

25 Билет.
Жи́дкие криста́ллы - это фазовое состояние, в которое переходят некоторые вещества при определенных условиях (температура, давление, концентрация в растворе). Жидкие кристаллы обладают одновременно свойствами как жидкостей (текучесть), так и кристаллов (анизотропия). По структуре ЖК представляют собой вязкие жидкости, состоящие из молекул вытянутой или дискообразной формы, определённым образом упорядоченных во всем объёме этой жидкости. Наиболее характерным свойством ЖК является их способность изменять ориентацию молекул под воздействием электрических полей, что открывает широкие возможности для применения их в промышленности. По типу ЖК обычно разделяют на две большие группы: нематики и смектики. В свою очередь нематики подразделяются на собственно нематические и холестерические жидкие кристаллы.
ДИАГРАММА СОСТОЯНИЯ (фазовая диаграмма), графич. изображение всех возможных состояний термодинамическое системы в пространстве основные параметров состояния температуры Т, давления р и состава х (обычно выражаемого молярными или массовыми долями компонентов). Для сложных систем, состоящих из многих фаз и компонентов, построение ДИАГРАММА СОСТОЯНИЯ с. является единственным методом, позволяющим на практике установить, сколько фаз и какие конкретно фазы образуют систему при данных значениях параметров состояния.
Твёрдые растворы - фазы переменного состава, в которых атомы различных элементов расположены в общей кристаллической решётке.
На диаграмме различают три фазовые области:
1. Выше линии ликвидуса АDВ находится область жидкой фазы Ж.
2. Под ней до линии солидуса АDВ расположена двухфазная область б + Ж. Фаза б представляет твердый раствор компонентов А и В, зерна имеют единую кристаллическую решетку. Однако у сплавов разного состава число атомов компонентов А и В в элементарных ячейках решетки различно.
3. Область, расположенная под линией солидуса, является однофазной (фаза б).
Типы диаграмм состояния. На диаграмме состояния двойных систем в координатах температура-состав образованию непрерывных твердых растворов отвечают три типа линий ликвидуса и солидуса.
Билет 26
Термодинамическая система - это некая физическая система, состоящая из большого количества частиц, способная обмениваться с окружающей средой энергией и веществом. Также обычно полагается, что такая система подчиняется статистическим закономерностям.
Термодинамические системы подразделяются на однородные по составу (например, газ в сосуде) и неоднородные (вода и пар или смесь газов в сосуде).
Выделяют также изолированные системы, то есть системы, которые не обмениваются с окружающей средой ни энергией, ни веществом, и закрытые системы, которые обмениваются со средой только энергией, но не обмениваются веществом. Если же в системе происходят обменные процессы с окружающей средой, то её называют открытой.
Закон:" число фаз, сосуществующих в равновесии, не превосходит числа независимых компонентов более чем на 2."
Правило фаз Гиббса определяет соотношение между числом фаз (Ф), компонентов (К), внешних переменных (П) и числом степеней свободы или вариантности (С) термодинамической системы, находящейся в равновесии и записывается следующим образом:
С = К + 2 - Ф;
Цифра 2 в правиле фаз связана с существованием 2-х переменных (температуры и давления), одинаковых для всех фаз. Например, правило фаз Гиббса для постоянного давления запишется как:
С = К +1 – Ф
Билет 27
Слово "термодинамика" происходит от греческих слов "термос" - тепло и "динамос" - сила, мощь.
Термодинамика изучает законы превращения энергии из одной формы в другую в различных процессах. Выделяют общую или физическую термодинамику (изучает общие вопросы превращения энергии); техническую термодинамику (изучает взаимопревращение между теплотой и механической работой) и химическую термодинамику (изучает превращение различных форм энергии в ходе химической реакции и при фазовых переходах, а также способность химических систем выполнять полезную работу).
Химическая термодинамика используется для решения таких задач, как
1) предсказание о возможности протекания химической реакции;
2) о направленности химической реакции;
3) о характере химического процесса.
Основным объектом исследования термодинамики является система. Это понятие означает ту часть материального мира, которая является предметом наблюдения или исследования. Это тело или группа тел, выделенных мысленно из материального мира (газ в баллоне, образец вещества, тепловая машина и т.д.). Остальная часть материального мира - за пределами условно выделенной из него системы - называется окружением или окружающей средой. Между средой и системой возможен обмен веществом и энергией.
Первый закон термодинамики является обобщением закона сохранения и превращения энергии для термодинамической системы. Он формулируется следующим образом:
Изменение ΔU внутренней энергии неизолированной термодинамической системы равно разности между количеством теплоты Q, переданной системе, и работой A, совершенной системой над внешними телами.
ΔU = Q - A.
Соотношение, выражающее первый закон термодинамики, часто записывают в другой форме:
Q = ΔU + A.
Количество теплоты, полученное системой, идет на изменение ее внутренней энергии и совершение работы над внешними телами.
Первый закон термодинамики является обобщением опытных фактов. Согласно этому закону, энергия не может быть создана или уничтожена; она передается от одной системы к другой и превращается из одной формы в другую.
В изохорном процессе (V = const) газ работы не совершает, A = 0. Следовательно,
Q = ΔU = U (T2) - U (T1).
Здесь U (T1) и U (T2) - внутренние энергии газа в начальном и конечном состояниях. Внутренняя энергия идеального газа зависит только от температуры (закон Джоуля). При изохорном нагревании тепло поглощается газом (Q > 0), и его внутренняя энергия увеличивается. При охлаждении тепло отдается внешним телам (Q < 0).
В изобарном процессе (p = const) работа, совершаемая газом, выражается соотношением
A = p (V2 - V1) = p ΔV.
Первый закон термодинамики для изобарного процесса дает:
Q = U (T2) - U (T1) + p (V2 - V1) = ΔU + p ΔV.
При изобарном расширении Q > 0 - тепло поглощается газом, и газ совершает положительную работу. При изобарном сжатии Q < 0 - тепло отдается внешним телам. В этом случае A < 0. Температура газа при изобарном сжатии уменьшается, T2 < T1; внутренняя энергия убывает, ΔU < 0.
Билет 28
Энтальпи́я, также тепловая функция и теплосодержание — термодинамический потенциал, характеризующий состояние системы в термодинамическом равновесии при выборе в качестве независимых переменных давления, энтропии и числа частиц.
Проще говоря, энтальпия — это та энергия, которая доступна для преобразования в теплоту при определенных температуре и давлении.
Понятие энтропии впервые было введено Клаузиусом в термодинамике в 1865 году для определения меры необратимого рассеивания энергии, меры отклонения реального процесса от идеального. Определённая как сумма приведённых теплот, она является функцией состояния и остаётся постоянной при обратимых процессах, тогда как в необратимых — её изменение всегда положительно.
![]() ,
,
где ![]() —
приращение энтропии;
—
приращение энтропии; ![]() —
минимальная теплота, подведенная к
системе; T — абсолютная температура
процесса;
—
минимальная теплота, подведенная к
системе; T — абсолютная температура
процесса;
Мерой изменения упорядоченности системы служитизменение энтропии ΔS.
