

38. Термодинамические понятия, используемые в теории растворов полимеров.
Растворы нужно делать ТД устойчивыми. Важна природа растворителя.
Функция Гиббса (равновесность системы). Р,Т = const. ΔG=ΔH-TΔS, если система однокомпонентная. ΔG<0, то процесс самопроизвольный. Раствор не расслоится и будет существовать долго. ΔG>0,то раствор прекратит существование. Он расслоится. ΔH и ΔS влияют на концентрацию раствора.
Изменения энтропии смешенного идеального р-ра: ΔSсмеш.ид= -К(N1lnx1+N2lnx2), где х1,х2- мольные доли компонента, N1,N2-число молекул компонента, К- постоянная Больцмана.
Х |
о |
О |
о |
О |
О |
Х |
Х |
х |
О |
х |
О |
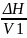 ,
где
,
где
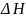 -
плотность энергии когезии (взаимодействия
между веществами внутри системы),
-
плотность энергии когезии (взаимодействия
между веществами внутри системы),
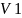 -
удельный объём данного компонента.
-
удельный объём данного компонента.
δ=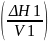 1/2-параметр
растворимости Гельдебранда.
Условие ΔЕ=Vφ1φ2*[(
1/2-параметр
растворимости Гельдебранда.
Условие ΔЕ=Vφ1φ2*[( )1/2-(
)1/2-(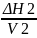 )1/2]2
или ΔЕ=Vφ1φ2
(δ1-δ2)2.
Параметр растворимости: бери растворитель,
значение которого ближе к значению
Гильдебрана полимера.
)1/2]2
или ΔЕ=Vφ1φ2
(δ1-δ2)2.
Параметр растворимости: бери растворитель,
значение которого ближе к значению
Гильдебрана полимера.
Основные уравнения ТД:
ΔGсмеси= Gр-ра – Gкомпонентов
ΔGсмеси= G р-ра – (Gрас-ль – Gраств. в-во).
ΔGсмеси= ΔН – ТΔSсмеси
1). ΔНсмеш<0, ΔSсмеш>0, то растворение экзотермическое, ΔS увеличивается. Энергия взаимодействия разнородных молекул больше, чем между однородными.
2). ΔНсмеш<0, ΔSсмеш<0, ΔНсмеш>ТΔS, то растворение экзотермическое. Сопровождается уменьшением энтропии. Она уменьшается за счёт захватывания растворителя. Такие системы расслаиваются при высоких Т. Они обладают низшей критической Тсмеш.
3). ΔНсмеш>0, ΔSсмеш>0, ΔНсмеш<ТΔS, то растворение эндотермическое. Расслаивание происходит при пониженных Т.
4). ΔНсмеш=0, ΔS<0, то атермическое растворение. Наблюдается при растворении полимера в гидрированном или собственном раст-ле.
В ТД рас-ов обычно пользуются парциальными величинами (хар-ют изменение экспериментальных (зависящих от массы) при добавлении бесконечно малого количества одного из компонентов).
Ğ1=(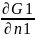 )P,T-
химический потенциал (μi);
Ğ2=(
)P,T-
химический потенциал (μi);
Ğ2=(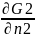 )P,T;
Ği=(
)P,T;
Ği=(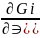 )P,T=
μi
)P,T=
μi
Для двухкомпонентной системы G=ΣGini; G=G1n1+ G2n2; n1>>n2-разб.рас-р.
d μi=RT(dlnxi)-изменение химического потенциала.xi- мольная доля i-ого ком-та в рас-ре. μi= μi0+ RTlnxi; при введении другого компонента Δμi=μi- μ0= RTlnxi.
39. Особенности термодинамики полимерных растворов. Энергетика растворения полимеров. Набухание полимеров. Фазовые диаграммы систем полимер-растворитель.
Фракционирование полимеров.
Уравнение связывающее ТД функцию молекулярными характеристиками раствора в равновесном состоянии π=f(М1, М2….). Модель Флори и Хагинса-решёточная модель регулярного р-ра (квазикристаллическая модель). Модель представляет объём р-ра как совокупность ячеек одинакового размера (двумерная решётка). Все макромолекулы считаются гибкими. Каждую ячейку решётки может занимать либо молекула растворителя, либо сегмент цепи. Они могут обмениваться местами между собой. n1-число молекул растворителя,n2-число цепей,S-число сегментов в цепи. Общее число ячеек n1+ S* n2.
Объёмная
доля р-ля:φ1=
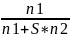 ;φ2=
;φ2= .
Координационное числоυ – число контактов
одной структурной единицы с окружающими.
