

Образование связи по донорно-акцепторному механизму
Кроме изложенного в предыдущем разделе гомогенного механизма образования ковалентной связи, существует гетерогенный механизм — взаимодействие разноименно заряженных ионов — протона H+ и отрицательного иона водорода H-, называемого гидрид-ионом:
H+ + H- → H2
При сближении ионов двухэлектронное облако (электронная пара) гидрид-иона притягивается к протону и в конечном счёте становится общим для обоих ядер водорода, то есть превращается в связывающую электронную пару. Частица, поставляющая электронную пару, называется донором, а частица, принимающая эту электронную пару, называется акцептором. Такой механизм образования ковалентной связи называется донорно-акцепторным.[7]
Распределение электронной плотности между ядрами в молекуле водорода одно и то же, независимо от механизма образования, поэтому называть химическую связь, полученную по донорно-акцепторному механизму, донорно-акцепторной связью некорректно.
В качестве донора электронной пары, кроме гидрид-иона, выступают соединения элементов главных подгрупп V—VII групп периодической системы элементов в низшей степени окисления элемента. Так, ещё Йоханнес Брёнстед установил, что протон не существует в растворе в свободном виде, в воде он образует катион оксония:
H+ + H2O → H3O+
Протон атакует неподелённую электронную пару молекулы воды и образует устойчивый катион, существующий в водных растворах кислот.[8]
Аналогично происходит присоединение протона к молекуле аммиака с образованием комплексного катиона аммония:
NH3 + H+ → NH4+
Таким путём (по донорно-акцепторному механизму образования ковалентной связи) получают большой класс ониевых соединений, в состав которого входят аммониевые, оксониевые, фосфониевые, сульфониевые и другие соединения.[9]
В качестве донора электронной пары может выступать молекула водорода, которая при контакте с протоном приводит к образованию молекулярного иона водорода H3+:
H2 + H+ → H3+
Связывающая электронная пара молекулярного иона водорода H3+ принадлежит одновременно трём протонам.
Комплексные соединения (лат. complexus — сочетание, обхват) или координационные соединения (лат. co — «вместе» и ordinatio — «упорядочение») — частицы (нейтральные молекулы или ионы), которые образуются в результате присоединения к данному иону (или атому), называемому комплексообразователем, нейтральных молекул или других ионов, называемых лигандами. Теория комплексных соединений (координационная теория) была предложена в 1893 г. А. Вернером.
Комплексные соединения мало диссоциируют в растворе (в отличие от двойных солей). Комплексные соединения могут содержать комплексный малодиссоциирующий анион ([Fe(CN)6]3−), комплексный катион ([Ag(NH3)2]+), либо вообще не диссоциировать на ионы (соединения типа неэлектролитов, например карбонилы металлов). Комплексные соединения разнообразны и многочисленны.
21.Первый закон термодинамики
Первый закон термодинамики (закон сохранения энергии для тепловых процессов) определяет количественное соотношение между изменением внутренней энергии системы дельта U, количеством теплоты Q, подведенным к ней, и суммарной работой внешних сил A, действующих на систему.
Первый закон термодинамики - Изменение внутренней энергии системы при ее переходе из одного состояния в другое равно сумме количества теплоты, подведенного к системе извне, и работы внешних сил, действующих на нее:
![]()
Первый закон термодинамики - количество теплоты, подведенное к системе, идет на изменение ее внутренней энергии и на совершение системой работы над внешними телами:
![]() Внутренняя
энергия и энтальпия
Внутренняя
энергия и энтальпия
1. Внутренней энергией U называется энергия системы, зависящая только от ее термодинамического состоянии. Для системы, нe подверженной действию внешних сил и находящейся в состоянии макроскопического покоя, внутренняя энергия представляет собой полную энергию системы. В некоторых простейших случаях внутренняя энергия равна разности между полной энергией W системы и суммой кинетической энергии WK ее макроскопического движения и потенциальной энергии Wп, обусловленной действием на систему внешних силовых полей: U = W - (Wk + Wп) 2. Внутренняя энергия является однозначной функцией состояния системы: ее изменение DU при переходе системы из состояния 1 в состояние 2 не зависит от вида процесса и равно DU = U2 - U1 3 Внутренняя энергия может быть определена только с точностью до постоянного слагаемого Я/0, которое не может быть найдено методами термодинамики. Однако это несущественно, так как при термодинамическом анализе системы приходится иметь дело не с абсолютными зна-чениями ее внутренней энергии, а с не зависящими от Ua изменениями этой энергии в различных процессах. По-этому часто полагают f/0 = 0, а под внутренней энергией системы понимают только тс ее составляющие, которые изменяются в рассматриваемых процессах. Например, при не слишком высоких температурах внутреннюю энер-гию идеального газа можно считать равной сумме кине-тических энергий хаотического движения его молекул. 4. Энтальпией H (теплосодержанием, тепловой функцией) называется функция состояния термодинамической системы, равная сумме ее внутренней энергии и произведения давления на объем системы, выраженного в тех же единицах: H = U + pV
22.