Энтропия системы тем выше, чем больше степень неупорядоченности (беспорядка) системы. Таким образом, если процесс идет в направлении увеличения неупорядоченности системы (а повседневный опыт показывает, что это наиболее вероятный процесс), ΔS — величина положительная. Для увеличения степени порядка в системе (ΔS > 0) необходимо затратить энергию. Оба этих положения вытекают из фундаментального закона природы — второго закона термодинамики. Количественно зависимость между изменениями энтальпии, энтропии и свободной энергии описываетсяуравнением Гиббса-Гельмгольца:
ΔG = ΔH - T · ΔS
Стандартные энтальпия и энтропия даются в таблицах.
Закон Гесса и следствия из него
В основе термохимических расчетов лежит закон, открытый русским ученым Г. И. Гессом в 1840 г. Закон гласит: тепловой эффект реакции зависит от вида и состояния исходных веществ и конечных продуктов, но не зависит от пути перехода (из начального состояния в конечное).
Или иначе: тепловое эффект реакции равен алгебраической сумме тепловых эффектов всех его промежуточных стадий:
ΔН = ΔН1 + ΔН2.
Рассмотрим пример получения диоксида углерода СО2 из графита, которую можно представить в виде ряда последовательных реакций, каждая из которых сопровождается своим тепловым эффектом (изменением энтальпии):
С(графит) + 1/2О2(г) = СО(г) |
|
ΔН1 I стадия. |
СО(г)+1/2О2(г)= СО2(г) |
|
ΔН2 II стадия. |
Или суммарно:
С(графит)
+ О2(г)
= СО2(г)![]() ΔН.
ΔН.
Представим в виде схемы:

Очевидно:
ΔН = ΔН1 + ΔН2 или ΔН1 + ΔН2 + (–ΔН) = 0.
Это обозначает, что если все три процесса удовлетворяют требованию Тисх.= Тконечн. и Рисх. = Рконечн., то не зависимо от того, сгорает графит сразу в СО2 или сначала в СО, а затем СО в СО2, тепловой эффект будет одним и тем же.
Следствия из закона Гесса:
Если в результате последовательных химических реакций система приходит в состояние, полностью совпадающее с исходным (круговой процесс), то сумма тепловых эффектов этих реакций будет равна нулю.
Тепловой эффект реакций (ΔНх.р.) равен сумме теплот образования (или ΔНобр.) конечных веществ (ΔНконеч. в-в) за вычетом суммы теплот образования исходных веществ (ΔНисх. в-в):
ΔНх.р. = Σ ΔНпрод. р-ции – Σ ΔНисх. в-в.
Билет 29
Изобарно-изотермический потенциал (энергия Гиббса)
Из рассмотренного ранее следует, что в химических процессах одновременно действуют две тенденции:
Стремление частиц объединиться в более сложные, что уменьшает энтальпию.
Стремление частиц разъединиться, увеличить беспорядок, что увеличивает энтропию.
Суммарный эффект этих двух противоположных тенденций в процессах, протекающих при постоянной температуре и постоянном давлении, отражает изменение изобарно-изотермического (или для краткости - изобарного) потенциала ΔG:
ΔG = ΔН – TΔS,
где ΔН - изменение эптальпии (теплосодержания системы), кДж/моль;
ΔS - изменение энтропии (меры беспорядка систем), Дж/моль•К;
T - температура, К.
Изобарный потенциал измеряется в ккал/моль или кДж/моль.
Характер изменений ΔG позволяет судить о принципиальной возможности или невозможности протекания процесса:
Если ΔG < 0, т. е. изобарный потенциал в ходе реакции уменьшается, то процесс возможен и, начавшись, он протекает самопроизвольно (спонтанно).
Если ΔG > 0, то невозможно осуществить процесс в данных условиях.
Если ΔG = 0, наблюдается состояние химического равновесия.
Не весь ответ!! На часть нет!
Билет 30
Скорость химической реакции можно выразить как изменение количества вещества (n, по модулю) в единицу времени (t) — сравните скорость движущегося тела в физике как изменение координат в единицу времени: υ = Δx/Δt. Чтобы скорость не зависела от объема сосуда, в котором протекает реакция, делим выражение на объем реагирующих веществ (v), т.е. получаем изменение количества вещества в единицу времени в единице объема, или изменение концентрации одного из веществ в единицу времени:
n2 − n1 Δn Δс υ = –––––––––– = –––––––– = –––––––– (1) (t2 − t1) • v Δt • v Δt
где c = n/v — концентрация вещества,
Условия, влияющие на скорость химических реакций
1) Скорость реакции зависит от природы реагирующих веществ. Проще говоря, разные вещества реагируют с разной скоростью. Например, цинк бурно реагирует с соляной кислотой, а железо довольно медленно.
2) Скорость реакции тем больше, чем выше концентрация веществ. С сильно разбавленной кислотой цинк будет реагировать значительно дольше.
3) Скорость реакции значительно повышается с повышением температуры. Например, для горения топлива необходимо его поджечь, т.е. повысить температуру. Для многих реакций повышение температуры на 10° C сопровождается увеличением скорости в 2–4 раза.
4) Скорость гетерогенных реакций увеличивается с увеличением поверхности реагирующих веществ. Твердые вещества для этого обычно измельчают. Например, чтобы порошки железа и серы при нагревании вступили в реакцию, железо должно быть в виде мелких опилок.
Обратите внимание, что в данном случае подразумевается формула (1)! Формула (2) выражает скорость на единице площади, следовательно не может зависеть от площади.
5) Скорость реакции зависит от наличия катализаторов или ингибиторов.
Катализаторы — вещества, ускоряющие химические реакции, но сами при этом не расходующиеся. Пример — бурное разложение перекиси водорода при добавлении катализатора — оксида марганца (IV):
2H2O2 = 2H2O + O2↑
Оксид марганца (IV) остается на дне, его можно использовать повторно.
Ингибиторы — вещества, замедляющие реакцию. Например, для продления срока службы труб и батарей в систему водяного отопления добавляют ингибиторы коррозии. В автомобилях ингибиторы коррозии добавляются в тормозную, охлаждающую жидкость.
Активность катализатора резко уменьшается в присутствии веществ, называемых каталитическими ядами. Если катализатор и реагирующие вещества находятся в одном агрегатном состоянии, обычно газообразном или жидком, то катализ называется гомогенным. В роли катализаторов в гомогенном катализе часто выступают растворы кислот, оснований, солей d-элементов, растворители. Катализ является гетерогенным, если катализатор и реагирующие вещества находятся в разных агрегатных состояниях или образуют самостоятельные фазы. В роли катализаторов в этом случае чаще всего выступают твердые вещества, обычно d-элементы или их соединения. Катализаторы не изменяют энтальпию и энергию Гиббса реакции и не влияют на положение химического равновесия реакции. Катализаторы только увеличивают в равной мере скорость прямой и обратной реакций.
Билет 31
Различают гомогенные и гетерогенные системы. Гомогенной называется система, состоящая из одной фазы, гетерогенной - система, состоящая из нескольких фаз. Фазой называется часть системы, отделенная от других ее частей поверхностью раздела, при переходе через которую свойства изменяются скачком.
Химическое равновесие в гомогенных системах
При равенстве энтальпийного и энтропийного факторов ΔН = ТΔS ΔG = 0, что является термодинамическим условием химического равновесия. Химическое равновесие имеет динамический характер. Скорость реакции (число частиц образующихся в единицу времени в единице объема) в прямом направлении равна скорости реакции в обратном направлении. В этот момент концентрации исходных веществ и продуктов реакции не изменяются во времени и называются равновесными концентрациями. Они обозначаются символом вещества в квадратных скобках.
