Особенности
органических соединений, их многообразие
определяются, прежде всего, электронным
строением атома углерода, который в
органических соединениях проявляет
валентность равную четырем и может
находиться в sp3-,
sp2-
и sp-гибридном состоянии. Поэтому связь
между атомами может осуществляться
одной, двумя и тремя электронными парами,
т.е. быть одинарной (σ - связь), двойной
(1σ - связь и 1π - связь), тройной (1σ - связь
и 2 π - связи). Исключительное свойство
углерода – способность образовывать
цепи атомов различной длины и циклические
структуры.Стереохимия
циклических соединенийПри
замыкании цепи углеродных атомов в
плоский цикл валентные углы атомов
углерода вынуждены отклоняться от
своего нормального тетраэдрического
значения, причем величина этого отклонения
зависит от числа атомов в цикле. Чем
больше угол отклонения валентных связей,
тем больше должен быть запас энергии
молекулы, тем меньше устойчивость цикла.
Однако, плоское строение имеет только
трехчленный циклический углеводород
(циклопропан); начиная с циклобутана
молекулы циклоалканов имеют неплоское
строение, что понижает "напряжение"
в системе.Молекула циклогексана может
существовать в виде нескольких
конформаций, в которых сохраняются
"нормальные" валентные углы (для
упрощения показаны только атомы
углерода):![]() Энергетически
наиболее выгодной является конформация
I - так называемая форма "кресла".
Конформация II - "твист"
- занимает промежуточное положение: она
менее выгодна, чем конформация кресла
(из-за наличия в ней заслоненно
расположенных атомов водорода), но более
выгодна, чем конформация III. Конформация
III - "ванна"
- наименее выгодна из трех вследствие
значительного отталкивания направленных
верх атомов водорода.Рассмотрение
двенадцати связей С-Н в конформации
кресла позволяет разделить их на две
группы: шесть аксиальных
связей, направленных поочередно то
вверх, то вниз, и шесть экваториальных
связей, направленных в стороны. В
монозамещенных циклогексанах заместитель
может находиться либо в экваториальном,
либо в аксиальном положении. Эти две
конформации обычно находятся в равновесии
и быстро переходят друг в друга через
конформацию твист:
Энергетически
наиболее выгодной является конформация
I - так называемая форма "кресла".
Конформация II - "твист"
- занимает промежуточное положение: она
менее выгодна, чем конформация кресла
(из-за наличия в ней заслоненно
расположенных атомов водорода), но более
выгодна, чем конформация III. Конформация
III - "ванна"
- наименее выгодна из трех вследствие
значительного отталкивания направленных
верх атомов водорода.Рассмотрение
двенадцати связей С-Н в конформации
кресла позволяет разделить их на две
группы: шесть аксиальных
связей, направленных поочередно то
вверх, то вниз, и шесть экваториальных
связей, направленных в стороны. В
монозамещенных циклогексанах заместитель
может находиться либо в экваториальном,
либо в аксиальном положении. Эти две
конформации обычно находятся в равновесии
и быстро переходят друг в друга через
конформацию твист:![]() а-
е-
а-
е-
Экваториальная конформация (е) обычно беднее энергией и поэтому более выгодна, чем аксиальная (а).При появлении в циклах заместителей (боковых цепей) кроме проблемы конформации самого цикла перед исследователем встают и проблемы конфигурации заместителей: так, в случае наличия двух одинаковых или различных заместителей появляются цис-транс-изомера. Отметим, что говорить о цис-транс-конфигурации заместителей имеет смысл только в приложении к насыщенным малым и средним циклам (до С8): в кольцах с большим числом звеньев подвижность становится уже столь значительной, что рассуждения о цис- или транс- положении заместителей теряют смысл.Так, классическим примером являются стереоизомерные циклопропан-1,2-дикарбоновые кислоты. Существуют две стереоизомерные кислоты: одна из них, имеющая т.пл. 139 оС, способна образовывать циклический ангидрид и является, следовательно, цис-изомером. Другая стереоизомерная кислота с т.пл. 175 оС, циклического ангидрида не образует; это транс-изомер
![]()
В таких же отношениях друг с другом находятся две стереоизомерные 1,2,2-триметилциклопентан-1,3-дикарбоновых кислоты. Одна из них, камфорная кислота, т.пл. 187 оС, образует ангидрид и, следовательно, является цис-изомером. Другая - изокамфорная кислота, т.пл. 171 оС, - ангидрида не образует, это транс-изомер:
![]() цис-
транс-
цис-
транс-
Хотя молекула циклопентана на самом деле неплоская, для наглядности удобно изображать ее в плоском виде, как на приведенном выше рисунке, имея в виду, что в цис-изомере два заместителя находятся по одну сторону цикла, а в транс-изомере - по разные стороны цикла. Дизамещенные производные циклогексана также могут существовать в цис- или транс-форме:. а) 1,2-дизамещенные циклогексаны:
![]()
![]()
б) 1,3-дизамещенные циклогексаны:
![]()
![]()
в) 1,4-дизамещенные циклогексаны:
![]()
![]()
Обратим внимание на то обстоятельство, что понятия цис- и транс- в производных циклогексана относительны: вместо двугранных углов между связями, равных нулю (цис-форма) или 180о (транс-форма) в ряду циклогексана наблюдаются иные углы. Например, у 1,2-замещенных в обеих конфигурациях, как цис-, так и транс-, угол между валентными связями одинаков: он составляет всего 60о, т.е. ближе к истинной цис-форме.
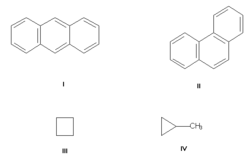
Билет 31.Виды структурной изомерии органических соединений.Структурная изомерия — результат различий в химическом строении. К этому типу относят:Изомерия углеводородной цепи (углеродного скелета)
Изомерия углеродного скелета, обусловленная различным порядком связи атомов углерода. Простейший пример — бутан СН3—СН2—СН2—СН3 и изобутан (СН3)3СН. Другие примеры: антрацен и фенантрен (формулы I и II, соответственно), циклобутан и метилциклопропан (III и IV).Валентная изомерия
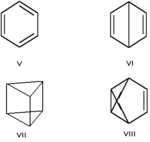
Валентная — особый вид структурной изомерии, при которой изомеры можно перевести друг в друга лишь за счёт перераспределения связей. Например, валентными изомерами бензола (V) являются бицикло[2.2.0]гекса-2,5-диен (VI, «бензол Дьюара»), призман (VII, «бензал Ладенбурга»), бензвален (VIII).Изомерия функциональной группыРазличается характером функциональной группы; например, этанол (CH3—CH2—OH) и диметиловый эфир (CH3—O—CH3).Изомерия положенияТип структурной изомерии, характеризующийся различием положения одинаковых функциональных групп или кратных связей при одинаковом углеродном скелете. Пример: 2-хлорбутановая кислота и 4-хлорбутановая кислота.
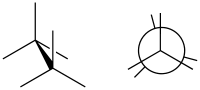
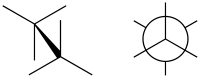
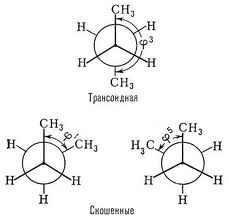
Билет 32. Энергетическая характеристика конформационных состояний: заслонённые, частично заслонённые (заторможенные), скошенные конформации.Различают следующие виды конформаций [8]:
Заторможенная (или трансоидная конформация) (англ. staggered conformation) — заместители одного атома на проекции размещены между заместителями другого атома, деля валентные углы, т.е. заместители расположены наиболее далеко друг от друга в пространстве. Такие конформации обладают наименьшей энергией.
Заслоненная конформация (или цисоидная конформация) (англ. eclipsing conformation) — конформация, в которой заместители как бы налагаются друг на друга или находятся друг относительно друга в наиболее близком положении. Такие конформации обладают наиболее высокой энергией.
(англ. bisecting conformation) — для структуры, содержащей группы R3C–C(Y) =X (с идентичными или различными группами R) структура, в которой угол вращения таков, что X антиперипланарный одной из групп R, и, в проекции Ньюмана, двойная связь, C=X делит пополам один из углов R–C–R. В этой структуре связь C–Y заслоняет одну из связей C–R. В заслоненной конформации X синперипланарный одной из групп R. [9]
Виды конформаций
![]()
Заслоненная конформация
Заторможенная конформация
Скошенная конформация
Билет 33.Конформации (кресло, ванна) циклических соединений (на примере циклогексана). Аксиальные и экваториальные связи.Шестичленным циклом является циклогексан, существуют также пятичленные и восьмичленные. Циклогексан служит удобной моделью для изучения конформаций шестичленных циклов. Среди нескольких возможных конформаций циклогесана наименьшей энергией будет обладать конформация «кресло». Но различают также и другие:Конформация "кресло" Конформация "ваннаКонформация "твист" — любые два соседние атомы смещены в разные стороны от плоскости, построенной по 3 оставшимсяКонформация "корона" Конформация "конверт" — 4 атома из пяти находятся на одной плоскости, а пятый выходит из неё.

1 – кресло; 2- конверт; 3,5 – твист-конформации; 4 – ванна.
Билет 34.Стереоизомерия с одним центром хиральности (энантиомерия). Понятие оптическая активность. Среди органических соединений встречаются вещества, способные вращать плоскость поляризации света. Это явление называют оптической активностью, а соответствующие вещества - оптически активными. Оптически активные вещества встречаются в виде пар оптических антиподов - изомеров, физические и химические свойства которых в обычных условиях одинаковы, за исключением одного - знака вращения плоскости поляризации. (Если один из оптических антиподов имеет, например, удельное вращение +20о, то другой - удельное вращение -20о).Оптическая изомерия появляется тогда, когда в молекуле присутствует асимметрический атом углерода. Так называют атом углерода, связанный с четырьмя различными заместителями. Возможны два тетраэдрических расположения заместителей вокруг асимметрического атома. Обе пространственные формы нельзя совместить никаким вращением; одна из них является зеркальным изображением другой.