Закон Гесса
Тепловой эффект химической реакции, проводимой в изобарно-изотермических или изохорно-изотермических условиях, зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания.
Энтальпия образования (теплота образования), энтальпия реакции образования данного вещества (или раствора) из заданных исходных веществ. Энтальпией образования химических соединения называют энтальпию реакции образования данного соединения из простых веществ.
Энтальпия
образования может быть определена при
любой температуре. По определению, для
всех простых веществ при любой температуре
![]() =
0. Для большинства хим. соед. энтальпия
образования- отрицат. величины.
=
0. Для большинства хим. соед. энтальпия
образования- отрицат. величины.
Единицы измерения энтальпии образования - Дж/г, Дж/моль.
В термодинамике растворов (расплавов) под энтальпией образования понимают изменение энтальпии при изотермо-изобарном образовании 1 моля (1 г) раствора (расплава) данного состава из чистых компонентов. Так, в двухкомпонентной системе KF-A1F3 энтальпия образования расплава 50 %-ного молярного состава соответствует процессу: 1/2КF(жидкий) + 1/2А1F3(жидкий) = КF-А1F3(расплав, 50 мол. %).
Определение стандартных энтальпий образования смесей возможно при любой температуре, как и энтальпия образования химических соединений, энтальпию образования смеси данного состава называют часто энтальпией смешения. Неидеальные растворы характеризуются избыточной энтальпией образования, которая представляет собой разницу между реальной энтальпией образования фазы данного состава и энтальпией образования гипотетического идеального раствора того же состава.
Энтальпию образования определяют экспериментально калориметрическими измерениями. Второе и третье начала термодинамики позволяют получать этнальпию образования из экспериментальных данных. Теоретические расчеты энтальпии образования химических соединений пока неосуществимы. Возможны эмпирические оценки энтальпии образования, например, из энтальпии образования родственных соединений.
энтальпия образования хим. соединений табулированы в термодинамических справочниках и банках информации.
23.
Энтропи́я (от др.-греч. ἐντροπία - поворот, превращение) — в естественных науках мера беспорядка системы, состоящей из многих элементов. В частности, в статистической физике — мера вероятности осуществления какого-либо макроскопического состояния; в теории информации — мера неопределённости какого-либо опыта (испытания), который может иметь разные исходы, а значит, и количество информации; в исторической науке, для экспликации феномена альтернативности истории (инвариантности и вариативности исторического процесса).
![]() ,
,
где
![]() —
приращение энтропии;
—
приращение энтропии;
![]() —
минимальная теплота, подведенная к
системе; T — абсолютная температура
процесса
—
минимальная теплота, подведенная к
системе; T — абсолютная температура
процесса
* 2-й закон — второе начало термодинамики: Второй закон термодинамики исключает возможность создания вечного двигателя второго рода. Имеется несколько различных, но в то же время эквивалентных формулировок этого закона.
1 — Постулат Клаузиуса. Процесс, при котором не происходит других изменений, кроме передачи теплоты от горячего тела к холодному, является необратимым, то есть теплота не может перейти от холодного тела к горячему без каких-либо других изменений в системе. Это явление называют рассеиванием или диссипацией энергии.
Приведем второе
начало термодинамики в ещё одной
формулировке Рудольфа
Юлиуса Клаузиуса
(1865): для любой квазиравновесной
термодинамической системы существует
однозначная функция термодинамического
состояния
![]() ,
называемая энтропией, такая, что ее
полный дифференциал
,
называемая энтропией, такая, что ее
полный дифференциал
![]() .
[3]
.
[3]
2 — Постулат Кельвина. Процесс, при котором работа переходит в теплоту без каких-либо других изменений в системе, является необратимым, то есть невозможно превратить в работу всю теплоту, взятую от источника с однородной температурой, не проводя других изменений в системе.
Свободная энергия Гиббса (или просто энергия Гиббса, или потенциал Гиббса, или термодинамический потенциал в узком смысле) — это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на вопрос о принципиальной возможности протекания химической реакции; это термодинамический потенциал следующего вида:
![]()
Энергию Гиббса можно понимать как полную химическую энергию системы (кристалла, жидкости и т. д.)