Химическое равновесие в гетерогенных системах
Химические реакции, протекающие на границе раздела фаз, называются гетерогенными химическими реакциями.
При равенстве скоростей прямой и обратной реакции наступает химическое равновесие в гетерогенной системе. Примерами гетерогенных процессов является пароводяная конверсия углерода, или восстановление оксидов металлов водородом:
С(к) + 2Н2О = СО2 + 2Н2 ,
МеО(к) + Н2 = Ме(к) + Н2 О.
Как и для любого равновесия, условием гетерогенного химического равновесия является равенство энергии Гиббса нулю, ΔG = 0.
Константа химического равновесия
При равновесии химической реакции:
bB + dD = lL + mM
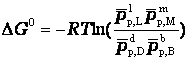
или
 ,
,
где pp,L, ppM, pp,D, ppB –равновесные парциальные давления веществ, а [L], [M],[D],[B] –равновесные концентрации веществ; l, m, d, b - показатели степени, равные стехиометрическим коэффициентам.
Отношения произведений парциальных давлений или концентраций получили названия констант химического равновесия соответственно Кр или Кс:
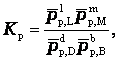
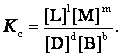
Эти уравнения являются математическими выражениями закона действующих масс, открытого норвежскими учеными К. Гульдбергом и П. Вааге в 1867 г.:
отношение произведения равновесных концентраций продуктов реакции в степенях, равных стехиометрическим коэффициентам, к произведению равновесных концентраций исходных веществ в степенях, равных стехиометрическим коэффициентам, при Т = соnst, является величиной постоянной.
Например, для реакции синтеза аммиака:
N2 + 3H2 = 2NH3
закон действующих масс имеет вид:
Кс = [NH3]2 / [N2][H2]3
Подставляя выражение константы в уравнения, получаем
ΔG0 = - RTlnKc = - 2,3RTlgKp,
ΔG0 = - RTlnKp = - 2,3RTlgKc.
Рассчитав величину ΔG0 химической реакции, можно определить константу химического равновесия. Используя закон действующих масс, можно рассчитать равновесные концентрации реагирующих веществ.
Билет 32
Смещение химического равновесия
Положение химического равновесия зависит от следующих параметров реакции: температуры, давления и концентрации. Влияние, которое оказывают эти факторы на химическую реакцию, подчиняются закономерности, которая была высказана в общем виде в 1885 году французским ученым Ле-Шателье.
Факторы влияющие на химическое равновесие:
1) температура
При увеличении температуры химическое равновесие смещается в сторону эндотермической (поглощение) реакции, а при понижении в сторону экзотермической (выделение) реакции.
CaCO3=CaO+CO2 -Q t↑ →, t↓ ←
N2+3H2↔2NH3 +Q t↑ ←, t↓ →
2) давление
При увеличении давления химическое равновесие смещается в сторону меньшего объёма веществ, а при понижении в сторону большего объёма. Этот принцип действует только на газы, т.е. если в реакции участвуют твердые вещества, то они в расчет не берутся.
CaCO3=CaO+CO2 P↑ ←, P↓ →
1моль=1моль+1моль
3) концентрация исходных веществ и продуктов реакции
При увеличении концентрации одного из исходных веществ химическое равновесие смещается в сторону продуктов реакции, а при повышении концентрации продуктов реакции-в сторону исходных веществ.
S2+2O2=2SO2 [S],[O]↑ →, [SO2]↑ ←
Катализаторы не влияют на смещение химического равновесия!
Принцип Ле Шателье
При изменении равновесных концентраций исходных веществ и продуктов реакции путем воздействия на систему происходит смещение химического равновесия. Если увеличиваются равновесные концентрации продуктов реакции, то говорят о смещении равновесия вправо. Если при внешнем воздействии увеличиваются концентрации исходных веществ, то говорят о смещении равновесия влево.
Характер смещения равновесия можно прогнозировать, применяя принцип французского ученого Ле Шателье:
если на систему, находящуюся в равновесии, оказывается внешнее воздействие, то равновесие смещается в том направлении, которое ослабляет внешнее воздействие.
Принцип Ле Шателье следует из закона действующих масс. Если система находится при постоянной температуре, то константа равновесия при внешних воздействиях остается постоянной. Поэтому любое изменение равновесных концентраций веществ должно приводить к такому изменению равновесных концентраций других веществ, чтобы соблюдалось постоянство константы равновесия.
Рассмотрим процесс конверсии метана:
CH4 + 2H2O = CO2 + 4H2,
Константа равновесия этого процесса имеет вид:
Кс = [CO2] [H2]4/[CH4] [H2O]2.
1. Рассмотрим, как влияет изменение концентраций на смещение равновесия. При увеличении концентрации метана СН4 равновесие системы нарушается, идет прямая реакция. Концентрации продуктов реакции СО2 и Н2 увеличиваются, а концентрации Н2О уменьшается. Процесс будет протекать до тех пор, пока не установится новое равновесие. Новые равновесные концентрации компонентов будут такими, что константа равновесия не изменится. Если увеличить концентрацию СО2, то по принципу Ле Шателье равновесие сместится влево.
2. Если в результате реакции изменяется число молей газообразных веществ, то изменяется общее давление в системе и происходит смещение равновесия. В соответствии с принципом ЛеШателье увеличение общего давления вызывает смещение равновесия в сторону уменьшения числа молей газообразных веществ, т.е. в сторону уменьшения давления. Для рассматриваемой реакции увеличение давления должно смещать равновесие влево (слева- 3 моля, справа – 5 молей).
3. С увеличением температуры равновесие смещается в сторону эндотермических реакций, т.е. реакций протекающих с поглощением теплоты, понижение – в сторону экзотермических реакций.
Итак, принцип Ле Шателье позволяет создавать такие условия протекания реакции, которые обеспечивают максимальный выход продуктов реакции.
33.
ЦЕПНЫЕ РЕАКЦИИ,
хим. превращения и ядерные процессы, в
к-рых появление промежуточной активной
частицы (свободного
радикала, атома,
возбужденной молекулы в
хим. превращениях, нейтрона -
в ядерных процессах) вызывает цепь
превращений исходных в-в. Примеры хим.
цепных реакций- радикальная
полимеризация, окисление, пиролиз и галогенирование углеводородов и
др. орг. соед.; ядерные цепные процессы
- цепное деление атомных
ядер.
Данная статья посвящена в основном
химическим цепным реакциям.
Термин
"цепные реакции"
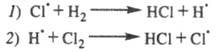
был предложен М. Боденштейном, обнаружившим
(1913), что в ряде фотохим. р-ций (напр., Н2 +
+ С12![]() 2НС1,
СО + С12
2НС1,
СО + С12![]() СОС12)
один поглощенный фотон вызывает
превращение сотен тысяч молекул.
Поскольку согласно закону квантовой
эквивалентности Штарка-Эйнштейна при
поглощении одного фотона в р-цию вступает
лишь одна частица, остальныемолекулы реагируют
без непосредственного светового
воздействия. Боденштейн предположил,
что активной частицей, вызывающей цепь
превращений, является возбужденная молекула
СОС12)
один поглощенный фотон вызывает
превращение сотен тысяч молекул.