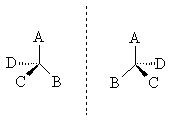
Рассмотренный вид изомерии называют оптической изомерией, зеркальной изомерией илиэнантиомерией. Обе зеркальные формы составляют пару оптических антиподов или энантиомеров.
Оптическая активность. способность в-ва- твердого, жидкого или газа-вращать плоскость поляризации проходящего через него света. Такие в-ва наз. оптически активными. Поворот происходит либо вправо (по часовой стрелке), либо влево (против часовой стрелки), если смотреть навстречу ходу лучей света. Оптической активностью обладают энантиомеры (см. Изомерия), а также энантиоморфные формы кристаллов (см. Энантиоморфизм)ахиральных в-в при хиральном расположении их молекул в кристаллич. решетке (напр., кварц, мочевина). От этой естественной оптической активности хиральных сред отличают наведенную оптическая активность ахиральных в-в, к-рая появляется в них в магн. поле (Фарадея эффект)или при контакте с хиральными молекулами (эффект Пфейфера).
Мера оптической активности-оптич. вращение a, к-рое измеряют при помощи поляриметров, спектрополяриметров и дихрогра-фов. Уд. вращение для жидкости вычисляют по ф-ле , где - угол поворота плоскости поляризации луча (в град) в кювете длиной l (в дм), d~ плотн. в-ва (в г/см3), и t означают длину волны света и т-ру р-ра, они влияют на величину . Для р-ра линейно зависит от толщины слоя р-ра и концентрации оптически активного в-ва (закон Б и о) и ф-ла имеет вид , где с-концентрация в-ва (г в 100 см3 р-ра). Уд. вращение зависит, кроме того, от типа р-рителя. и его также необходимо указывать. Напр., для 20% р-ра правовращающей винной к-ты в воде для D-линии натрия (= 589 нм) и 20 °С записывают: + 11,98° (вода, с 20). Часто вместо уд. вращения указывают молярное вращение , где М-мол. масса оптически активного в-ва. Совр. поляриметры позволяют измерять оптическая активность с высокой точностью (до 0,001°).
Билет 35. Проекционные формулы Фишера. D и L – система стереохимической номенклатуры. Правила R S номенклатуры.
Для изображения оптических изомеров (энантиомеров) обычно используют проекционные формулы Фишера (1а) – (1г), показанные ниже на примере одного из энантиомеров молочной кислоты. Для перехода от модели одного из энантиомеров молочной кислоты к формуле Фишера, надо ориентировать тетраэдр таким образом, чтобы горизонтальная пара связей была обращена к наблюдателю, а вертикальная – удалена от него. Очевидно, что при этом возможны разные ориентации: на тетраэдр можно смотреть с разных сторон. В результате одна модель может дать двенадцать (!) внешне непохожих друг на друга проекционных формул Фишера. Существуют, однако, определенные правила: в случае гидроксикислот вверху помещают карбоксильную группу, а углеродную цепь располагают сверху вниз. Это расположение переносят на плоскость бумаги.
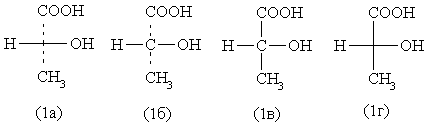
Из разных вариантов записи проекционных формул наиболее целесообразен вариант (1а), столь же ясное изображение дает вариант (1б): в обоих случаях видно, какие группы приближены к наблюдателю, какие удалены от него. В вариантах (1в) и (1г) такой определенности нет, что создает почву для недоразумений; увидев изображение типа (1в) или (1г), надо еще разобраться, имел ли в виду автор подлинную проекцию Фишера (к наблюдателю приближены боковые заместители).
Правила преобразования формул ФишераПроекционные формулы можно вращать в плоскости чертежа на 180°, не меняя их стереохимического смысла. Пользуясь записью типа (1а), можно повернуть формулу в плоскости чертежа и на 90°: изменившаяся ориентация пунктирной линии покажет, что теперь боковые заместители лежат дальше от наблюдателя, но конфигурация по-прежнему изображена правильно. В случае формул типа (1в) и (1г) поворот на 90° приведет к обратной конфигурации.
Одна (или любое нечетное число) перестановок заместителей у асимметрического центра приводит к формуле оптического антипода; любое четное число перестановок не изменяет стереохимического смысла фомулы.
Вместо перестановок проекционные формулы можно прообразовывать вращая три заместителя по часовой стрелке или против нее.
Проекционные формулы нельзя выводить из плоскости чертежа; нельзя, например, рассматривать их “на просвет”, с обратной стороны бумаги, так как при этом мы увидели бы формулу антипода.
Билет 36.Среди органических соединений встречаются вещества, способные вращать плоскость поляризации света. Это явление называют оптической активностью, а соответствующие вещества - оптически активными. Оптическая изомерия появляется тогда, когда в молекуле присутствует асимметрический атом углерода. Так называют атом углерода, связанный с четырьмя различными заместителями. Возможны два тетраэдрических расположения заместителей вокруг асимметрического атома. Обе пространственные формы нельзя совместить никаким вращением; одна из них является зеркальным изображением другой.
Рассмотренный вид изомерии называют оптической изомерией, зеркальной изомерией илиэнантиомерией. Обе зеркальные формы составляют пару оптических антиподов или энантиомеров. Диастереомеры- пространственные изомеры, не составляющих друг с другом оптических антиподов. Диастереомеры отличаются друг от друга не только оптическим вращением, но и всеми другими физическими константами: у них разные температуры плавления и кипения, разные растворимости и др. Примером соединения рассматриваемого типа может случить хлоряблочная кислота.
![]()
Если в формуле вещества есть асимметрический атом, это отнюдь не означает, что такое вещество будет обладать оптической активностью. Если асимметрический центр возникает в ходе обычной реакции (замещение в группе СН2, присоединение по двойной связи и т.п.), то вероятность создания обеих антиподных конфигураций одинакова. Несмотря на асимметрию каждой отдельной молекулы, получающееся вещество оказывается оптически неактивным. Такого рода оптически неактивные модификации, состоящие из равного количества обоих антиподов, называются рацематами.Мезоформа является диастереоизомером по отношению к каждому из зеркальных изомеров и поэтому отличается от них по температуре плавления и некоторым другим свойствам. Ее следует отличать от оптически неактивного рацемического соединения: последнее может быть разделено на оптически активные изомеры, тогда как для мезоформы этого сделать нельзя, ибо ее оптическая неактивность является свойством ее молекул. Мезоформа- оптически неактивная модификация стрериоизомера. В отличие от рацемата, который может быть расщеплен на оптические антиподы, мезоформа принципиально нерасщепляема: каждая ее молекула имеет один асимметрический центр одной конфигурациии, второй - противоположной. В итоге происходит внутримолекулярная компенсация вращения обоих асимметрических центров.
Билет 37. Цис-транс-Изомеры – стереоизомеры, различающиеся взаимным пространственным расположением заместителей относительно плоскости двойной связи или цикла.В цис-изомерах заместители находятся по одну сторону от плоскости двойной связи или кольца, в транс-изомерах – по разные. цис-транс-Изомерия (часто этот вид стереоизомерии называют геометрической изомерией) проявляется в случае, когда каждый из двух атомов углерода двойной связи C=C (или цикла) имеет различные заместители.
Пи-диастереомеры. К ним относят конфигурационные изомеры, содержащие пи-связь. Этот вид диастереомерии характерен, в частности, для алкенов. Относительно плоскости пи-связи одинаковые заместители у двух атомов углерода могут располагаться по одну (цис) или по разные (транс) стороны. Это приводит к существованию стереоизомеров, известных также под названием цис- и транс-изомеров. Основная причина существования цис- и транс-изомеров заключается в невозможности вращения вокруг пи-связи без ее нарушения. Цис- и транс-изомерыимеют одинаковую последовательность связывания атомов, но отличаются друг от друга пространственным расположением заместителей и потому являются стереоизомерами. В то же время их молекулы ахиральны (в них нет хиральных центров). Таким образом, цис- и трансизомеры алкенов относительно друг друга являются диастереомерами и обладают разными свойствами.

Представителями ненасыщенных дикарбоновых кислот с одной двойной связью служат пи-диастереомерные малеиновая и фумаровая кислоты.
Билет 38.
Поляризация химической связи — асимметрия (смещение) электронной плотности, связывающей молекулярной орбитали ковалентной связи. Полярная связь — химическая связь, обладающая постоянным электрическим дипольным моментом вследствие несовпадения центров тяжести отрицательного заряда электронов и положительного заряда ядер. Большинство ковалентных, а также донорно-акцепторные связи являются полярными.
Индуктивный эффект (I-эффект) -- смещение электронной плотности по цепи -связей, которое обусловлено различиями в электроотрицательностях (ЭО) атомов.
Из-за слабой поляризуемости -связей I-эффект быстро затухает с удалением от заместителя и через 3-4 связи становится практически равным нулю.
Индуктивный эффект заместителя является отрицательным (-I), если заместитель уменьшает электронную плотность на данном атоме углерода, индуцируя на нем частичный положительный заряд (сигма «+»)
Индуктивный эффект заместителя является положительным (+I), если заместитель увеличивает электронную плотность на данном атоме углерода, индуцируя на нем частичный отрицательный заряд (сигма «-»).
–I эффект проявляют заместители, которые содержат более атомы с большей ЭО, чем у углерода:
+I эффект проявляют заместители, содержащие атомы с низкой ЭО:
Электронные эффекты заместителей
Если заместитель подаѐт электронную плотность, то он является электронодонорным (ЭД), если заместитель оттягивает электронную плотность на себя, то он является электроноакцепторным (ЭА). Один и тот же заместитель может проявлять разные электронные эффекты в зависимости от того с каким радикалом он связан.