Самопроизвольное протекание изобарно-изотермического процесса определяется двумя факторами: энтальпийным, связанным с уменьшением энтальпии системы (ΔH), и энтропийным T ΔS, обусловленным увеличением беспорядка в системе вследствие роста ее энтропии. Разность этих термодинамических факторов является функцией состояния системы, называемой изобарно-изотермическим потенциалом или свободной энергией Гиббса (G, кДж)
Энергия Гиббса и направление протекания реакции
В
химических процессах одновременно
действуют два противоположных фактора —
энтропийный
(![]() )
и энтальпийный
(
)
и энтальпийный
(![]() ).
Суммарный эффект этих противоположных
факторов в процессах, протекающих при
постоянном давлении и температуре,
определяет изменение энергии
Гиббса (
).
Суммарный эффект этих противоположных
факторов в процессах, протекающих при
постоянном давлении и температуре,
определяет изменение энергии
Гиббса (![]() ):
):![]()
24.
Скорость химической реакции — изменение количества одного из реагирующих веществ за единицу времени в единице реакционного пространства. Является ключевым понятием химической кинетики. Скорость химической реакции — величина всегда положительная, поэтому, если она определяется по исходному веществу (концентрация которого убывает в процессе реакции), то полученное значение умножается на −1.
Закон действия масс.
25.
Факторы влияющие на величину скорости химической реакции
Для элементарных реакций показатель степени при значении концентрации каждого вещества часто равен его стехиометрическому коэффициенту, для сложных реакций это правило не соблюдается. Кроме концентрации на скорость химической реакции оказывают влияние следующие факторы:
природа реагирующих веществ,
наличие катализатора,
температура (правило Вант-Гоффа),
давление,
площадь поверхности реагирующих веществ.
Если мы рассмотрим самую простую химическую реакцию A + B → C, то мы заметим, что мгновенная скорость химической реакции величина непостоянная.
26.
Химическое равновесие — состояние химической системы, в котором обратимо протекает одна или несколько химических реакций, причём скорости в каждой паре прямая-обратная реакция равны между собой. Для системы, находящейся в химическом равновесии, концентрации реагентов, температура и другие параметры системы не изменяются со временем.[1]
А2 + В2 ⇄ 2AB
Конста́нта равнове́сия — величина, определяющая для данной химической реакции соотношение между термодинамическими активностями (либо, в зависимости от условий протекания реакции, парциальными давлениями, концентрациями или фугитивностями) исходных веществ и продуктов в состоянии химического равновесия (в соответствии с законом действующих масс). Зная константу равновесия реакции, можно рассчитать равновесный состав реагирующей смеси, предельный выход продуктов, определить направление протекания реакции.
27.
Смещение химического равновесия
Положение химического равновесия зависит от следующих параметров реакции: температуры, давления и концентрации. Влияние, которое оказывают эти факторы на химическую реакцию, подчиняются закономерности, которая была высказана в общем виде в 1885 году французским ученым Ле-Шателье.
Факторы влияющие на химическое равновесие:
1) температура
При увеличении температуры химическое равновесие смещается в сторону эндотермической (поглощение) реакции, а при понижении в сторону экзотермической (выделение) реакции.
CaCO3=CaO+CO2 -Q t↑ →, t↓ ←
N2+3H2↔2NH3 +Q t↑ ←, t↓ →
2) давление
При увеличении давления химическое равновесие смещается в сторону меньшего объёма веществ, а при понижении в сторону большего объёма. Этот принцип действует только на газы, т.е. если в реакции участвуют твердые вещества, то они в расчет не берутся.
CaCO3=CaO+CO2 P↑ ←, P↓ →
1моль=1моль+1моль
3) концентрация исходных веществ и продуктов реакции
При увеличении концентрации одного из исходных веществ химическое равновесие смещается в сторону продуктов реакции, а при понижении концентрации продуктов реакции-в сторону исходных веществ.
S2+2O2=2SO2 [S],[O]↑ →, [SO2]↑ ←
Катализаторы не влияют на смещение химического равновесия!
28.
Раство́р — гомогенная (однородная) смесь, состоящая из частиц растворённого вещества, растворителя и продуктов их взаимодействия. "Гомогенный" - значит, каждый из компонентов распределён в массе другого в виде своих частиц, то есть атомов, молекул или ионов.[1].
Раствор — однофазная система переменного, или гетерогенного, состава, состоящая из двух или более компонентов.