Поскольку согласно закону квантовой
эквивалентности Штарка-Эйнштейна при
поглощении одного фотона в р-цию вступает
лишь одна частица, остальныемолекулы реагируют
без непосредственного светового
воздействия. Боденштейн предположил,
что активной частицей, вызывающей цепь
превращений, является возбужденная молекула![]() В
1916 В. Нернст высказал предположение об
атомарной природе активных частиц и
предложил след, механизм цепных реакций с
участием Сl2.
Поглощение фотона приводит к образованию
из молекулы С12 двухатомов
В
1916 В. Нернст высказал предположение об
атомарной природе активных частиц и
предложил след, механизм цепных реакций с
участием Сl2.
Поглощение фотона приводит к образованию
из молекулы С12 двухатомов![]() к-рые
вступают последовательно в р-ции:
к-рые
вступают последовательно в р-ции:
ФОТОХИМИЧЕСКИЕ РЕАКЦИИ, хим. р-ции, протекающие под действием света. Поглощение фотона с длиной волны ~ 100-1500 нм, чему соответствует энергия 0,8-12,4 эВ (80-1200 кДж/моль), вызывает квантовый переход молекулы в-ва из основного электронного состояния в одно из возбужденных состояний или фотоионизацию - отщепление электрона и образование катион-радикала.Возбужденные состояния молекул имеют отличную от основного состояния электронную структуру и, как правило, более высокуюреакционную способность. Молекулы вступают в хим. р-ции, первичные продукты к-рых (ионы, радикалы, изомеры) чаще всего оказываются нестабильными. Конечные продукты фотохимических реакций появляются в результате обычных термич. р-ций, к-рые протекают либо непосредственно с участием первичных частиц, либо как ряд последовательных хим. превращений.
Как правило, для молекул с четным числом электронов при фотовозбуждении первоначально образуется возбужденное синглетное состояние (с мультиплетностъю, равной 1). Фотохимическая реакция обычно протекает из нижнего возбужденного синглетного состояния или из триплетного состояния (мультиплетность 3), к-рое получается из возбужденного синглетного состояния путем интеркомбинационной конверсии (изменения спина одного из электронов).
С хим. р-циями возбужденных молекул конкурируют фо-тофиз. процессы: испускание фотона (флуоресценция или фосфоресценция), внутренняя и интеркомбинационная конверсия в нижележащие электронные состояния (триплетное или основное). Вследствие этих процессов времена жизни возбужденных синглетных состояний обычно не превышают 10-8-10-9с. Триплетные состояния в жидких р-рах обычно "гибнут" в результате безызлучат. перехода и дезактивации (тушения) примесями (напр., кислородом); времена их жизни не превышают 10-5 с. В "жестких" средах (замороженных р-рах, полимерных матрицах), где процессы дезактивации замедляются, времена жизни триплетных состояний могут достигать десятка секунд.
РАДИАЦИОННО-ХИМИЧЕСКИЕ РЕАКЦИИ, совокупность хим. и физ.-хим. превращений в-в под действием ионизирующего излучения. Предшествующие этим превращениям физ. процессы взаимод. излучения с в-вом обычно также рассматривают как стадию радиационно-химических реакций. Нек-рые из этих процессов и превращений могут осуществляться при действии на в-во квантов света УФ диапазона, в электрич. разряде (см. Плазмохимия), при электронном ударе, поглощении мощности СВЧ, в кавитац. полостях, создаваемых ультразвуковым полем внутри жидкости (см. Механохимия), и т. п. Механизм радиационно-химических реакций. Теоретич. рассмотрение взаимод. излучения с в-вом, диссипации поглощенной энергии с учетом электрич. релаксации, а также их мат.моделирование показали, что длительность этих процессов составляет не более 10-18-10-11 с. Эксперим. данные о диффузииобразовавшихся частиц и кинетике хим. р-ций с их участием, полученные методом импульсного радиолиза, позволяют выделить процессы, длительность к-рых превышает 10-11 с. В табл. сопоставлены времена процессов, составляющих радиационно-химическиереакции. Условно различают неск. последоват. стадий взаимод. излучения с в-вом: физическую (процессы продолжительностью 10-18-10-15 с), физико-химическую (завершается спустя 10-11 с после прохождения ионизирующей частицы или кванта излучения через в-во), собственно химическую (процессы длительностью более 10-10 с).
Физическая стадия определяет потери энергии ионизирующего излучения при неупругих столкновениях с частицами среды. Эти потери характеризуются т. наз. линейной передачей энергии (ЛПЭ)-энергией, переданной среде ионизирующей частицей в заданной окрестности ее траектории на единицу длины пролета. Значения ЛПЭ варьируются в зависимости от природы излучения и его энергии в широких пределах: от 0,2 эВ/нм для высокоэнергетич. квантов и ускоренных электронов до 104 эВ/нм для осколков деления тяжелых ядер.
ГОРЕНИЕ, физ.-хим. процесс, при к-ром превращение в-ва сопровождается интенсивным выделением энергии и тепло-и массообменомс окружающей средой. В отличие от взрыва и детонации протекает с более низкими скоростями и не связано с образованием ударной волны. В основе горения лежит хим. р-ция, способная протекать с прогрессирующим самоускорением вследствие накопления выделяющегося тепла (тепловое горение) или активных промежут. продуктов (цепное горение). наиб. распространено тепловое горение; цепное горение в чистом виде встречается сравнительно редко, гл. обр. в случае нек-рых газофазных р-ций при низкихдавлениях.
Условия термич. самоускорения м. б. обеспечены для всех р-ций с достаточно большими тепловыми эффектами и энергиями активации. наиб. обширный класс р-ций горения-окисление углеводородов, напр. при горении прир. топлив, водорода, металлов и т.п.; окислители-кислород, галогены, нитросоединения, перхлораты. В режиме горения могут происходить: разложение озона, ацетилена,гидразина, динитрогликоля, метилнитрата и др.; окислит.-восстановит. р-ции, в к-рых восстановители-элементы с высоким сродством ккислороду (Са, Al, Si, Mg и др.); синтез из элементов оксидов, галогенидов, халькогенидов, гидридов, интерметаллидов, тугоплавкихнитридов и карбидов.
Горение может начаться самопроизвольно в результате самовоспламенения либо быть инициированным зажиганием (см.Воспламенение). При фиксиров. внеш. условиях (давление, т-ра, размеры реактора, параметры тепло- и массопереноса и др.) непрерывное горение может протекать в стационарном режиме, когда осн. характеристики процесса - скорость р-ции, кол-во тепла, выделяющегося в единицу времени (мощность тепловыделения), т-ра и состав продуктов - не изменяются во времени, либо в периодич. режиме, когда эти характеристики колеблются около своих средних значений. Вследствие сильной нелинейной зависимости скорости р-ции от т-ры горение отличается высокой чувствительностью к внеш. условиям: при их незначит. изменении медленная р-ция может перейти в режим горения или, наоборот, развитое горение может прекратиться. Это же св-во горения обусловливает существование неск. стационарных режимов при одних и тех же условиях (гистерезисный эффект).
34.