Рассмотрим фенол и бензиловый спирт:
Группа OH в феноле проявляет оба эффекта, причѐм +М больше чем –I. Следовательно, группа OH в феноле является электронодонором.
Группа OH в бензиловом спирте проявляет только –I эффект (+М эффекта нет, так как нет сопряжения с кольцом). Следовательно, в данном случае группа OH является электроноакцептором.
Билет 39.
Сопряжение (мезомерия,
от греч. mesos —
средний) — это выравнивание связей и
зарядов в реальной молекуле (частице)
по сравнению с идеальной, но несуществующей
(резонансной) структурой.Пи-пи-сопряжение.
Простейшим представителем пи-пи-сопряженных
систем с углеродной цепью служит
бутадиен- 1,3. Атомы углерода и водорода
и все сигма-связи в его молекуле лежат
в одной плоскости, образуя плоский
сигма-скелет. Атомы углерода находятся
в состоянии sp2-гибридизации.
Негибридизованные р-АО каждого атома
углерода расположены перпендикулярно
плоскости сигма-скелета и параллельно
друг другу, что является необходимым
условием для их перекрывания. Перекрывание
происходит не только между р-АО С-1 и
С-2, С-З и С-4, а также между р-АО С-2 и С-З.
В результате образуется охватывающая
четыре атома угле рода единая пи-система,
т. е. возникает делокализованная
ковалентная связь.
р-пи-сопряжение. В р-пи-сопряженных системах с углеродной цепью сопряжение может осуществляться при наличии рядом с пи-связью атома углерода с негибридизованной р-орбиталью. Очевидно, что такими системами не могут быть нейтральные молекулы, а могут быть только промежуточные частицы — карбанионы, карбокатионы, свободные радикалы, например аллильной структуры. В аллил-анионе СН2=СН—СН2 sp2-гибридизованный атом углерода С-З поставляет в общую сопряженную систему два электрона, в аллильном радикале СН2=СН—СН2 — один, а в аллильном карбокатионе СН2=СН—СН2 не поставляет ни одного. В результате при перекрывании р-АО трех sp2-гибридизованных атомов углерода образуется делокализованная трехцентровая связь, содержащая четыре (в карбанионе), три (в свободном радикале) и два (в карбокатионе) электрона соответственно. Формально атом С-З в аллил-катионе несет положительный заряд, в аллильном радикале — неспаренный электрон, а в аллил-анионе — отрицательный заряд. В действительности в таких сопряженных системах имеет место делокализация (рассредоточение) электронной плотности, что приводит к выравниванию связей и зарядов. Атомы углерода С-1 и С-З в этих системах равноценны. Например, в аллил-катионе каждый из них несет положительный заряд + 1/2 и связан полуторной связью с атомом углерода С-2.
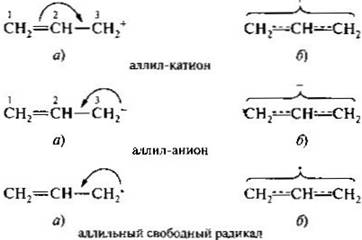
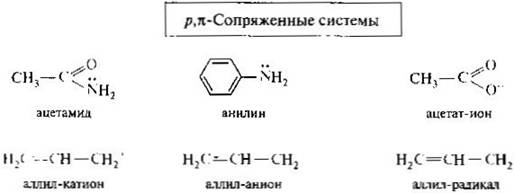
Сопряжение в открытых системах возникает при следующих условиях:
а) все атомы, участвующие в образовании сопряженной системы, находятся в sр2-гиб-ридизации;
б) рZ-орбитали всех атомов, образующих сопряженную систему, перпендикулярны плоскости -скелета, т. е. параллельны друг другу.
В замкнутых сопряженных системах создаются условия для круговой делокализации р-электронов, и они обладают особыми ароматическими свойствами.
Под ароматичностью понимается способность плоскостных циклических систем с замкнутой системой сопряжения, охватывающей все атомы цикла, вступать в обычных условиях в реакции замещения, а не присоединения и обладать повышенной устойчивостью цикла к разрыву и окислению.
Типичной карбоциклической ароматической системой является молекула бензола.
Критерии ароматичности были установлены Хюккелем в 1931 году и получили название правило Хюккеля. Система ароматична, если она обладает совокупностью следующих признаков:
а) все атомы в цикле находятся в sр2-гибридизации (следовательно -скелет плоскостной);
б) молекула имеет циклическую систему сопряжения;
в) в сопряжении участвует (4n+2) р-электрона, где n — целое число (n = 0, 1, 2, 3, 4...).
Бензол отвечает правилу Хюккеля при n = 1, т. е. в сопряжении участвует 6 р-элек-тронов.
Ароматические свойства характерны и для конденсированных систем. Конденсированными называются системы, у которых два и более циклов имеют общие пары углеродных атомов.
Билет 40.Мезомерный эффект (М-эффект)Мезомерный эффект - смещение электронной плотности по цепи делокализованных (сопряженных) π-связей Этот эффект проявляют заместители, связанные с sp2- или sp-гибридизованным атомом. Благодаря подвижности π-электронов, М-эффект передается по цепи сопряжения без затухания. +М-эффектом обладают заместители, повышающие электронную плотность в сопряженной системе. К ним относятся группы, которые содержат атомы с неподеленной парой электронов, способные к передаче этой пары электронов в общую систему сопряжения. +М-эффект характерен для групп -OH и -NH2.При образовании ковалентной связи в молекулах органических соединений общая электронная пара заселяет связывающие молекулярные орбитали, имеющие более низкую энергию. В зависимости от формы МО – σ-МО или π-МО – образующиеся связи относят к σ- или p-типу.σ-Связь – ковалентная связь, образованная при перекрывании s-, p- и гибридных АО вдоль оси, соединяющей ядра связываемых атомов (т.е. при осевом перекрывании АО).π-Связь – ковалентная связь, возникающая при боковом перекрывании негибридных р-АО. Такое перекрывание происходит вне прямой, соединяющей ядра атомов.π-Связи возникают между атомами, уже соединенными σ-связью (при этом образуются двойные и тройные ковалентные связи). π-Связь слабее σ-связи из-за менее полного перекрывания р-АО.Для определения знака М-эффекта полезно строить атомно-орбитальные модели, отражающие пространственную ориентацию орбиталей и возможности их перекрывания.
Билет 41.Заместители электронодонорные, повышающие общую электронную плотность в ядре, облегчают реакцию электрофильногс замещения и являются, таким образом, активирующими, а заместители электроноакцепторные, понижающие электронную плот ность в кольце, этой реакции препятствуют, т.К электронодонорным (активирующим) заместителям относятся следующие атомы и группы атомов (в скобках указаны основные электронные эффекты, проявляемые этими заместителями): алкилы (+1), —ОН, —OR, —NH2, — NHR, —NR2, (+M > — I), —0~, (+M и + I).К электроноакцепторным (дезактивирующим) заместителям относятся: —COR, —СООН, -COOR, -CN, -N02 (—М и -I), галогены (+М < —I), NRf (—I).Если заместитель подаёт электронную плотность, то он является электронодонорным (ЭД), если заместитель оттягивает электронную плотность на себя, то он является электроноакцепторным (ЭА). Один и тот же заместитель может проявлять разные электронные эффекты в зависимости от того с каким радикалом он связан.
Билет 42. Классификация органических реакций:
ОВР- изменение степени окисления атомов. Окисление- процесс отдачи электрона атомом в-ва. Восстановление- процесс присоединения электрона в-ом. Любая ОВР- конкуренция за электрон междя сопряженными ОВ-парами.
Типы ОВР:
межмолекулярные- в которых степень окисления изменяется у атомов разных молекул;
Внутримолекулярные- изменение СО атома, входящего в состав одно молекулы;
Дисмутации (диспрапорционирование) где окислитель и восстановитель- атом одного в-ва.
Замещение- Реакции замещения — химические реакции, в которых одни функциональные группы, входящие в состав химического соединения, меняются на другие группы.
Типы: В реакциях нуклеофильного замещения атакующей частицей является нуклеофил, то есть отрицательно заряженная частица или частица со свободной электронной парой. Уходящая группа носит название нуклеофуг.Реакции нуклеофильного замещения более характерны для алифатических систем.В реакция электрофильного замещения атакующей частицей является электрофил, то есть положительно заряженная частица или частица с дефицитом электронов. Уходящая частица носит название электрофуг.Реакции электрофильного замещения более характерны для ароматических систем.В реакция радикального замещения атакующей частицей являются свободные радикалы.Реакции присоединения- химические реакции, в которых одни химические соединения присоединяются к кратным (двойным илитройным) связям другого химического соединения. Присоединение может осуществляться как по связи углерод-углерод, так и по связи углерод-гетероатом. Типы:В реакциях нуклеофильного присоединения атакующей частицей является нуклеофил, то есть отрицательно заряженная частица или частица со свободной электронной парой.В реакциях электрофильного присоединения атакующей частицей является электрофил, то есть положительно заряженная частица, чаще всего протон H+, или частица с дефицитом электронов.В реакциях радикального присоединения атакующей частицей являются свободные радикалы. Реакции радикального присоединения обычно протекают вместо реакций электрофильного присоединения в присутствии источника свободных радикалов.
ЭЛИМИНИРОВАНИЯ РЕАКЦИИ (р-ции
отщепления), отщепление от молекулы орг.
соед. частиц (атомов или
атомных групп) без замены их другими.
Различают![]() и d-элиминирования реакции.
При
и d-элиминирования реакции.
При![]() -элиминировании
(отщепление частиц от одного атома)
образуются валентно-ненасыщ. соед.