Растворитель — компонент, агрегатное состояние которого не изменяется при образовании раствора. В случае же растворов, образующихся при смешении газа с газом, жидкости с жидкостью, твёрдого вещества с твёрдым, растворителем считается компонент, количество которого в растворе преобладает[1].
Образование того или иного типа раствора обусловливается интенсивностью межмолекулярного, межатомного, межионного или другого вида взаимодействия, то есть, теми же силами, которые определяют возникновение того или иного агрегатного состояния. Отличия: образование раствора зависит от характера и интенсивности взаимодействия частиц разных веществ[1].
По сравнению с индивидуальными веществами по структуре растворы сложнее[1].
Растворы бывают газовыми, жидкими и твёрдыми[1].
Способы выражения концентрации растворов
Концентрацию веществ в растворах можно выразить разными способами. На этой страничке вы с ними познакомитесь. Наиболее часто используют массовую долю растворённого вещества, молярную и нормальную концентрацию.
Массовая доля растворённого вещества w(B) - это безразмерная величина, равная отношению массы растворённого вещества к общей массе раствора m :
w(B)= m(B) / m

Массовую долю растворённого вещества w(B) обычно выражают в долях единицы или в процентах. Например, массовая доля растворённого вещества – CaCl2 в воде равна 0,06 или 6%. Это означает,что в растворе хлорида кальция массой 100 г содержится хлорид кальция массой 6 г и вода массой 94 г.
Нормальность раствора обозначает число грамм-эквивалентов данного вещества в одном литре раствора или число миллиграмм-эквивалентов в одном миллилитре раствора. Грамм - эквивалентом вещества называется количество граммов вещества, численно равное его эквиваленту. Для сложных веществ - это количество вещества, соответствующее прямо или косвенно при химических превращениях 1 грамму водорода или 8 граммам кислорода. Эоснования = Моснования / число замещаемых в реакции гидроксильных групп Экислоты = Мкислоты / число замещаемых в реакции атомов водорода Эсоли = Мсоли / произведение числа катионов на его заряд Диспе́рсная систе́ма — это система, образованная из двух или более фаз (тел), которые совершенно или практически не смешиваются и не реагируют друг с другом химически. Первое из веществ (дисперсная фаза) мелко распределено во втором (дисперсионная среда). Если фаз несколько, их можно отделить друг от друга физическим способом (центрифугировать, сепарировать и т. д.).
Обычно дисперсные системы — это коллоидные растворы, золи. К дисперсным системам относят также случай твёрдой дисперсной среды, в которой находится дисперсная фаза.
Диспе́ргаторы бывают рециркуляционного, встроенного и погружного типа.
29.
Коллигативные свойства растворов — это те свойства, которые при данных условиях оказываются равными и независимыми от химической природы растворённого вещества; свойства растворов, которые зависят лишь от количества кинетических единиц и от их теплового движения.
Первый закон Рауля
Пар, находящийся в равновесии с жидкостью, называют насыщенным. Давление такого пара над чистым растворителем (p0) называют давлением или упругостью насыщенного пара чистого растворителя.
В 1886 (1887) году Ф. М. Рауль сформулировал закон:
Давление пара раствора, содержащего нелетучее растворенное вещество, прямо пропорционально мольной доле растворителя в данном растворе:
p = p0 · χр-ль, где
p — давление пара над раствором, ПА;
p0 — давление пара над чистым растворителем;
χр-ль —— мольная доля растворителя.
Для растворов электролитов используют несколько другую форму уравнения, позволяющую добавить в неё изотонический коэффициент:
Δp = i · p0 · χв-ва, где
Δp — собственно изменение давления по сравнению с чистым растворителем;
χв-ва — мольная доля вещества в растворе.
Второй закон Рауля
Тот же Рауль экспериментально доказал, что
повышение температуры кипения раствора по сравнению с температурой кипения растворителя, а равно и понижение температуры замерзания раствора по сравнению с аналогичным характеризующей величиной для растворителя прямо пропорциональна моляльности раствора, то есть,
ΔTкип/зам= Kэб/кр · mв-ва, где
Kэб/кр — соответственно эбулиоскопическая (от лат. ebullire — «кипеть» и др.-греч. σκοπέω — «наблюдаю») и криоскопическая (относится к замерзанию) константы, характерные для данного растворителя;
mв-ва — моляльность вещества в растворе.
Зависимость скорости химической реакции от температуры описывает эмпирическое правило Вант-Гоффа. ЗАКОН ВАНТ-ГОФФА описывает зависимость ОСМОТИЧЕСКОГО ДАВЛЕНИЯ разбавленных растворов от температуры и молярной концентрации раствора: Вант-Гофф пришел к заключению, что закон Авогадро справедлив и для разбавленных растворов.