ДИСПЕРСНЫЕ СИСТЕМЫ, гетерог. системы из двух или большего числа фаз с сильно развитой пов-стъю раздела между ними. Обычно одна из фаз образует непрерывную дисперсионную среду, в объеме к-рой распределена дисперсная фаза (или неск. дисперсных фаз) в виде мелких кристаллов, твердых аморфных частиц, капель или пузырьков. Дисперсные системы могут иметь и более сложное строение, напр., представлять собой двухфазное образование, каждая из фаз к-рого, будучи непрерывной, проникает в объем др. фазы. К таким системам относятся твердые тела, пронизанные разветвленной системой каналов-пор, заполненных газом или жидкостью, нек-рые микрогетерогенные полимерные композиции и др. Нередки случаи, когда дисперсионная среда "вырождается" до тончайших слоев (пленок), разделяющих частицы дисперсной фазы.
Классификация дисперсных систем по агрегатным состояниям фаз.
Дисперсион-ная среда |
Дисперс-ная фаза |
Примеры дисперсных систем |
Твердая |
Твердая |
Рубиновое стекло; пигментированные волокна;сплавы; рисунок на ткани, нанесенный методом пигментной печати |
Твердая |
Жидкая |
Жемчуг, вода в граните, вода в бетоне, остаточныймономер в полимерно-мономерных частицах |
Твердая |
Газо- образная |
Газовые включения в различных твердых телах: пенобетоны, замороженные пены, пемза, вулканическая лава, полимерные пены,пенополиуретан |
Жидкая |
Твердая |
Суспензии, краски, пасты, золи, латексы |
|
|
|
Жидкая |
Жидкая |
Эмульсии: молоко, нефть, сливочное масло, маргарин, замасливатели волокон |
Жидкая |
Газо- образная |
Пены, в том числе для пожаротушения и пенных технологий замасливания волокон, беления и колорирования текстильных материалов |
Газообразная |
Твердая |
Дымы, космическая пыль, аэрозоли |
Газообразная |
Жидкая |
Туманы, газы в момент сжижения |
Газообразная |
Газо- образная |
|
Образование дисперсных систем. Возможно двумя путями: диспергационным и конденсационным. Диспергирование макрофаз с образованием лиофильных дисперсных систем происходит самопроизвольно - для этого достаточно энергии теплового движения. Такой процесс осуществляется при значениях поверхностного натяжения ниже нек-рого критич. значения кр = kТ/2, где - размер частицдисперсной фазы, Т - абс. т-ра, k - постоянная Больцмана, - безразмерный коэф., принимающий значения примерно 10-30.Образование лиофобных дисперсных систем путем диспергирования стабильной макрофазы требует значительных энергетич. затрат, определяемых суммарной площадью пов-сти частиц дисперсной фазы. В реальных условиях на образование пов-сти при измельчениитвердых тел или при распылении и эмульгировании жидкостей приходится лишь небольшая часть (доли процента) подводимой к системе энергии; остальное расходуется на побочные процессы и рассеивается в окружающем пространстве (см. Диспергирование).Конденсационный путь образования дисперсных систем связан с зарождением новой фазы (или новых фаз) в пересыщенной метастабильной исходной фазе - будущей дисперсионной среде. Для возникновения высокодисперсной системы необходимо, чтобы число зародышей новой фазы было достаточно большим, а скорость их роста не слишком велика. Кроме того, требуется наличие факторов, ограничивающих возможности чрезмерного разрастания и сцепления частиц дисперсной фазы. Переход первоначально стабильной гомог. системы в метастабилъное состояние может произойти в результате изменения термодинамич. параметров состояния (давления, т-ры, состава). Так образуются, напр., природные и искусственные аэрозоли (туман - из переохлажденных водяных паров, дымы - из парогазовых смесей, выделяемых при неполном сгорании топлива), нек-рые полимерные системы - из р-ров при ухудшении "термодинамич. качества" р-рителя, органозоли металлов путем конденсации паров металла совместно с парами орг.жидкости или при пропускании первых через слой орг. жидкости, коллоидно-дисперсные поликристаллич. тела (металлич. сплавы, нек-рые виды горных пород и искусств. неорг. материалов). Возможно также образование дисперсных систем в результате хим. р-ции в гомог. среде, если продукт р-ции при данных условиях находится в агрегатном состоянии, отличном от "материнской" фазы, или практически не растворяется в ней. Примерами подобных систем могут служить аэрозоли с твердыми частицами NH4Cl (образуются при взаимод. газообразных NH3 и НСl), аэрозоли с капелъно-жидкими частицами H2SO4 (при взаимод. SO3 и водяного пара). В природе и технол. процессах часто образуются гидрозоли разного состава при гидролизе солей и др. соед., неустойчивых к действию воды. Окислит.-восстановит. р-ции используют для получения золей Аu и Ag, разложение Na2S2O3 разб. серной или соляной к-той - для получения гидрозоля элементарной серы. Хим. или термохим. разложения карбонатов, орг. порофоров (порообразователей,вспенивающих агентов) и др. соед. с выделением газообразных в-в в первоначально жидких средах лежит в основе пром. произ-ва мн. пеноматериалов.
35.
Грубодисперсные системы, к которым относятся суспензии, эмульсии, пены и аэрозоли, различающиеся по фазовому составудисперсной фазы и дисперсионной среды, рассматриваются в одном разделе курса коллоидной химии благодаря общности таких свойств, которые определяются размером частиц дисперсной фазы. Вместе с тем каждая из этих систем обладает и присущими только ей свойствами. В этой связи в данной главе свойства каждого типа систем рассматриваются самостоятельно.
Суспензии – это грубодисперсные системы, в которых дисперсная фаза – твердое тело, а дисперсионная среда - жидкость. Размер частиц в суспензиях – 10-6-10-5 м.
Суспензии имеют большое практическое значение, даже значительно большее, чем золи. Только лакокрасочная, полиграфическая и текстильная промышленность потребляют огромное количество суспензий пигментов и красящих веществ, диспергированных в водеили углеводородах. Угольные и графитовые суспензии широко применяются для предотвращения образования накипи в котлах. Здесь частички используются как центры кристаллизации солей. Бурение скважин сопровождается закачкой в них глинистых суспензий. Концентрированные суспензии (пасты) - основа технологического использования цементов, керамики, кирпичного производства и производства других строительных материалов. Сельское хозяйство также связано с использованием суспензий, так как почвы с водойдают типичные суспензии. Препараты для обработки семян и растений в борьбе с вредителями также часто применяются в виде суспензий.
36.
Коллоидные системы, коллоидно-дисперсные системы, коллоиды, традиционные названия предельно высокодисперсных (микрогетерогенных) систем. Частицы дисперсной фазы в коллоидных системах, коллоидные частицы, обычно имеют размер от 10-7 до 10-5 см. В газе или жидкости они свободно и независимо одна от другой участвуют в интенсивном броуновском движении, равномерно заполняя весь объем дисперсионной среды. Такие свободнодисперсные коллоидные системы (дымы, золи) седиментационно устойчивы, то есть их частицы не выпадают в осадок. В процессе броуновского движения и при перемешивании коллоидные частицы сталкиваются. Если при этом не происходит укрупнения частиц вследствие их слипания (коагуляции) или слияния (коалесценции), токоллоидные системы являются агрегативно устойчивыми (например, лиофильные и стабилизованные лиофобные коллоидные системы, см. Лиофильные и лиофобные коллоиды).