(напр., карбены,нитрены),
при
-элиминировании
(отщепление частиц от одного атома)
образуются валентно-ненасыщ. соед.
(напр., карбены,нитрены),
при![]() -элиминировании
(отщепление частиц от соседних атомов)
- соед. с кратными
связями (С
= С, С = С, С = N, C = N), при
-элиминировании
(отщепление частиц от соседних атомов)
- соед. с кратными
связями (С
= С, С = С, С = N, C = N), при![]() либо
либо![]() -элиминировании
(отщепление частиц от атомов,
разделенных одним или двумя атомами)
- циклич. соед. Разновидность
элиминирования реакций -
выброс фрагмента из углеродной цепи
или цикла с образованием новой
-элиминировании
(отщепление частиц от атомов,
разделенных одним или двумя атомами)
- циклич. соед. Разновидность
элиминирования реакций -
выброс фрагмента из углеродной цепи
или цикла с образованием новой![]() -связи
(такие р-ции иногда наз. р-циями экструзии).
-связи
(такие р-ции иногда наз. р-циями экструзии).
Билет 43. Реакционная способность предельных углеводородов. Механизм реакции свободнорадикального замещения. Галогенирование, сульфирование, нитрование. Окисление.
Инертны при обычных условиях:
Не способны к реакциям присоединения
С большинством хим. реагентов не реагируют или мало
Реакции замещения при наличии высокой температуры или облучении:
Окисление O2, галогенами, HNO3, сульфоокисление.
Изомеризация
Пиролиз
Механизм свободнорадикального замещения

Галогенирование (см. картинку выше).Сульфирование. (Нашел в конспектах сульфохлорирование) и нитрование.

Окисление.
Горение алканов при t > 300 градусов Цельсия – воспламенение.
Полное окисление: C4H10 + 6.5O2 = 4CO2 + 5H2O + (дельта)H
Неполное окисление:C4H10 + 4.5O2 = 4CO + 5H2O + (дельта)H
Билет 44. Реакционная способность непредельных углеводородов. Механизм реакции электрофильного присоединения к алкенам. Влияние статических и динамических факторов на региоселективность реакций.
Благодаря электронам π-связи в молекулах алкенов имеется область повышенной электронной плотности, поэтому они склонны подвергаться атаке электрофильными реагентами. Относительно плоскости π-связи одинаковые заместители у двух атомов углерода могут располагаться по одну(цис) или по разные (транс) стороны. Это приводит к существованию в ряду алкенов пространственных изомеров (стереоизомеров), известных под названием цис- и транс-изомеров. Основная причина существованияцис- и транс-изомеров заключается в невозможности вращения вокруг π-связи без ее нарушения.
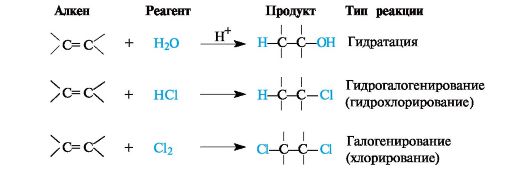
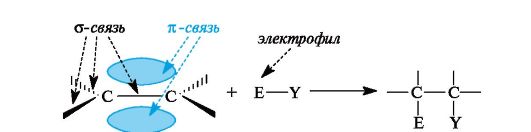
Общее описание механизма реакции электрофильного присоединения AE. Присоединение к алкенам электрофильных реагентов НХ (Н2О, ННа1 и т.п.) протекает по гетеролитическому механизму. Электрофильной частицей в данном процессе служит простейший электрофил - протон.В реакции выделяют две основные стадии:• атаку алкена протоном с образованием карбокатиона (медленная стадия, определяющая скорость процесса в целом);• атаку образовавшегося карбокатиона нуклеофилом; в реакции гидратации это молекула Н2О (быстрая стадия).Так, при гидратации пропена (несимметричного алкена) в соот ветствии с правилом Марковникова преимущественным продукто реакции является пропанол-2:
![]() Такое
направление реакции объясняется
совокупностью двух факторов.
В статическом, т.
е. нереагирующем, состоянии в несимметричных
алкенах электронная плотность π-связи
смещена под влиянием заместителя.
Возникшие частичные заряды определяют
место будущей атаки протоном. В пропене
таким местом будет атом С-1 с частичным
отрицательным зарядом (как следствие
+/-эффекта метильной группы). Таким
образом, статическийфактор
благоприятствует элек-трофильной атаке
по группе СН2 (путь а), что
приводит к вторичному карбокатиону
(I). При атаке по атому С-2 должен был бы
образоваться менее стабильный первичный
карбокатион (II) (путь б).
Такое
направление реакции объясняется
совокупностью двух факторов.
В статическом, т.
е. нереагирующем, состоянии в несимметричных
алкенах электронная плотность π-связи
смещена под влиянием заместителя.
Возникшие частичные заряды определяют
место будущей атаки протоном. В пропене
таким местом будет атом С-1 с частичным
отрицательным зарядом (как следствие
+/-эффекта метильной группы). Таким
образом, статическийфактор
благоприятствует элек-трофильной атаке
по группе СН2 (путь а), что
приводит к вторичному карбокатиону
(I). При атаке по атому С-2 должен был бы
образоваться менее стабильный первичный
карбокатион (II) (путь б).
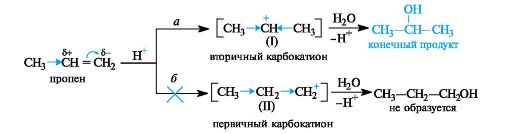
В динамическом состоянии, т. е. в ходе реакции, из двух возможных карбокатионов (I) или (II) будет образовываться более устойчивый. Во вторичном карбокатионе положительно заряженный атом угле- рода связан с двумя электронодонорными алкильными группами, в первичном - с одной. В результате во вторичном карбокатионе за счет +/-эффекта двух алкильных групп осуществляется более эффективное уменьшение положительного заряда. Таким образом, качественная оценка относительной устойчивости промежуточных частиц также говорит в пользу образования вторичного карбокатиона (путь а).
Правило Марковникова применяется без оговорок только к алкенам. Однако ненасыщенные соединения часто содержат при двойной связи электроноакцепторные группы (карбоксильную, альдегидную и др.). Учитывая поляризацию связи С=С под влиянием заместителя (статический фактор), можно предсказать иной характер присоединения, а именно, против правила Марковникова. Например, при гидратации α,β-ненасыщенных карбоновых кислот в сильно кислой среде образуются β-гидроксикарбоновые кислоты.
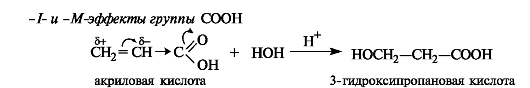 Присоединение
галогеноводородов и серной кислоты. Алкены
и циклоалкены в достаточно мягких
условиях взаимодействуют с
галогеноводородами, серной и другими
сильными кислотами, способными к
диссоциации с образованием протона.В
результате присоединения галогеноводородов
образуются галогенопроизводные алканов
и циклоалканов.
Присоединение
галогеноводородов и серной кислоты. Алкены
и циклоалкены в достаточно мягких
условиях взаимодействуют с
галогеноводородами, серной и другими
сильными кислотами, способными к
диссоциации с образованием протона.В
результате присоединения галогеноводородов
образуются галогенопроизводные алканов
и циклоалканов.
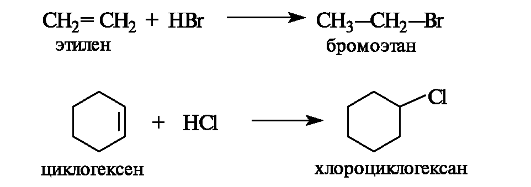
В реакции с концентрированной серной кислотой алкены образую гидросульфаты, в результате гидролиза которых получаются спирты.
![]() Галогенирование. Алкены
в обычных условиях легко присоединяют
галогены. Так, быстрое обесцвечивание
бромной воды без выделения бромоводорода
служит качественной пробой на двойную
связь. Еще легче проходит присоединение
хлора.
Галогенирование. Алкены
в обычных условиях легко присоединяют
галогены. Так, быстрое обесцвечивание
бромной воды без выделения бромоводорода
служит качественной пробой на двойную
связь. Еще легче проходит присоединение
хлора.
![]()
Восстановление и окисление связей C=C. Восстановление двойных связей алкенов осуществляется каталитическим гидрированием - присоединением водорода в присутствии металлов (никеля - при нагревании, платины или палладия - при обычной температуре). Результатом является образование насыщенного продукта - алкана (отсюда, кстати, и возникли термины «насыщенные» и «ненасыщенные» соединения).
![]()
Окисление двойных углерод-углеродных связей в зависимости от условий может приводить к эпоксидам, 1,2-диолам (гликолям) или карбонильным соединениям - продуктам расщепления двойной связи.
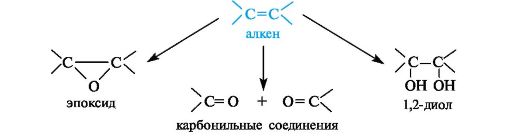
Окисление р-ом KMnO4 в нейтр. или слабощелочной среде (реакция Вагнера):3 CH3=CH2 + 2 KMnO4 = 3 CH2OH-CH2OH + 2 MnO2 + 2 KOH (обесцвечивание KMnO4)Окислительное расщепление кислотным р-ом KMnO4 или K2Cr2O7, O3(озон):

Правило Марковникова:При присоединении протонных кислот или воды к несимметричным алкенам или алкинам атом водорода присоединяется к наиболее гидрированному атому углерода.
Билет 45.
Билет 46.