30. теория электр.диссоциации
Электролитическая диссоциация — процесс распада электролита на ионы при его растворении или плавлении.
Сильные
и слабые электролиты.
Степень
электролитической диссоциации
При
растворении одних электролитов
равновесие диссоциации значительно
смещено вправо, в растворах
таких электролитов диссоциация происходит
практически полностью (сильные
электролиты). При растворении
других электролитов диссоциация
происходит в незначительной мере
(слабые
электролиты).
С позиций современной
электростатической теории сильные
электролиты диссоциируют
необратимо, а слабые электролиты
– обратимо.
Для количественной
оценки силы электролита введено
понятие степени электролитической
диссоциации.
Степень
электролитической диссоциации
– отношение количества вещества
электролита распавшегося на ионы
(Vрасп.)
к количеству вещества электролита,
поступившего в раствор
(Vобщ.):
![]() ,
Степень
электролитической диссоциации
зависит от природы электролита, его
концентрации в растворе и температуры.
С разбавлением и с повышением
температуры степень электролитической
диссоциации возрастает.
Оценить
силу различных электролитов можно,
сравнивая степень их электролитической
диссоциации при одинаковых условиях.
Электролиты, степень диссоциации которых
при 18 0С
в растворах с концентрацией
0,1 моль/л электролита больше 30% относят
к сильным
электролитам.
Это щелочи, большинство солей, некоторые
неорганические кислоты (HClO4,
HI, HBr, HCl, HNO3,
H2SO4).
Электролиты, степень диссоциации которых
при 18 0С
в растворах с концентрацией
0,1 моль/л электролита меньше
3% относят к слабым
электролитам. Это
многие неорганические кислоты:
H2S,
HCN, HСlO, практически все органические
кислоты (например, HCOOH, CH3COOH,
CH3CH2COOH),
водный раствор аммиака NH3•H2O,
вода. Электролиты, степень диссоциации
которых при 18 0С
в 0,1М растворах от 3 до 30% относят к
электролитам
средней силы.
Это, например, ортофосфорная кислота
H3PO4,
фтороводородная кислота HF, азотистая
кислота HNO2.
Константа электролитической
диссоциации
Как уже было сказано выше, ЭД
слабых электролитов – обратимый
процесс. Поэтому силу электролита
также можно охарактеризовать с
помощью константы химического
равновесия процесса диссоциации
электролита – константы
диссоциации. Так, например,
диссоциация уксусной кислоты
протекает по уравнению:
СН3СООН
↔ СН3СОО–+Н+
характеризуется
константой диссоциации:
,
Степень
электролитической диссоциации
зависит от природы электролита, его
концентрации в растворе и температуры.
С разбавлением и с повышением
температуры степень электролитической
диссоциации возрастает.
Оценить
силу различных электролитов можно,
сравнивая степень их электролитической
диссоциации при одинаковых условиях.
Электролиты, степень диссоциации которых
при 18 0С
в растворах с концентрацией
0,1 моль/л электролита больше 30% относят
к сильным
электролитам.
Это щелочи, большинство солей, некоторые
неорганические кислоты (HClO4,
HI, HBr, HCl, HNO3,
H2SO4).
Электролиты, степень диссоциации которых
при 18 0С
в растворах с концентрацией
0,1 моль/л электролита меньше
3% относят к слабым
электролитам. Это
многие неорганические кислоты:
H2S,
HCN, HСlO, практически все органические
кислоты (например, HCOOH, CH3COOH,
CH3CH2COOH),
водный раствор аммиака NH3•H2O,
вода. Электролиты, степень диссоциации
которых при 18 0С
в 0,1М растворах от 3 до 30% относят к
электролитам
средней силы.
Это, например, ортофосфорная кислота
H3PO4,
фтороводородная кислота HF, азотистая
кислота HNO2.