Коллоидные системы образуются при конденсации вещества в гомогенной среде (пересыщенном растворе, паре, переохлажденнойжидкости), если возникающие в ней зародыши новой дисперсной фазы, то есть мельчайшие капли или кристаллики, не получают возможности расти до размеров, превышающих 10-5 — 10-4 см. Конденсация часто сопровождает химические реакции, в результате которых образуются трудно-растворимые соединения. Другой путь получения коллоидных систем — диспергирование, самопроизвольное в случае лиофильных и принудительное в случае лиофобных систем. Существование жидких агрегативно устойчивых лиофобных коллоидных систем всегда обусловлено наличием в дисперсионной среде поверхностно-активных веществ —стабилизаторов. Эти вещества создают на поверхности частиц адсорбционно-сольватный защитный слой, препятствующий их сближению и коагуляции под влиянием близкодействующих сил молекулярного притяжения. Препятствием к сближению частиц могут быть расклинивающее давление жидкой дисперсионной среды, сольватно связанной (см. Сольватация) в адсорбционном слоемолекулами или ионами стабилизатора; электростатическое отталкивание одноименно заряженных ионов, адсорбированных на поверхности частиц; повышенная структурная вязкость поверхностного защитного слоя, так называемый структурно-механический барьер.
Неструктурированные жидкие коллоидные системы, золи, по комплексу свойств занимают промежуточное положение междугрубодисперсными системами и растворами. При ослаблении в лиофобных золях защитного действия стабилизаторов (астабилизация, или дестабилизация, системы) возникают структурированные коллоидные системы — гели (см. также Дисперсная структура). Кколлоидным системам относят и твёрдые микрогетерогенные системы — мелкозернистые тела (некоторые минералы, сплавы, закристаллизованные стекла и др.), в которых величина структурных элементов или включений не выходит за пределы коллоидных размеров.
Характерной чертой дисперсных систем является их исключительное многообразие, которое можно объяснить изменениями:
· степени раздробленности дисперсной фазы;
· агрегатного состояния дисперсионной среды и дисперсной фазы;
· химического состава фаз.
37.
Раздел «Устойчивость и коагуляция дисперсных систем» считается одним из центральных в курсе коллоидной химии. Связано это с тем, что устойчивость дисперсий и нарушение ее (коагуляция) играют большое значение во многих технологических процессах, связанных с их применением. Например, при синтезе латексов синтетических полимеров дисперсная система должна быть устойчивой во избежание потерь полимера в результате образования коагулюма. Целевое выделение полимера из латекса возможно, наоборот, только при нарушении устойчивости, т.е. при управляемой коагуляции. Использование латексов синтетических полимеров для модификации тканей с целью придания им новых свойств (формоустойчивости, масло-, кислото-, водоотталкивания), связано с процессом гетерокоагуляции частиц полимеров на поверхности волокон в тканях. Устойчивость дисперсных систем (пигментов в поликонденсационных и полимеризационных системах) позволяет получать пигментированные волокна. Нарушение устойчивости дисперсий пигментов в растворах или расплавах полимеров и образование агрегатов частиц способны вызвать разрыв при формовании волокон в результате разрыва целостности текущего пигментированного раствора или расплава.
Стабильность печатных пигментированных красок, используемых для колорирования тканей, также влияет на процесс печати, на оттенок и даже на цвет пигментов в полимерных пленках на поверхности тканей.
Очевидно, что знание закономерностей создания устойчивых систем и их коагуляции имеет не только теоретический, но и практический интерес для специалистов, занимающихся проблемами получения химических волокон, производства текстильных материалов и применения латексов. В этой связи данный раздел будет полезным для студентов – будущих инженеров, химиков-технологов, специалистов по производству химических волокон, текстильных и нетканых материалов. Особенно важно знание этих проблем для будущих специалистов в области инженерной охраны окружающей среды методами очистки сточных вод и газовых выбросов от вредныхвеществ, так как широкий ряд способов очистки основан именно на коагуляции дисперсных загрязнений.
38.
Общая характеристика растворов.
Растворами называются гомогенные системы переменного состава, в которых растворенное вещество находится в виде атомов, ионов или молекул, равномерно окруженных атомами, ионами или молекулами растворителя. Любой раствор состоит по меньшей мере из двух веществ, одно из которых считается растворителем, а другое - растворенным веществом. Растворителем считается компонент, агрегатное состояние которого такое же, как и агрегатное состояние раствора. Деление это довольно условно, а для веществ, смешивающихся в любых соотношениях (вода и ацетон, золото и серебро), лишено смысла. В этом случае растворителем считается компонент, находящийся в растворе в большем количестве. Состав растворов может меняться в довольно широких пределах, в этом растворы сходны с механическими смесями. По другим признакам, таким как однородность, наличие теплового эффекта и окраски растворы сходны с химическими соединениями. Растворы могут существовать в газообразном, жидком или твердом агрегатном состоянии. Воздух, например, можно рассматривать как раствор кислорода и других газов в азоте; морская вода - это водный раствор различных солей в воде. Металлические сплавы относятся к твердым растворам одних металлов в других. Растворение веществ является следствием взаимодействия частиц растворяемого вещества и растворителя. В начальный момент времени растворение идет с большой скоростью, однако по мере увеличения количества растворенного вещества возрастает скорость обратного процесса - кристаллизации. Кристаллизацией называется выделение вещества из раствора и его осаждение. В какой-то момент скорости растворения и осаждения сравняются и наступит состояние динамического равновесия. Раствор, в котором вещество при данной температуре уже больше не растворяется, или иначе, раствор, находящийся в равновесии с растворяемым веществом, называется насыщенным. Для большинства твердых веществ растворимость в воде увеличивается с повышением температуры. Если раствор, насыщенный при нагревании, осторожно охладить так, чтобы не выделялись кристаллы, то образуется пересыщенный раствор. Пересыщенным называется раствор, в котором при данной температуре содержится большее количество растворенного вещества, чем в насыщенном растворе. Пересыщенный раствор крайне нестабилен и при изменении условий (энергичное встряхивание или внесение активных центров кристаллизации - кристалликов соли, пылинок) образуется насыщенный раствор и кристаллы соли. Раствор, содержащий меньше растворенного вещества, чем насыщенный, называется ненасыщенным раствором.
Согласно второму началу термодинамики при р, Т = = const вещества самопроизвольно могут растворяться в каком-либо растворителе, если в результате этого процесса энергия Гиббса системы уменьшается, т. е.
?G = (?Н – T?S) < 0.
Величину ?Н называют энтальпийным фактором, а величину T?S – энтропийным фактором растворения.
При растворении жидких и твердых веществ энтропия системы обычно возрастает (?S > 0), так как растворяе–мые вещества из более упорядоченного состояния пе–реходят в менее упорядоченное. Вклад энтропийного фактора, способствующий увеличению растворимости, особенно заметен при повышенных температурах, по–тому что в этом случае множитель Т велик и абсолютное значение произведения T?S также велико, соответст–венно возрастает убыль энергии Гиббса.