Ароматичность, критерии ароматичности, правило Хюккеля.Ароматичность — особое свойство некоторых химических соединений, благодаря которому сопряженное кольцо ненасыщенных связей проявляет аномально высокую стабильность; большую чем та, которую можно было бы ожидать только при одном сопряжении.Ароматичность не имеет непосредственного отношения к запаху органических соединений, и является понятием, характеризующим совокупность структурных и энергетических свойств некоторых циклических молекул, содержащих систему сопряженных двойных связей. Термин «ароматичность» был предложен потому, что первые представители этого класса веществ обладали приятным запахом.К ароматическим соединениям относят обширную группу молекул и ионов разнообразного строения, которые соответствуют критериям ароматичности.
склонность к реакциям замещения, а не присоединения (определяется легче всего, исторически первый признак, пример — бензол, в отличие от этилена не обесцвечивает бромную воду)
выигрыш по энергии, в сравнении с системой несопряженных двойных связей. Также называется Энергией Резонанса (усовершенствованный метод — Энергией Резонанса Дьюара) (выигрыш настолько велик, что молекула претерпевает значительные преобразования для достижения ароматичного состояния, например циклогексадиен легко дегидрируется до бензола, двух и трехатомные фенолы существуют преимущественно в форме фенолов (енолов), а не кетонов и.т.д.).
наличие кольцевого магнитного тока (наблюдение требует сложной аппаратуры), этот ток обеспечивает смещение хим-сдвигов протонов, связанных с ароматическим кольцом в слабое поле (7-8 м.д. для бензольного кольца), а протонов расположенных над/под плоскостью ароматической системы — в сильное поле (спектр ЯМР).
наличие самой плоскости (минимально искаженной), в которой лежат все (либо не все — гомоароматичность) атомы образующие ароматическую систему. При этом кольца пи-электронов, образующиеся при сопряжении двойных связей (либо электронов входящих в кольцо гетероатомов) лежат над и под плоскостью ароматической системы.
практически всегда соблюдается Правило Хюккеля: ароматичной может быть лишь система, содержащая (в кольце) 4n+2 электронов (где n = 0, 1, 2, …). Система, содержащая 4n электронов является антиароматичной (в упрощенном понимании это обозначает избыток энергии в молекуле, неравенство длин связей, низкая стабильность — склонность к реакциям присоединения). В то же время, в случае пери-сочленения (есть атом(ы), принадлежащий(е) одновременно 3 циклам, то есть возле него нет атомов водорода или заместителей), общее число пи-электронов не соответствует правилу Хюккеля (фенален, пирен, коронен). Также предсказывается, что если удастся синтезировать молекулы в формеленты Мёбиуса (кольцо достаточно большого размера, дабы закручивание в каждой паре атомных орбиталей было мало), то для таких молекул система из 4n электронов будет ароматичной, а из 4n+2 электронов — антиароматичной.
Билет 47.
Ароматичные бензольные соединения (бензол, нафталин, фенантрен).
Бензол
Нафталин
![]()
Фенантрен

Билет 48.
Механизм реакции элетрофильного замещения ароматических соединений. П-комплекс и
Механизм реакции SEAr или реакции ароматического электрофильного замещения (англ. Electrophilic aromatic substitution) является самым распространенным и наиболее важным среди реакций замещения ароматических соединений и состоит из двух стадий. На первом этапе происходит присоединение электрофила, на втором — отщепление электрофуга:

В ходе реакции образуется промежуточный положительно заряженный интермедиат (на рисунке — 2b). Он носит название интермедиат Уэланда, арониевый ион или σ-комплекс. Этот комплекс, как правило, очень реакционноспособен и легко стабилизируется, быстро отщепляя катион.
Лимитирующей стадией в подавляющем большинстве реакций SEAr является первый этап.
Скорость реакции SEAr обычно представляется в следующем виде[2]:
Скорость реакции = k*[ArX]*[E+] |
В качестве атакующей частицы обычно выступают относительно слабые электрофилы, поэтому в большинстве случаев реакция SEAr протекает под действием катализатора — кислоты Льюиса. Чаще других используются AlCl3, FeCl3, FeBr3, ZnCl2.
В этом случае механизм реакции выглядит следующим образом (на примере хлорирования бензола, катализатор FeCl3)[3]:
1.На первом этапе катализатор взаимодействует с атакующей частицей с образованием активного электрофильного агента:
![]()
2. На втором этапе, собственно, и реализуется механизм SEAr:
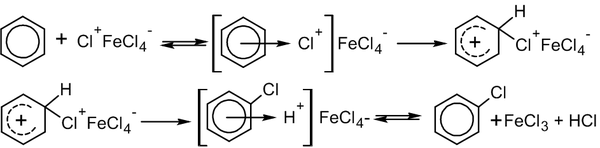
Типовые реакции ароматического электрофильного замещения
1. Нитрование ароматических систем азотной кислотой в присутствии серной кислоты с получением нитросоединений:
![]()
Образование активной частицы[2]:
![]()
Скорость реакции = k*[ArH]*[NO2+] |
2. Сульфирование бензола с получением сульфокислоты:
![]()
Активной частицей в реакции является SO3.
3. Галогенирование бензола бромом, хлором или йодом приводит к образованию арилгалогенидов. Катализатором реакции выступает галогенид железа (III):
![]()
Образование активной частицы[2]:
![]()
Скорость реакции = k*[ArH]*[X2]*[FeX3] |
4. Реакция Фриделя-Крафтса — ацилирование или алкилирование с использованием ацил- или алкилгалогенидов. Типичным катализатором реакции служит хлорид алюминия, но может использоваться любая другая сильная кислота Льюиса.
![]()
Скорость реакции = k*[ArH]*[RX]*[AlCl3] |
Про п комплекс только в тетради нашел
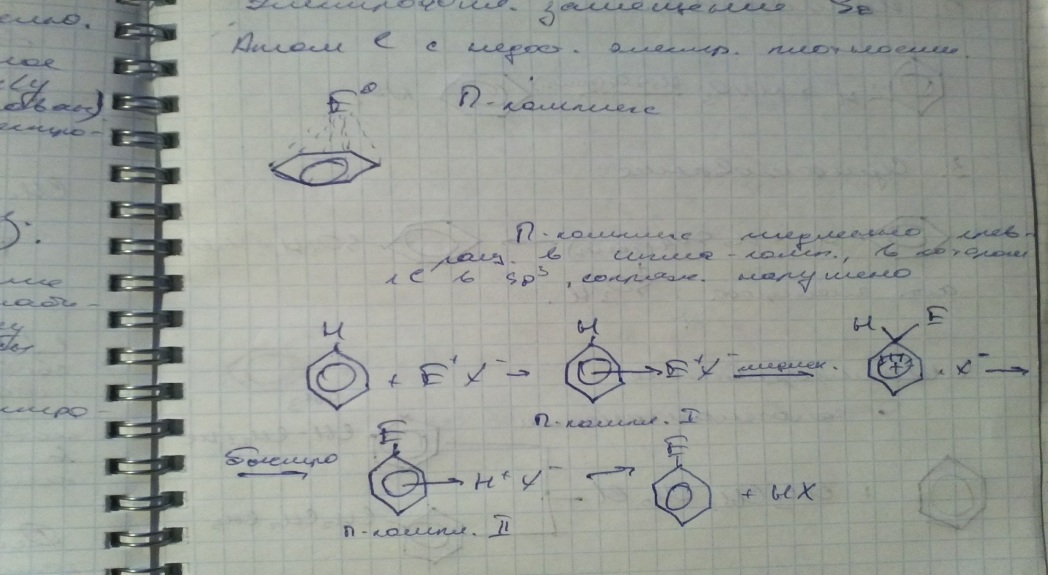
Билет 49-54
Билет 56.
Многоатомные спирты. Кач реакции на фенолы и виц диолы.Многоатомные спирты - органические соединения, содержащие в своём составе более одной гидроксильной группы -ОН.Важнейшие из многоатомных спиртов - этиленгликоль и глицерин:
![]()
этиленгликоль глицери. Это — вязкие жидкости, сладкие на вкус, хорошо растворимые в воде и плохо растворимые в органических растворителях.Получение.
1. Гидролиз алкилгалогенидов (аналогично одноатомным спиртам):
ClCH2-CH2Cl + 2NaOH → НОСН2-СН2ОН + 2NaCl.
2. Этиленгликоль образуется при окислении этилена водным раствором перманганата калия:
СН2=СН2 + [О] + Н2О → НOСН2-СН2ОН.
3. Глицерин получают гидролизом жиров.
Химические свойства. Для двух- и трехатомных спиртов характерны основные реакции одноатомных спиртов. В реакциях могут участвовать одна или две гидроксильные группы. Взаимное влияние гидроксильных групп проявляется в том, что многоатомные спирты — более сильные кислоты, чем одноатомные спирты. Поэтому многоатомные спирты, в отличие от одноатомных, реагируют со щелочами, образуя соли. По аналогии с алкоголятями соли двухатомных спиртов называют гликолятами, а трехатомных — глицератами.
Качественной реакцией на многоатомные спирты, содержащие группы ОН при соседних атомах углерода, является ярко, синее окрашивание при действии свежеосажденного гидроксида меди (II). Цвет раствора обусловлен образованием комплексного гликолята меди:
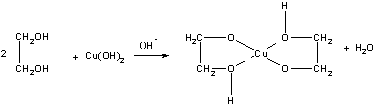
Для многоатомных спиртов характерно образование сложных эфиров. В частности, при реакции глицерина с азотной кислотой в присутствии каталитических количеств серной кислоты образуется тринитрат глицерина, известный под названием нитроглицерин (последнее название неверно с химической точки зрения, поскольку в нитросоединениях группа -NO2непосредственно связана с атомом углерода):
![]()
Применение. Этиленгликоль применяют для синтеза полимерных материалов и в качестве антифриза. В больших количествах он используется также для получения диоксана, важного (хотя и токсичного) лабораторного растворителя. Диоксан получают межмолекулярной дегидратацией этиленгликоля:
![]()
Диоксан.Глицерин находит широкое применение в косметике, пищевой промышленности, фармакологии, производстве взрывчатых веществ. Чистый нитроглицерин взрывается даже при слабом ударе; он служит сырьем для получения бездымных порохов и динамита ― взрывчатого вещества, которое в отличие от нитроглицерина можно безопасно бросать. Динамит был изобретен Нобелем, который основал известную всему миру Нобелевскую премию за выдающиеся научные достижения в области физики, химии, медицины и экономики. Нитроглицерин токсичен, но в малых количествах служит лекарством, так как расширяет сердечные сосуды и тем самым улучшает кровоснабжение сердечной мышцы.