Необходимо рассчитать ΔG,ΔH,ΔS.
КомбинаториальнаяS
смешения ΔSк.
.
Координационное числоυ – число контактов
одной структурной единицы с окружающими.
Необходимо рассчитать ΔG,ΔH,ΔS.
КомбинаториальнаяS
смешения ΔSк.
S=KlnW;ΔSк= -R(n1lnφ1+n2lnφ2)- комб. S растворения ВМС
Для НМС ΔSк= -K(n1lnN1+n2lnN2). При одинаковых мольных долях ВМС и НМС ΔSк ВМС намного больше ( ввиду большего объёма макромолекулы).
При переходе макромолекулы в раствор реализуется броуновское движение сегментов, которое неосущ. в ТВ агрегатном состоянии- физ. смысл большой ΔSк для ВМС. Аномально высокие значения ΔS смешения обуславливают отклонения р-ров ВМС от законов Рауля и Вант- Гоффа.
Определение
величины ТД сегмента. Определяют
Р(давление) пара рас-ля над рас-ом и Р
пара чистого растворителя. Закон
Рауля
 =N1*-
эффективная мольная доля для полимеров
рас-ля. Мольная доля рас-ля в рас-ре
N1*=
=N1*-
эффективная мольная доля для полимеров
рас-ля. Мольная доля рас-ля в рас-ре
N1*=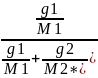 -
кажущаяся Mr
полимера- величина, эквивалентная Mr
полимер гомолога, который образовался
бы с данным растворителем.
-
кажущаяся Mr
полимера- величина, эквивалентная Mr
полимер гомолога, который образовался
бы с данным растворителем.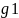 -
масса навески.
-
масса навески.
Идеальный раствор. Расчёт энтальпии смешения ΔН= КTχn1φ2, где χ-число молекул растворителя, φ2-объёмная доля полимера.
Параметр
Флори-Хагинса(константа Хагинса,
коэффициент взаимодействия ) χ=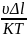 ,
где
,
где
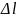 -
алгебраическая сумма энергий различных
контактов. Константа характеризует
энергию взаимодействия одной молекулы
рас-ля, отнесенную к тепловой энергии.
-
алгебраическая сумма энергий различных
контактов. Константа характеризует
энергию взаимодействия одной молекулы
рас-ля, отнесенную к тепловой энергии.
Δе= ,
где е11,е22,е12- энергии одного контакта
между одинаковыми и разными частицами
соответственно.
,
где е11,е22,е12- энергии одного контакта
между одинаковыми и разными частицами
соответственно.
Изменение свободной энергии смешения
ΔG=ΔH-TΔS
ΔG=KTχn1φ2+ KT(n1lnφ1+n2lnφ2)=KT(χn1lnφ2+ n1lnφ1+ n2lnφ2), где χn1lnφ2- энергетический терм, n1lnφ1+ n2lnφ2- комбинаториальный терм.
ΔG→π
(осмотическое Р)
 .
Показатель ТД сродства между полимером
и растворителем отнесённый к еденице
объёма π=
.
Показатель ТД сродства между полимером
и растворителем отнесённый к еденице
объёма π=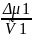 ,
где Ṽ=парциальный
мольный объём рас-ля. Чем меньше π, тем
меньше
,
где Ṽ=парциальный
мольный объём рас-ля. Чем меньше π, тем
меньше
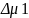 =>
тем выше ТД сродство растворителя к
полимеру.
=>
тем выше ТД сродство растворителя к
полимеру.
Уравнение состояния полимера в р-ре (разбавл. р-р)
Приведенное
осмотическое давление
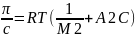 ,
где
,
где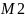 -Mr
ВМС. Второй вириальный коэфициент А2=
-Mr
ВМС. Второй вириальный коэфициент А2=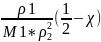 .
.
Зависимость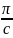 от С.
=RT(A1+A2C+A3C2+……),
A1=
от С.
=RT(A1+A2C+A3C2+……),
A1=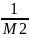 .
При масых С зависимость считают линейной
(берут первые 2 члена). Определяется
средняя Mr.
Mrембранная
осмометрия позволяет определить Mr
в диапазоне 3*104-1*106.
Другой метод концевых групп.
.
При масых С зависимость считают линейной
(берут первые 2 члена). Определяется
средняя Mr.
Mrембранная
осмометрия позволяет определить Mr
в диапазоне 3*104-1*106.
Другой метод концевых групп.
В ТД хорошем растворителе α большое, в плохом α невелико.
В
определённых растворителях при
определённой Т межсегментальные,
когезионные силы уравновешиваются
дисперсионными силами взаимодействия
между сегментами и частицами растворителя.