При растворении газов в жидкости энтропия системы обычно уменьшается (?S < 0), так как растворяемое вещество из менее упорядоченного состояния (боль–шого объема) переходит в более упорядоченное (ма–лый объем). Снижение температуры благоприятствует растворению газов, потому что в этом случае множи–тель Т мал и абсолютное значение произведения T?S будет тем меньше, а убыль энергии Гиббса тем больше, чем ниже значение Т.
В процессе образования раствора энтальпия систе–мы также может как увеличиваться (NaCI), так и умень–шаться (КОН). Изменение энтальпии процесса раство–рения нужно рассматривать в соответствии с законом Гесса как алгебраическую сумму эндо– и экзотермиче–ских вкладов всех процессов, сопровождающих про–цесс растворения. Это эндотермические эффекты раз–рушения кристаллической решетки веществ, разрыва связи молекул, разрушения исходной структуры рас–творителя и экзотермические эффекты образова–ния различных продуктов взаимодействия, в том числе сольватов.
Для простоты изложения приращение энтальпии раст–ворения ?Нраств можно представить как разность энер–гии Екр, затрачиваемой на разрушение кристаллической решетки растворяемого вещества, и энергии Есол, выде–ляющейся при сольватации частиц растворенного веще–ства молекулами растворителя. Иначе говоря, измене–ние энтальпии представляет собой алгебраическую сумму изменения энтальпии ?Нкр в результате разруше–ния кристаллической решетки и изменения энтальпии ?Нсол за счет сольватации частицами растворителя:
?Нраств = ?Нкр + ?Нсол,
где ?Нраств – изменение энтальпии при растворении.
Однако растворение благородных газов в органичес–ких растворителях нередко сопровождается поглоще–нием теплоты, например гелия и неона в ацетоне, бен–золе, этаноле, циклогексане.
При растворении твердых веществ с молекулярной кристаллической структурой и жидкостей молекуляр–ные связи не очень прочные, и поэтому обычно ?Нсол > ?Нкр Это приводит к тому, что растворение, например, спиртов и сахаров представляет собой экзотермиче–ский процесс (?Нраств < 0).
При растворении твердых веществ с ионной решет–кой соотношение энергий Екр и Есолмогут быть различ–ным. Однако в большинстве случаев энергия, выделяе–мая при сольватации ионов, не компенсирует энергию, затрачиваемую на разрушение кристаллической решет–ки, следовательно, и процесс растворения является эн–дотермическим.
Таким образом, термодинамические данные позво–ляют прогнозировать самопроизвольное растворение различных веществ на основе первого и второго начал термодинамики.
Процесс растворения газов в металлах можно разделить на несколько элементарных кинетических стадий:
1) поступление частиц из газовой фазы к поверхности металла;
2) адсорбция газа на этой поверхности, сопровождающаяся диссоциацией, а в соответствующих условиях и образованием химического соединения газа с одним из компонентов металла;
3) переход адсорбированных атомов с поверхности в раствор;
4) перенос растворенных атомов с поверхности металла в его объем в результате выравнивания концентрации газа в металле, осуществляемого атомарной диффузией частиц газа в твердых металлах и конвективной диффузией в расплавах.
Эти же кинетические стадии, но в обратном порядке характеризуюткинетику процесса десорбции. В обоих случаях скорость суммарного процесса будет лимитироваться и определяться скоростями наиболее медленных стадий процесса. Из-за отсутствия каких-либо экспериментальных данных в настоящее время не представляется возможным выделить в самостоятельные кинетические стадии процессы: переход адсорбированных газов с поверхности металла в растворенное состояние при сорбции газа металлом и обратный их переход при десорбции.
Скорость перемещения частиц газа к поверхности металла и от нее, рассчитываемая по известным уравне-
ниям молекулярно-кинетической теории газа, велика и обычно не определяет кинетику растворения. Поэтому анализ кинетики сорбционных и десорбционных процессов по существу сводится к разделению скорости пронес» сов, протекающих на поверхности раздела, — адсорбции или десорбции, и скорости массопереноса газа с поверхности или к ней в объеме металла.
Уравнение Ленгмюра для кинетики мономолекулярной адсорбции выведено исходя из предположения, что энергия связи газа с поверхностью не зависит от ее заполнения. Как отмечалось выше, это предположение не оправдывается для твердой поверхности, по, по-видимому, справедливо для жидкого металла. В последнем случае характер лимитирующей стадии можно определить по виду зависимости начальной скорости процесса от давления растворяющегося газа; скорость адсорбции по уравнению (1-6) пропорциональна давлению в первой степени, а скорости последующих стадий процесса растворения— корню квадратному из его парциального давления.
Как было показано в первой главе, для твердой энергетически неоднородной поверхности этот критерий не является достаточно надежным, так как адсорбция может в свою очередь лимитироваться активированной миграцией адсорбированных атомов газа на поверхности, также пропорциональной корню квадратному из давления.
Массоперенос или диффузия атомов газа в твердых металлах протекает иначе, чем в жидких. В твердом металле атомы растворенного газа перемещаются из одних междоузлий кристаллической решетки в другие вследствие их перераспределения в объеме металла; в жидких расплавах выравнивание концентрации растворенного газа достигается главным образом перемещением отдельных объемов абсорбента. Поэтому кинетикурастворения (и десорбции) газов твердыми и жидкими металлами следует рассматривать раздельно.
ГИДРАТЫ, продукты присоединения воды (гидратации)к молекулам, атомам или ионам. М. б. газообразными, жидкими и твердыми; последние наз. кристаллогидратами.
В р-ре многозарядные ионы (А13+, Сг3+, Be2+ и др.) и небольшие однозарядные (Li+) достаточно прочно связывают ближайшие к ниммолекулы воды, образуя аквакатионы, напр. [А1(Н2О)6]3+ . Кристаллогидраты солей обычно образуются в тех случаях, когда катионы в их кристаллич. решетке образуют более прочные связи с молекулами Н2О, чем с анионами в решетке безводной соли. При низких т-рахвода в кристаллогидратах м. б. связана и с катионом, и с анионом соли. Напр., у MgCl2*12Н2О ниже — 25 °С в кристаллич. решетке находятся акваионы [Mg(H2O)6]2+ и [С1(Н2О)6]-. Воду, входящую в состав кристаллогидратов, наз. кристаллизационной. Существуют также псевдогидраты-соед., в к-рых все или часть молекул воды превращаются в гидроксид-ионы или ионы гидроксония, напр. Sr(BO2)2*4H2O (по существу это Sr[B(OH)4]2) и НС1О4*Н,О [или (Н3О)С1О4]. Воду, входящую в состав псевдогидратов, наз. конституционной. В каждом кристаллогидрате молекулы воды располагаются определенным образом. Так, в [А1(Н2О)6]С13 вода входит в состав аквакатиона, в гидратах цеолитов размещается в пустотах сетчатой ионной структуры. В соед. типа Mg2(OH)2(H2O)3CO3цепочки молекул воды находятся в каналах структуры. Вода может связывать аквакатион с анионом водородными связями, напр. CuSO4*5H2O или [Cu(H2O)4](H2O)SO4. Существуют кристаллогидраты, в к-рых вода образует слои, объединяемые ионами соли(CaSO4*2H2O и др.). Известны тектогидраты, в к-рых молекулы воды образуют непрерывную фазу со структурой, подобной структурельда, включающую др. молекулы или ионы (Na2S04*10H2O, H3[PW12O40]*29H2O и др.). Если посторонняя молекула в структуру льда не встраивается, то образуется эвтектич. тонкая мех. смесь льда и соли, иногда наз. криогидратом. При удалении воды мн.кристаллогидраты солей превращаются в низшие кристаллогидраты или в безводные соли с иной (по сравнению с исходной) кристаллич. структурой, но часто в результате гидролиза при обезвоживании образуются гидроксосоли, гидроксиды или оксиды. Кристаллич. структура кристаллогидратов солей, содержащих воду в пустотах или каналах структуры, при обезвоживании часто не изменяется. Каждый кристаллогидрат устойчив лишь в определенном температурном интервале, поэтому кривые их р-римости имеют изломы, соответствующие превращениям одного кристаллогидрата в другой. Для каждого кристаллогидрата характерно определенное равновесное давление водяного пара, зависящее от т-ры. Если это давление над кристаллогидратом больше, чем парциальноедавление водяных паров в окружающей среде при той же температуре, то кристаллогидрат постепенно теряет воду ("выветривается"); в противоположном случае кристаллогидрат поглощает воду и даже может в ней раствориться ("расплывается")
Существуют кристаллогидраты [Fe(NO3)2*9Н2О, СаС12*6Н2О и др.], к-рые самопроизвольно не кристаллизуются ни при каком пересыщении. С увеличением числа молекул кристаллизац. воды энтальпия растворения кристаллогидратов уменьшается по абс. величине и становится даже положит. величиной. О гидратах газов см. Газовые гидраты.