Кач реакция на фенолы:Взаимодействие фенолов с треххлорным железом (FeCl3) приводит к об-разованию комплексного соединения с чернильной окраской.
Билет 55. Реакции элиминирования
Элиминирование— это отщепление от молекулы органического соединения атомов или атомных групп без замены их другими.
Реакция элиминирования может проходит в одну стадию (по механизму E2), либо в две стадии, по механизму E1. Исходными веществами могут служить представители разных классов органических соединений.
Элиминирования протекает под действием сильных
оснований – концентрированных растворов гидроксидов щелочных металлов в спирте,
алкоголятов или амидов щелочных металлов. Основания отщепляют протон из b -положения,
одновременно из молекулы уходит галоген в виде галогенид-аниона:
Если возможно образование двух разных продуктов
элиминирования, то преимущественно образуется наиболее замещенный у двойной
связи алкен, который является термодинамически более стабильным (правило
Зайцева):
Механизм реакций элиминирования
Реакции элиминирования могут протекать, по мономолекулярному (Е1) или
бимолекулярному (Е2) механизмам
Е1-реакции протекают параллельно с реакциями SN1 и включают две стадии. Сначала образуется карбокатион,
от которого затем отщепляется под действием основания протон:
Реакции Е2 протекают параллельно с реакциями
SN2 и включают одну стадию, в
ходе которой одновременно происходит разрыв старых и образование новых
связей:
Механизм элиминирования (Е1 или Е2) определяется
теми же факторами, что и соответствующие процессы нуклеофильного замещения
(SN1 и SN2).
Конкуренция реакций нуклеофильного замещения
и элиминирования
Процессы элиминирования и нуклеофильного замещения всегда протекают
параллельно, так как все нуклеофилы одновременно являются и основаниями:
Соотношение продуктов элиминирования и замещения
зависит от природы реагентов и условий проведения реакции. Подбирая условия
реакции и реагент, можно добиться преимущественного протекания реакции в нужном
направлении.
Факторы, способствующие протеканию
элиминирования:
1) Высокая основность реагента.
Сильные основания будут атаковать в первую
очередь атом водорода в b -положении, а не углерод. Так, под действием
алкоголят-анионов, которые являются сильными основаниями, протекает в основном
элиминирование, в то время как менее основные тиолят-анионы реагируют по атому
углерода и дают продукты замещения:
CH3-CHI-CH3 +
C2H5O-Na+® CH3-CH=CH2 + C2H5OH + NaI
CH3-CHI-CH3 +
C2H5S-Na+® (CH3)2CH-S-C2H5 +NaI
Протеканию элиминирования способствует не только
высокая основность, но и большой объем реагента, что затрудняет его атаку по
атому углерода. Поэтому третичные алкоголят-анионы дают в основном продукты
элиминирования.
2) Малополярные растворители.
Один и тот же реагент – гидроксид калия, в водном растворе реагирует как
нуклеофил с образованием продуктов замещения, а в менее полярном растворителе –
спирте дает в основном продукты элиминирования:
3) Высокая температура.
Реакции элиминирования имеют большую энергию активации, чем реакции
замещения, и поэтому их скорость возрастает в большей степени при увеличении
температуры.
Билет 57.
Окислители – KMnO4, K2Cr2O7+H2SO4, CuO, O2+катализатор. Легкость окисления спиртов уменьшается в ряду:
первичные ≥ вторичные >> третичные.
Первичные спирты при окислении образуют альдегиды, которые затем легко окисляются до карбоновых кислот.

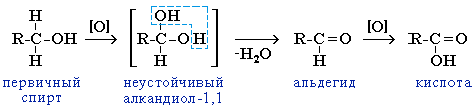
При окислении вторичных спиртов образуются кетоны.
![]()
Третичные спирты более устойчивы к действию окислителей. Они окисляются только в жестких условиях (кислая среда, повышенная температура), что приводит к разрушению углеродного скелета молекулы и образованию смеси продуктов (карбоновых кислот и кетонов с меньшей молекулярной массой).
Процесс идет через стадию дегидратации спирта с последующим деструктивным (жестким) окислением алкена. Например:
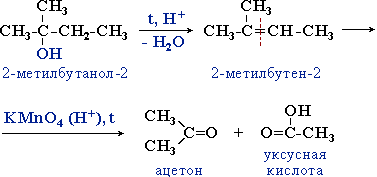
Предельное окисление гидроксисоединений до CO2 и Н2О происходит при их горении, например: 2CH3OH + 3O2 = 2CO2 + 4H2O
Полное окисление метанола идет схеме:
![]()
При сгорании спиртов выделяется большое количество тепла.
C2H5OH + 3O2 = 2CO2 + 3H2O + 1370 кДж
Благодаря высокой экзотермичности реакции горения этанола, он считается перспективным и экологически чистым заменителем бензинового топлива в двигателях внутреннего сгорания. В лабораторной практике этанол применяется как горючее для "спиртовок".Применение спиртов:Способность спиртов участвовать в разнообразных химических реакциях позволяет их использовать для получения всевозможных органических соединений: альдегидов, кетонов, карбоновых кислот простых и сложных эфиров, применяемых в качестве органических растворителей, при производстве полимеров, красителей и лекарственных препаратов.Применение фенолов:Фенолы применяются в производстве синтетических смол, пластмасс, полиамидов и других полимеров, лекарственных препаратов, красителей, поверхностно-активных веществ, антиоксидантов, антисептиков, взрывчатых веществ.
Билет 58.
Органические соединения, в молекуле которых имеется карбонильная группа > С=О, называются карбонильными соединениями или оксосоединениями. Они делятся на две родственные группы — альдегиды и кетоны. В молекулах альдегидов карбонильная группа связана с атомами водорода или с одним углеводородным радикалом:
![]()
альдегиды формальдегид ацетальдегид (муравьиный альдегид)(уксусный альдегид)
а в молекулах кетонов — с двумя углеводородными радикалами:
![]()
кетоны ацетон метилэтилкетон (диметилкетон)
Углеводородные радикалы могут быть алифатическими (насыщенными или ненасыщенными), алициклическими и ароматическими.В молекуле кетона радикалы могут быть одинаковыми или разными. Поэтому кетоны, как и простые эфиры, делятся на симметричные и смешанные.Мы рассмотрим только алифатические карбонильные соединения. Общая формула предельных альдегидов и кетонов СnН2nО.Строение. В карбонильной группе связь между атомами углерода и кислорода — двойная. Атом углерода находится в состоянии sp2-гибридизации и образует 3 σ-связи (две связи С-Н и одну связь С-О), которые располагаются в одной плоскости под углом 120° друг к другу, p-связь С-О образована при перекрывании негибридных 2р-орбиталей атомов углерода и кислорода. Двойная связь С=О является сочетанием одной s- и одной p-связей. В силу большей электроотрицательности атома кислорода электронная плотность двойной связи смещена в сторону атома кислорода:
бутаналь 2-метилпропаналь
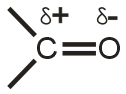
Полярность связи С=О сказывается на физических и химических свойствах карбонильных соединений. Изомерия альдегидов связана только со строением углеродного скелета, например:
СН3-СН2-СН2-СН=О СН3-СН(СН3)-СН=О.
Изомерия кетонов связана со строением углеродного скелета и с положением карбонильной группы, например:
![]()
пентанон-2 пентанон-3
Кроме того, альдегиды и кетоны с одинаковым числом атомов углерода изомерны друг другу, например ацетон и пропаналь или глюкоза и фруктоза.
Номенклатура. Для альдегидов часто используют тривиальные названия, например формальдегид Н2С=О. По международной номенклатуре названия альдегидов образуют, прибавляя окончание -аль к названию углеводорода с самой длинной углеродной цепью, включающей карбонильную группу, от которой и начинают нумерацию цепи.
Кетоны часто называют по наименованию радикалов, связанных с карбонильной группой, например метилэтилкетон СН3-СО-СН2-СН3. По международной номенклатуре: к названию предельного углеводорода добавляют окончание -он и указывают номер атома углерода, связанного с карбонильным кислородом. Нумерацию начинают с ближайшего к карбонильной группе конца цепи, например: метилэтилкетон — это то же самое, что бутанон-2.
Физические свойства. Карбонильные соединения не образуют водородных связей, поскольку в их молекулах нет атомом водорода с положительным зарядом. По этой причине температуры кипения альдегидов и кетонов значительно ниже, чем соответствующих спиртов. Низшие альдегиды и кетоны —легкокипящие жидкости (формальдегид — газ) с резким запахом, хорошо растворимы в воде.
Строение карбонильной группы C=O.
Свойства альдегидов и кетонов определяются строением карбонильной группы >C=O.
Атомы углерода и кислорода в карбонильной группе находятся в состоянии sp2-гибридизации. Углерод своими sp2-гибридными орбиталями образует 3 -связи (одна из них - связь С–О), которые располагаются в одной плоскости под углом около 120° друг к другу. Одна из трех sp2-орбиталей кислорода участвует в -связи С–О, две другие содержат неподеленнные электронные пары. p-Связь образована р-электронами атомов углерода и кислорода. |
Связь С=О сильно полярна. Ее дипольный момент (2,6-2,8D) значительно выше, чем у связи С–О в спиртах (0,70D). Электроны кратной связи С=О, в особенности более подвижные p-электроны, смещены к электроотрицательному атому кислорода, что приводит к появлению на нем частичного отрицательного заряда. Карбонильный углерод приобретает частичный положительный заряд.