Константа электролитической
диссоциации
Как уже было сказано выше, ЭД
слабых электролитов – обратимый
процесс. Поэтому силу электролита
также можно охарактеризовать с
помощью константы химического
равновесия процесса диссоциации
электролита – константы
диссоциации. Так, например,
диссоциация уксусной кислоты
протекает по уравнению:
СН3СООН
↔ СН3СОО–+Н+
характеризуется
константой диссоциации:
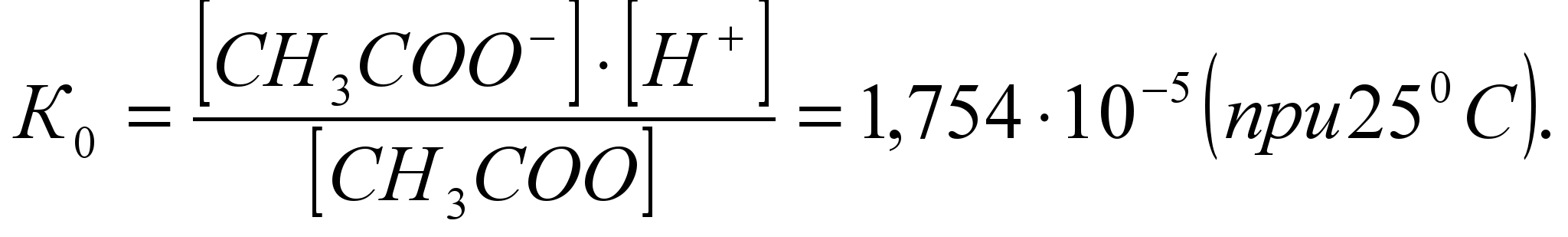 Зависимость
между степенью диссоциации и константой
диссоциации слабого электролита
определяется законом разбавления
Оствальда:
Константа диссоциации
зависит от температуры, но
не зависит от концентрации
электролита. В этом ее
преимущество по сравнению со
степенью электролитической диссоциации.
Чем больше значение константы
диссоциации, тем сильнее
электролит.
Зависимость
между степенью диссоциации и константой
диссоциации слабого электролита
определяется законом разбавления
Оствальда:
Константа диссоциации
зависит от температуры, но
не зависит от концентрации
электролита. В этом ее
преимущество по сравнению со
степенью электролитической диссоциации.
Чем больше значение константы
диссоциации, тем сильнее
электролит.
31.
Ионообменные реакции Реакции в растворах электролитов, при которых не происходит изменения зарядов ионов, входящих в соединения, называются ионообменными реакциями.
Обратимые и необратимые реакции
типы реакций химических (См. Реакции химические). Реакцию называют обратимой, если её направление зависит от концентраций веществ — участников реакции. Например, в случае гетерогенно-каталитической реакции
N2 + 3H2 = 2NH3 (1) при малой концентрации аммиака в газовой смеси и больших концентрациях азота и водорода происходит образование аммиака; напротив, при большой концентрации аммиака он разлагается, реакция идёт в обратном направлении. По завершении обратимой реакции, т. е. при достижении равновесия химического (См. Равновесие химическое), система содержит как исходные вещества, так и продукты реакции. Реакцию называют необратимой, если она может происходить только в одном направлении и завершается полным превращением исходных веществ в продукты; пример — разложение взрывчатых веществ. Одна и та же реакция в зависимости от условий (от температуры, давления) может быть существенно обратима или практически необратима.
Простая (одностадийная) обратимая реакция состоит из двух происходящих одновременно элементарных реакций, которые отличаются одна от другой лишь направлением химического превращения. Направление доступной непосредственному наблюдению итоговой реакции определяется тем, какая из этих взаимно-обратных реакций имеет большую скорость. Например, простая реакция
N2O4 ⇔ 2NO2 (2) складывается из элементарных реакций N2O4 →2NO2 и 2NO2 →N2O4.
Для обратимости сложной (многостадийной) реакции, например реакции (1), необходимо, чтобы были обратимы все составляющие её стадии.
32.
Ио́нное произведе́ние воды́ — произведение концентраций ионов водорода Н+ и ионов гидроксила OH− в воде или в водных растворах, константа автопротолиза воды.
Водоро́дный показа́тель, pH (произносится «пэ аш», английское произношение англ. pH — piː'eɪtʃ «Пи эйч») — мера активности (в очень разбавленных растворах она эквивалентна концентрации) ионов водорода в растворе, и количественно выражающая его кислотность, вычисляется как отрицательный (взятый с обратным знаком) десятичный логарифм активности водородных ионов, выраженной в молях на литр:
![]()
Теории кислот и оснований — совокупность фундаментальных физико-химических представлений, описывающих природу и свойства кислот и оснований. Все они вводят определения кислот и оснований -- двух классов веществ, реагирующих между собой. Задача теории -- предсказание продуктов реакции между кислотой и основанием и возможности её протекания, для чего используются количественные характеристики силы кислоты и основания. Различия между теориями лежат в определениями кислот и оснований, характеристики их силы и, как следствие -- в правилах предсказания продуктов реакции между ними. Все они имеют свою область применимости, каковые области частично пересекаются.