В результате размеры клубка остаются
невозмущёнными. α=1→(ĥ2)1/2=(ĥ02)1/2.
θ- растворитель занимает промежуточное
положение между плохим рас-ем и осадителем.
Т при которой α=1назыв. θ-температурой
(или Т
Флори).
Идеальный растворитель бывает только
при θ- температуре (это Т, при которой
разбавленные р-ры ВМС подчиняются
идеальным законам Рауля и Вант- Гоффа).
При θ-температуре определяется Mr
сегмента по закону Вант- Гоффа. В θ-усл.
А2=0, Тθ=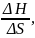 ΔG=0.
ΔG=0.
Набухание внутри кристалла.
Набухание – это процесс поглощения или сорбции низкомолекулярных жидкостей (или их паров) полимером. Набухание сопровождается увеличением массы, объёма и изменением структуры полимера. Скорость проникновения растворителя от поверхности в глубь полимера зависит от степени термодинамического сродства растворителя и полимера, уровня межмолекулярного взаимодействия в полимере, температуры и других условий процесса. На начальной стадии набухания распределение растворителя в объеме полимера неоднородно.
Различаются ограниченное и неограниченное набухание. Неограниченное набухание – это набухание, самопроизвольно переходящее в растворение. Если набухание не заканчивается растворением, оно называется ограниченным. Оно обусловленного ограниченным смешением линейного и разветвлённого строения с растворителем («плохой» растворитель), а так же наличием поперечных химических связей в полимере, что исключает разделение макромолекул и их переход в раствор.
Процесс
ограниченного набухания характеризуется
степенью
набухания
α=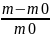 ; α=
; α=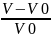 ;
где-масса набухшего ВМС,
;
где-масса набухшего ВМС, -масса
сухого ВМС,
-масса
сухого ВМС,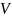 -
количество поглощённой ВМС жидкости,
отнесённое к еденице массы или объёма
ВМС.α зависит от продолжительности
процесса.
-
количество поглощённой ВМС жидкости,
отнесённое к еденице массы или объёма
ВМС.α зависит от продолжительности
процесса.
Скорость
набухания может также характеризоваться
углом наклона кривой зависимости степени
набухания от времени к оси абсцисс: чем
большеα, тем выше скорость. Уравнение
для ограниченного набухания ВМС, скорость
набухания
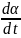 =
K(α∞-α)
K=constυ;
ln(α∞-α)
=lnα∞-Kt.
Уравнение справедливо, если исходный
полимер находится в высокоэластическом
состоянии.
=
K(α∞-α)
K=constυ;
ln(α∞-α)
=lnα∞-Kt.
Уравнение справедливо, если исходный
полимер находится в высокоэластическом
состоянии.
Степень набухания, которой соответствует появление горизонтального участка на кривой, называется максимальной или равновесной степенью набухания.
Форма кинетических кривых набухания зависит от структуры полимера, степени термодинамического сродства его к растворителю. При изменении внешних условий ограниченное набухание может перейти в неограниченное, и наоборот.
Образование соединения стехиометрического состава- «соединение набухания». Единицы растворителя образуют некоторые соединения с макромолекулами (целлюлоза-68% р-р HNO3.Образуется соединение оксониевого типа 2С6Н10О5* НNO3). В растворе щелочи образуются алкоголяты целлюлозы CHClO4 – 2C6H10O5*HClO4. Соединения набухания способны к кристаллизации.
Наряду с полиморфными превращениями происходит частичная декристаллизация ВМС. Возможно разрушение кристаллической структуры и неформирование новой( образуется аморфн. соед.) при внутри крист. набухании при нестехиометрическом набухании ВМС.
Микрокристаллическое набухание. Используют для пластификации ВМС и перевода аморфных зон из стеклообразного состояния в высокоэластичное.
Ацетилирование целлюлозы –медленно. Перед этим целлюлоза набухает в АсОН (межкристаллитное набухание). АсОНнаруш. ВС в молекуле целлюлозы и переводят ее в высокоэл. состояние.
Фракционирование. Разделение полимера на сравнительно узкие фракции и изучение их позволяет построить функцию распределения полимера в целом. Успех этого метода определяется правильным выбором условий фракционирования, позволяющих провести разделение полимолекулярного образца на возможно более узкие не перекрывающиеся фракции. Физический принцип фракционирования полимеров основан на зависимости их растворимости в критической области от величины молекулярной массы.
Два типа взаимодействия:
– взаимодействие «полимер - растворитель»;
– взаимодействие «полимер - полимер».
Разница в энергии взаимодействий «полимер - полимер» и «полимер - растворитель», зависящая от величины макромолекул, и позволяет проводить фракционирование.