Поэтому углерод подвергается атаке нуклеофильными реагентами, а кислород - электрофильными, в том числе Н+.
В молекулах альдегидов и кетонов отсутствуют атомы водорода, способные к образованию водородных связей. Поэтому их температуры кипения ниже, чем у соответствующих спиртов. Метаналь (формальдегид) - газ, альдегиды С2–C5 и кетоны С3–С4 - жидкости, высшие - твердые вещества. Низшие гомологи растворимы в воде, благодаря образованию водородных связей между атомами водорода молекул воды и карбонильными атомами кислорода. С увеличением углеводородного радикала растворимость в воде падает.
Модели простейших карбонильных соединений |
||
Название |
Формула |
Модель |
Формальдегид (метаналь) |
H2C=O |
|
Ацетальдегид (этаналь) |
СH3-CH=O |
|
Ацетон (пропанон) |
(СH3)2C=O |
|
|
Связь С = О ковалентная, поляризованная. Электронная плотность связи смещается к более электроотрицательному атому кислорода. На атоме углерода возникает частичный положительный заряд. На ру АО кислорода находится неподеленная пара электронов. Таким образом, для карбонильной группы будут характерны реакции нуклеофильного присоединения АN.
Реакционные центры:
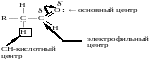
РАЗЛИЧИЯ В РЕАКЦИОННОЙ СПОСОБНОСТИ АЛЬДЕГИДОВ И КЕТОНОВ
Реакционная способность альдегидов и кетонов объясняется особенностями распределения электронной плотности, характером заместителей, связанных с карбонильной группой, пространственной доступностью реакционных центров.
2,2,2-трихлорэтаналь |
метаналь |
альдегид |
кетон |


Билет 59.
Присоединение воды к карбонильной группе — гидратация — обратимая реакция. Степень гидратации альдегида или кетона в водном растворе зависит от строения субстрата. Продукт гидратации, как правило, в свободном виде выделить с помощью перегонки не удается, так как он разлагается на исходные компоненты. Формальдегид в водном растворе гидратирован более чем на 99,9%, ацетальдегид — приблизительно наполовину, ацетон практически не гидратирован.
![]()
Присоединение спиртов. Спирты при взаимодействии с альдегидами легко образуют полуацетали. При обработке полуацеталей избытком спирта в кислой среде могут быть получены ацетали (реакция напоминает синтез простых эфиров из спиртов).

Присоединение аминов. Амины и другие азотсодержащие соединения общего вида NН2Х реагируют с альдегидами и кетонами в две стадии. Сначала образуются продукты нуклеофильного присоединения которые затем вследствие неустойчивости отщепляют воду. Поэтому данный процесс в целом классифицируют как реакциюприсоединения-отщепления.
Присоединение спиртов и тиолов.Альдегиды присоединяют спирты с образованием полуацеталей. При избытке спирта и в присутствии кислотного катализатора реакция идет дальше – до образования ацеталей
![]()
Реакция образования полуацеталя протекает как нуклеофильное присоединение и ускоряется в присутствии кислот или оснований.
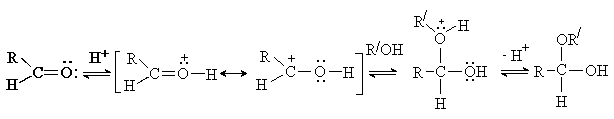
Процесс образования ацеталя идет как нуклеофильное замещение ОН группы в полуацетале и возможен только в условиях кислотного катализа, когда группа ОН превращается в хорошую уходящую группу (H2O).
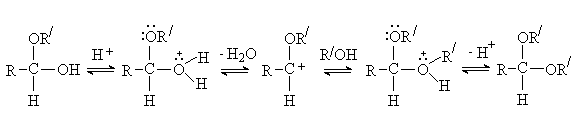
Образование ацеталей – обратимый процесс. В кислой среде полуацетали и ацетали легко гидролизуются. В щелочной среде гидролиз не идет. Реакции образования и гидролиза ацеталей играют важную роль в химии углеводов.Кетоны в аналогичных условиях кеталей не дают.Тиолы как более сильные нуклеофилы, чем спирты, образуют продукты присоединения и с альдегидами, и с кетонами.
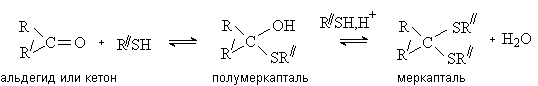
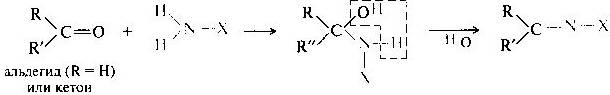
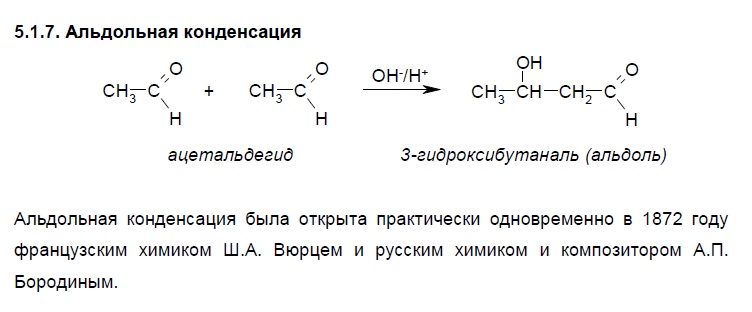
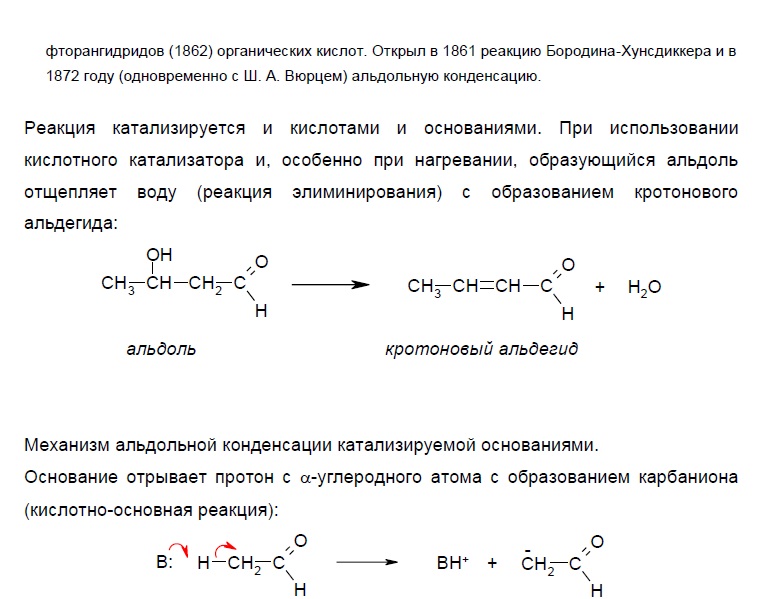

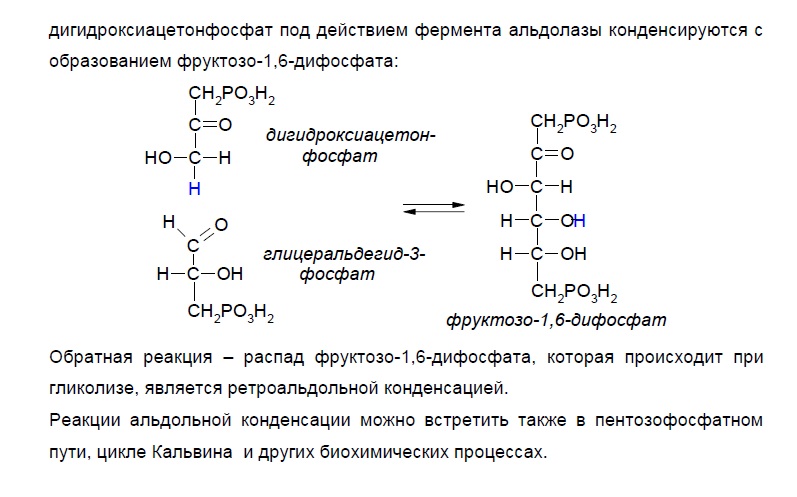
Билет 60.2.3. Реакции окисления и восстановленияВосстановление. Карбонильные соединения восстанавливаются до спиртов в результате каталитического гидрирования или под действием восстановителей, которые являются донорами гидрид-анионов.
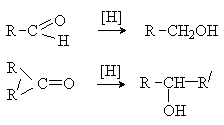
[H]: H2/кат., кат. – Ni, Pt, Pd;LiAlH4; NaBH4 .Восстановление карбонильных соединений комплексными гидридами металлов включает нуклеофильную атаку карбонильной группы гидрид-анионом. При последующем гидролизе образуется спирт.
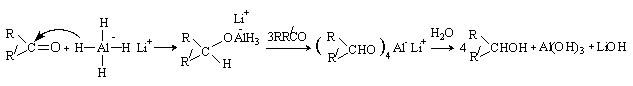
Аналогично происходит восстановление карбонильной группы in vivo под действием кофермента НАД Н, который является донором гидрид-иона (см. лек. №19).
ОкислениеАльдегиды окисляются очень легко практически любыми окислителями, даже такими слабыми, как кислород воздуха и соединения серебра (I) и меди (II).
![]()
Две последние реакции используются как качественные на альдегидную группу.В присутствии щелочей альдегиды, не содержащие a-водородных атомов диспропорционируют с образованием спирта и кислоты (реакция Канницаро).