Кислотно-основные взаимодействия чрезвычайно распространенены в природе и находят широкое применение в научной и производственной практике. Теоретические представления о кислотах и основаниях имеют важное значение в формировании всех концептуальных систем химии и оказывают разностороннее влияние на развитие многих теоретических концепций во всех основных химических дисциплинах.
33.
Типы гидролиза солей
Рассмотрим гидролиз солей разных типов.
Соли сильного основания и сильной кислоты гидролизу не подвергаются, так как нет связывания ионов, не происходит образования слабых электролитов. В этом случае реакция среды в растворе – нейтральная. Соли слабого основания и сильной кислоты подвергаются гидролизу по катиону, реакция среды в растворе, в таком случае, кислая: Гидролиз соли сильного основания и слабой кислоты происходит по аниону, реакция среды в растворе – щелочная:
Гидролиз соли слабого основания и слабой кислоты происходит как по аниону, так и по катиону. Реакция среды в этом случае зависит от соотношения констант диссоциации соответствующих основания и кислоты.
34.Индикаторы
Лакмус (от нидерл. lakmoes) — красящее вещество природного происхождения, один из первых и наиболее широко известных кислотно-основных индикаторов.
Наименование стандартного химического препарата «лакмусовая бумага» стало нарицательным в русском языке, как в химии для всех типов индикаторных бумаг, так и в повседневной жизни при описании знаковых явлений и событий.
Свойства
В чистом виде представляет собой тёмный порошок со слабым запахом аммиака. Хорошо растворяется в чистой воде, образуя растворы фиолетового цвета. В кислых средах (pH<4,5) лакмус приобретает красную окраску, в щелочных (pH>8,3) — синюю.
Состав
|
|
|
Лакмус (pH индикатор) |
||
нижний предел pH 4.5 |
|
верхний предел pH 8.3 |
red |
↔ |
blue |
азолитмин (англ. Azolitmin, сост. C9H10NO5) — может быть выделен из лакмуса экстракцией и использоваться как самостоятельный кислотно-щелочной индикатор;
эритролитмин (англ. Erythrolitmin или Orcein Erythrolein, сост. С13H22O6);
Также экстракционным разделением из лакмуса могут быть выделены:
спанолитмин (англ. Spaniolitmin);
лейкоорцеин (англ. Leucoorcein);
лейказолитмин (англ. Leucazolitmin).
Применение
Применяется как индикатор для определения реакции среды. На практике используется несколько препаративных форм лакмуса: водный раствор лакмуса, полоски и клочки ленты фильтровальной бумаги, пропитанные лакмусом — т. н. лакмусовая бумага, «лакмусовое молоко» (нем. Lackmusmilch).
Фенолфталеи́н (4,4'-диоксифталофенон или 3,3-бис-(4-гидроксифенил)фталид) — трифенилметановый краситель, кислотно-основный индикатор, изменяющий окраску от бесцветной (при pH < 8,2) до красно-фиолетовой, «малиновой» (в щелочной); но в концентрированной щелочи — вновь бесцветен. В концентрированной серной кислоте образует розовый катион.
Вещество представляет собой бесцветные кристаллы, плохо растворимые в воде, но хорошо — в спирте и диэтиловом эфире.
Химические свойства
При сплавлении фенолфталеина с NaOH образуется 4,4'-дигидроксибензофенон, при нагревании с конц. H2SO4 — фенол и 2-гидроксиантрахинон.
Применение
В качестве индикатора
Фенолфталеин меняет окраску в зависимости от уровня pH среды. Он способен существовать в нескольких формах, которые превращаются одна в одну при изменении кислотности.
Применение в медицине
До обнаружения определенных проканцерогенных свойств фенолфталеин более полутора веков использовался в медицине как слабительное средство (пурген), хотя обладает кумулятивными свойствами и может оказывать раздражающее действие на почки.
Метиловый оранжевый(метилоранж гелиантин, 4-(4-диметиламинофенилазо)бензолсульфонат натрия) — известный кислотно-основной индикатор. Метилоранж является органическим синтетическим красителем из группы азокрасителей
|
Внешний вид при обычных условиях: оранжево-жёлтые листочки или порошок, чешуйки. Метилоранж растворим в воде 0,2 г. на 100 г., лучше в горячей.
В растворах с рН 2 абсорбирует свет в λmax 505 нм.
Переход окраски в водных растворах от красной к оранжево-жёлтой наблюдается в области рН 3, 1 — 4, 4 (в кислой среде красный, в щелочной — жёлтый).