В соответствии с теорией растворов полимеров Флори-Хаггинса
![]() ,
(2.5.50)где φ2, φ1 = (1- φ2) - объёмные доли
полимера и растворителя
,
(2.5.50)где φ2, φ1 = (1- φ2) - объёмные доли
полимера и растворителя![]() ,и
,и![]() ,
где z – число звеньев в макромолекуле
(для гибких полимеров z соответствует
степени полимеризации, z = Р); n1,n2- число
молей растворителя и полимера; k–
параметр Флори-Хаггинса, характеризующий
разность энергий взаимодействия
«полимер-полимер» и «полимер-растворитель».
,
где z – число звеньев в макромолекуле
(для гибких полимеров z соответствует
степени полимеризации, z = Р); n1,n2- число
молей растворителя и полимера; k–
параметр Флори-Хаггинса, характеризующий
разность энергий взаимодействия
«полимер-полимер» и «полимер-растворитель».
Нарастание параметра k вплоть до некоторого критического значения kкр приводит к ограниченной растворимости полимера. При этом значении параметра взаимодействия начинается формирование второй фазы и при k >kкр раствор разделяется на две сосуществующие фазы. Такая критическая точка (экстремум) должна характеризоваться условиями
![]() (2.5.51)(как
любая экстремальная точка).
(2.5.51)(как
любая экстремальная точка).
Учитывая условия (2.5.51) после двойного дифференцирования уравнения (2.5.50) можем получить для параметра
![]() т.е.
условия, необходимые для выпадения
полимера из раствора с ухудшением
качества растворителя определяются
молекулярной массой полимера.
т.е.
условия, необходимые для выпадения
полимера из раствора с ухудшением
качества растворителя определяются
молекулярной массой полимера.
По мере уменьшения молекулярной массы kкр снижается, поэтому прежде всего из раствора будут выделяться фракции с большей молекулярной массой, а при экстракции будут растворяться фракции с меньшей молекулярной массой.
При
добавлении осадителя по мере ухудшения
качества растворителя раствор разделяется
на две фазы. Распределение полимерных
молекул между двумя растворами можно
описать уравнением
экспоненциального распределения
Бренстеда-Шульце:![]() ,
(2.5.53) где φ,φ¢- объемные доли полимера в
разбавленном и концентрированном
растворах; P – степень полимеризации
распределенного полимера; s -сложная
функция постоянной Флори-Хаггинса k,
температуры, концентрации и т.д.
,
(2.5.53) где φ,φ¢- объемные доли полимера в
разбавленном и концентрированном
растворах; P – степень полимеризации
распределенного полимера; s -сложная
функция постоянной Флори-Хаггинса k,
температуры, концентрации и т.д.
Эффективности фракционирования:
![]() ,
(2.5.54)где φ P – доля молекул полимера со
степенью полимеризации Р, остающихся
в разбавленном растворе; Vφ, V'φ' –
соответственно объемы разбавленного
и концентрированного растворов; φ ,φ' –
объемная доля полимера в разбавленном
и концентрированном растворе; R = V'φ'/
Vφ.
,
(2.5.54)где φ P – доля молекул полимера со
степенью полимеризации Р, остающихся
в разбавленном растворе; Vφ, V'φ' –
соответственно объемы разбавленного
и концентрированного растворов; φ ,φ' –
объемная доля полимера в разбавленном
и концентрированном растворе; R = V'φ'/
Vφ.
![]() .
(2.5.55) позволяет оценить эффективность
фракционирования и выявить ее зависимость
от степени полимеризации (молекулярной
массы).
.
(2.5.55) позволяет оценить эффективность
фракционирования и выявить ее зависимость
от степени полимеризации (молекулярной
массы).
Общие рекомендации по фракционированию.
-Доля полимера в разбавленном растворе тем больше, чем меньше отношение объемов осадка и раствора. Для качественного разделения необходимо снижать R.
-В осадке преимущественно будут накапливаться фракции с наибольшей молекулярной массой, так как чем больше R, тем меньше fP.
Однако это не означает, что другие фракции не будут переходить в осадок. Поэтому качественное разделение может быть достигнуто при повторном фракционировании первичных фракций.
-Для лучшего фракционирования следует применять минимальную концентрацию растворов (как правило, не более 1%).
-При выборе системы «растворитель-осадитель» необходимо подбирать такие компоненты, чтобы они смешивались и во всех соотношениях, во всем диапазоне рабочих температур. Необходимо следить, чтобы малые добавки осадителя не изменяли сильно качества растворителя и не выделяли больших количеств полимера.