2HCHO + NaOH ® HCOONa + CH3OHЭто является причиной того, что водный раствор формальдегида (формалин) при длительном хранении приобретает кислую реакцию.
Кетоны устойчивы к действию окислителей в нейтральной среде. В кислой и щелочной средах под действием сильных окислителей (KMnO4) они окисляются с разрывом связи С-С. Расщепление углеродного скелета происходит по двойной углерод-углеродной связи енольных форм карбонильного соединения, подобно окислению двойных связей в алкенах. При этом образуется смесь продуктов, содержащая карбоновые кислоты или карбоновые кислоты и кетоны.
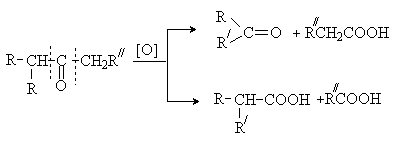
Качественная реакция на альдегиды с гидроксидом меди (II)
Одной из качественных реакций на альдегиды является реакция с гидроксидом меди (II). Получим гидроксид меди (II) сливанием растворов гидроксида натрия и сульфата меди (II). Прильем к полученному осадку раствор формальдегида. Нагреем смесь. На стенках пробирки выделяется металлическая медь.
Н-СОН + Cu(OH)2 = HCOOH + Cu + H2O
Однако чаще в результате этой реакции образуется красный осадок оксида меди (I)
Н-СОН + 2 Cu(OH)2 = HCOOH + Cu2O↓+ 2 H2O
Реакция серебренного зеркалаR-CH=O + 2[Ag(NH3)2]OH => RCOONH4 + 2Ag + 3NH3 + H2O
Серебряное зеркало образуется в том случае, если восстанавливающееся серебро осаждается на гладких стенках сосуда из не слишком концентрированных растворов. Малейшие загрязнения мешают восстанавливающемуся серебру «уцепиться» за стекло и заставляют его выделяться в виде рыхлого осадка.
Билет 61.
Применение альдегидов и кетонов.
Формальдегид Н2С=О (его водный раствор называют формалином) используют как дубитель кожи и консервант биологических препаратов.
Ацетон (СН3)2С=О – широко применяемый экстрагент и растворитель лаков и эмалей.
Ароматический кетон бензофенон (С6Н5)2С=О с запахом герани, используется в парфюмерных композициях и для ароматизации мыла.
Некоторые из альдегидов были сначала найдены в составе эфирных масел растений, а позже искусственно синтезированы.
Алифатический альдегид СН3(СН2)7С(Н)=О (тривиальное название – пеларгоновый альдегид) содержится в эфирных маслах цитрусовых растений, обладает запахом апельсина, его используют как пищевой ароматизатор.
Ароматический альдегид ванилин (рис. 10) содержится в плодах тропического растения ванили, сейчас чаще используется синтетический ванилин – широко известная ароматизирующая добавка в кондитерские изделия (рис. 10).
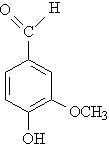
Рис. 10. ВАНИЛИН
Бензальдегид С6Н5С(Н)=О с запахом горького миндаля содержится в миндальном масле и в эфирном масле эвкалипта. Синтетический бензальдегид используется в пищевых ароматических эссенциях и в парфюмерных композициях.
Бензофенон (С6Н5)2С=О и его производные способны поглощать УФ-лучи, что определило их применение в кремах и лосьонах от загара, кроме того, некоторые производные бензофенона обладают противомикробной активностью и применяются в качестве консервантов. Бензофенон обладает приятным запахом герани, и потому его используют в парфюмерных композициях и для ароматизации мыла.
Способность альдегидов и кетонов участвовать в различных превращениях определила их основное применение в качестве исходных соединений для синтеза разнообразных органических веществ: спиртов, карбоновых кислот и их ангидридов, лекарственных препаратов (уротропин), полимерных продуктов (фенолоформальдегидные смолы, полиформальдегид), в производстве всевозможных душистых веществ (на основе бензальдегида) и красителей.
3.6. Применение альдегидов и кетонов
Метаналь (муравьиный альдегид) CH2=O
получение фенолформальдегидных смол;
получение мочевино-формальдегидных (карбамидных) смол;
полиоксиметиленовые полимеры;
синтез лекарственных средств (уротропин);
дезинфицирующее средство;
консервант биологических препаратов (благодаря способности свертывать белок).
Этаналь (уксусный альдегид, ацетальдегид) СН3СН=О
производство уксусной кислоты;
органический синтез.
Ацетон СН3-СО-СН3
растворитель лаков, красок, ацетатов целлюлозы;
сырье для синтеза различных органических веществ.
Применение альдегидов в медицине.
Формальдегид (формалин), прозрачная бесцветная жидкость со своеобразным острым запахом. Применяют как дезинфицирующее и дезодорирующее средство для мытья рук, обмывания кожи при повышенной потливости (0,5-1 %), для дезинфекции инструментов (0,5 %), для спринцеваний (1:2000 - 1:3000). Входит в состав лизоформа.
Формидрон - жидкость, содержащая раствора формальдегида 10 частей, спирта этилового 95 % 40 частей, воды 50 частей, одеколона 0,5 частей. Применяют для протирания кожи при повышенной потливости.
Мазь формальдегидная, белого цвета со слабым запахом формалина и отдушки. Применяют при повышенной потливости, втирают в подмышечняю впадины один раз в сутки, в межпальцевые складки.
Лизоформ мыльный раствор формальдегида. Состав: формалина 40 частей, мыла калийного 40 частей, спирта 20 частей. Оказывает дезинфицирующее и дезодорирующее действие. Применяют для спринцевания в гинекологической практике, для дезинфекции рук (1-3 % растворы).
Уротропин (гексаметилентетрамин), бесцветные кристаллы без запаха, легко растворимы в воде. Водные растворы имеют щелочную реакцию. Применяют главным образом при инфекционных процессах мочевыводящих путей (циститах, пиелитах).
Действие основано на способности препарата разлагаться в кислой среде с образованием формальдегида. Назначают препарат натощак. Показаниями для его применения служат холециститы, холангиты, аллергические заболевания кожи, глаз (кератиты, иридоциклиты и др.). Препарат может вызвать раздражение паренхимы почек, при этих признаках прием препарата прекращают.
Уросал, таблетки, содержащие по 0,3 г гексаметилентетрамина и фенилсалицилата.
Кальцекс - таблетки белого цвета, солено-горького вкуса, легко растворимы в воде.
Содержат 0,5 г комплексной соли гексаметилентетрамина и кальция хлорида. Применяют по 1-2 таблетки 3-4 раза в день при простудных заболеваниях.
Циминаль, подавляет (местно) грамположительные и грамотрицательные бактерии, способствует эпителизации и заживлению ран. Применяют наружно при лечении ран, пиодермии, трофических язв, ожогов.
Назначают в виде порошка (для припудривания) или 1-3 % суспензии, которую наносят на поврежденную поверхность, перевязки через 3-4 дня. При длительном применении препарата возможно возникновение дерматитов, чувства жжения и зуда.
Билет 62.
Карбоксильная группа (карбоксил) -СООН — функциональная одновалентная группировка, входящая в состав карбоновых кислот и определяющая их кислотные свойства.Карбоксильная группа сочетает в себе две функциональные группы - карбонил и гидроксил, взаимно влияющие друг на друга:
![]()
Кислотные свойства карбоновых кислот обусловлены смещением электронной плотности к карбонильному кислороду и вызванной этим дополнительной (по сравнению со спиртами) поляризации связи О–Н. В водном растворе карбоновые кислоты диссоциируют на ионы:
![]()
Растворимость в воде и высокие температуры кипения кислот обусловлены образованием межмолекулярных водородных связей.
![]()

С увеличением молекулярной массы растворимость кислот в воде уменьшается.
1. Диссоциация кислот. Все карбоновые кислоты, подобно неорганическим кислотам, обладают кислыми свойствами, окрашивая лакмус в красный цвет. Это обусловлено диссоциацией кислот:
RCOOH <-> RCOO- + Н+
Склонность карбоновых кислот к диссоциации обусловлена подвижностью водорода гидроксильной группы карбоксила. Мы уже знаем, что водород гидроксильной группы спиртов тоже подвижен и спирты в некоторой степени проявляют кислотные свойства. Однако в спиртах гидроксильная группа связана с насыщенным углеводородным остатком, и под его влиянием подвижность водорода в гидроксиле столь мала, что спирты являются более слабыми кислотами, чем вода (константы диссоциации спиртов ниже 10-16), и практически нейтральны. В кислотах же гидроксил непосредственно связан не с углеводородным остатком, а с карбонильной группой, под влиянием этой группы подвижность водорода в гидроксиле настолько увеличивается, что он способен к отщеплению в виде протона. Константы диссоциации карбоновых кислот значительно больше констант диссоциации спиртов и достигают порядка 10-4-105.
Билет 63.
Ацилирование – введение ацильной группы (ацила) RC в молекулу органического соединения путем замещения атома водорода. В широком смысле ацилирование это замещение любого атома или группы атомов на ацила. В зависимости от атома к которому присоединяют ацил различают C-, N-, O-, S – ацилирование.
Реакции ацилирования обладают очень многими полезными свойствами. Они позволяют вести в молекулу функциональную группу C=O путем реакций присоединения либо замещения, не подвергая исходную молекулу окислению (восстановлению). Таким образом, можно получать соединения различных классов: а) амиды; б) сложные эфиры; в) ангидриды карбоновых кислот; г) кетоны и другие полезные соединения. Неудивительно, что реакции ацилирования находят широкое применение в промышленности и в химических исследованиях. В своей курсовой работе я рассмотрю три наиболее важных типа реакций ацилирования C-ацилирование, O-ацилирование и N-ацилирование.