На интервал перехода окраски влияют: температура, наличие в растворе солей, органических растворителей, белковых веществ и других. Влияние температуры наиболее значительно для индикаторов, являющихся слабыми основаниями: например, для метилового оранжевого при комнатной температуре окраска изменяется в пределах рН 3, 1 — 4, 4, а при 100°С — в пределах рН 2, 5 — 3, 7.
|
|
|
Метиловый оранжевый
Применяется в качестве кислотно-основного индикатора, титранта при определении сильных окислителей, спектрофотометрическом определении окислителей (хрома, брома).
0,1%-ный водный раствор применяется в аналитической химии как индикатор.
Изменяет цвет от красного в кислотной среде (pH 3,1 до 4,4) к оранжевому в нейтральной и жёлтому в щелочной.
35.
Сте́пень окисле́ния (окислительное число, формальный заряд) — вспомогательная условная величина для записи процессов окисления, восстановления и окислительно-восстановительных реакций, численная величина электрического заряда, приписываемого атому в молекуле в предположении, что электронные пары, осуществляющие связь, полностью смещены в сторону более электроотрицательных атомов.
Представления о степени окисления положены в основу классификации и номенклатуры неорганических соединений.
Вале́нтность (от лат. valēns «имеющий силу») — способность атомов химических элементов образовывать определённое число химических связей с атомами других элементов
Понятия валентности и химической связи неразрывно связаны между собой и являются основополагающими для современной химии. В то же время понятие степени окисления, сыгравшее определенную роль при интерпретации окислительно-восстановительных процессов и классификации химических соединений, в действительности имеет отдаленную связь с валентностью и не имеет прямого отношения к химической связи.
степень окисления - условный заряд который приобретает атом.... Постоянные степени окисления: 1) F только (-1) 2) O2 только (-2) 3) H2 только (+1) 4) металл + № группы гл. подруппа....поб подгруппа +2 5) простое в-во КНС=0 ст.ок Сумма степеней окисления в любом соединении равна 0!!!!
36.
Окисли́тельно-восстанови́тельные реа́кции, ОВР, редокс (от англ. redox ← reduction-oxidation — окисление-восстановление) — это встречно-параллельные химические реакции, протекающие с изменением степеней окисления атомов, входящих в состав реагирующих веществ, реализующихся путём перераспределения электронов между атомом-окислителем и атомом-восстановителем.
Окисление
Окисление - процесс отдачи электронов, с увеличением степени окисления.
При окисле́нии вещества в результате отдачи электронов увеличивается его степень окисления. Атомы окисляемого вещества называются донорами электронов, а атомы окислителя — акцепторами электронов.
В некоторых случаях при окислении молекула исходного вещества может стать нестабильной и распасться на более стабильные и более мелкие составные части (см. Свободные радикалы). При этом некоторые из атомов получившихся молекул имеют более высокую степень окисления, чем те же атомы в исходной молекуле.
Окислитель, принимая электроны, приобретает восстановительные свойства, превращаясь в сопряжённый восстановитель:
окислитель + e− ↔ сопряжённый восстановитель.
Восстановление
При восстановлении атомы или ионы присоединяют электроны. При этом происходит понижение степени окисления элемента. Примеры: восстановление оксидов металлов до свободных металлов при помощи водорода, углерода, других веществ; восстановление органических кислот в альдегиды и спирты; гидрогенизация жиров и др.
Восстановитель, отдавая электроны, приобретает окислительные свойства, превращаясь в сопряжённый окислитель:
восстановитель — e− ↔ сопряжённый окислитель.
Несвязанный, свободный электрон является сильнейшим восстановителем.
Метод электронного баланса — один из методов уравнивания окислительно-восстановительных реакций (ОВР).Заключается в том чтобы на основании степеней окисления расставить коэффициенты в ОВР.Для правильного уравнивания следует придерживаться определённой последовательности действий:
Найти окислитель и восстановитель.
Составить для них схемы (полуреакции) переходов электронов, отвечающие данному окислительно-восстановительному процессу.
Уравнять число отданных и принятых электронов в полуреакциях.
Просуммировать порознь левые и правые части полуреакций.
Расставить коэффициенты в уравнении окислительно восстановительной реакции.
Теперь рассмотрим конкретный пример
Дана реакция: Li + N2 = Li3N
1. Находим окислитель и восстановитель:
Li0 + N20 = Li3+1N−3
N присоединяет электроны, он-окислитель
Li отдаёт электроны, он-восстановитель
2. Составляем полуреакции:
Li0 — 1e = Li+1
N20 + 6e = 2N−3
37.