39.
РАСТВОРЫ НЕЭЛЕКТРОЛИТОВ, бинарные или многокомпонентные мол. системы, состав к-рых может изменяться непрерывным образом (по крайней мере, в нек-рых пределах). В отличие от растворов электролитов, в растворах неэлектролитов (мол. р-рах) заряженные частицы в сколько-нибудь заметных концентрациях отсутствуют. Растворы неэлектролитов могут быть твердыми, жидкими и газообразными. В данной статье рассматриваются жидкие р-ры; см. также Твердые растворы.
Взаимная р-римость двух жидкостей при заданных т-ре Т и давлении р м. б. полной (неограниченной) или ограниченной. В последнем случае р-ры в нек-рой области составов расслаиваются, т. е. разделяются на две жидкие фазы, отличающиеся по концентрации. В многокомпонентных расслаивающихся р-рах число сосуществующих жидких фаз м. б. более двух. Если один (или более) из компонентовраствора неэлектролитов в чистом состоянии при заданных Т и р является газом или твердым телом, область существования растворанеэлектролитов простирается от чистой жидкости (смеси жидкостей), выступающей в роли р-рителя, до состава, отвечающего насыщ. р-ру.
Растворы неэлектролитов служат средой, в к-рой протекают многие прир. и пром. процессы. Изучение и прогнозирование св-в этих систем тесно связаны с такими практич. проблемами, как подбор р-рителей для реализации технол. процессов, получение систем с заданными св-вами, разделение прир. и пром. смесей (включая газы и нефти), глубокая очистка в-в.
Физ. химия изучает широкий диапазон св-в р-ров. Наиб. разработана и имеет практически важные применения равновеснаятермодинамика р-ров; дальнейший материал посвящен в осн. этому разделу физ. химии р-ров. Кроме того, изучаются транспортные св-ва р-ров-диффузия, теплопроводность, вязкость (см. Физико-химическая гидродинамика), а также спектроскопич., электрич., акустич. и др. физ. св-ва. Методы исследования макроскопич. св-в растворов неэлектролитов и их структурных характеристик во многом аналогичны методам исследования индивидуальных жидкостей, но осн. внимание уделяется рассмотрению концентрац. зависимостей св-в. Важнейшая задача физ.-хим. исследований - установление связи между наблюдаемыми на опыте св-вами, структурой р-ров и характеристиками межмолекулярных взаимодействий. Эксперим. информацию о структуре р-ров и межмолекулярных взаимод. в них дают методы оптической и радиоспектроскопии, дифракционные, электрич. и др. Важную роль в изучении растворов неэлектролитов играет физико-химический анализ, основанный на построении и исследовании фазовых диаграмм, концентрац. зависимостей термодинамич. и др. физ. св-в (показателя преломления, вязкости, теплопроводности, акустич. характеристик и др.). При этом одна из главных задач состоит в том, чтобы на основании анализа диаграмм состав - свойство устанавливать факт образования хим. соединений между компонентами растворов неэлектролитов и находить их характеристики.
Осмос (от греч. ōsmós — толчок, давление), диффузия вещества, обычно растворителя, через полупроницаемую мембрану, разделяющую раствор и чистый растворитель или два раствора различной концентрации. Полупроницаемая мембрана — перегородка, пропускающая малые молекулы растворителя, но непроницаемая для более крупных молекул растворённого вещества. Выравниваниеконцентраций по обе стороны такой мембраны возможно лишь при односторонней диффузии растворителя. Поэтому всегда идёт от чистого растворителя к раствору или от разбавленного раствора к концентрированному. Осмос, направленный внутрь ограниченного объёма жидкости, называется эндосмосом, наружу — экзосмосом. Перенос растворителя через мембрану обусловлен осмотическим давлением. Оно равно избыточному внешнему давлению, которое следует приложить со стороны раствора, чтобы прекратить осмос, т. е. создать условия осмотического равновесия. Превышение избыточного давления над осмотическим может привести к обращениюосмоса — обратной диффузии растворителя (см. Ультрафильтрация). В случаях, когда мембрана проницаема не только длярастворителя, но и для некоторых растворённых веществ, диффузия последних из раствора в растворитель позволяет осуществитьдиализ, применяемый как способ очистки полимеров и коллоидных систем от низкомолекулярных примесей, например электролитов. Впервые осмос наблюдал А. Нолле в 1748, однако исследование этого явления было начато спустя столетие. Осмос имеет важнейшее значение в биологических процессах (см. Осморегуляция), его широко используют в лабораторной технике: при определении молярных характеристик полимеров, концентрировании растворов, исследовании разнообразных биологических структур. Осмотические явления иногда используются в промышленности, например при получении некоторых полимерных материалов, очистке высоко-минерализованной воды методом «обратного» осмоса жидкостей.
смос играет важную роль во многих биологических процессах. Мембрана, окружающая нормальную клетку крови, проницаема лишь для молекул воды, кислорода, некоторых из растворённых в крови питательных веществ и продуктов клеточной жизнедеятельности; для больших белковых молекул, находящихся в растворённом состоянии внутри клетки, она непроницаема. Поэтому белки, столь важные для биологических процессов, остаются внутри клетки.
Осмос участвует в переносе питательных веществ в стволах высоких деревьев, где капиллярный перенос не способен выполнить эту функцию.
Осмос широко используют в лабораторной технике: при определении молярных характеристик полимеров, концентрировании растворов, исследовании разнообразных биологических структур. Осмотические явления иногда используются в промышленности, например при получении некоторых полимерных материалов, очистке высоко-минерализованной воды методом обратного осмоса жидкостей.
ДИФФУЗИЯ (от лат. diflusio - распространение, растекание, рассеивание), перенос частиц разной природы, обусловленный хаотич. тепловым движением молекул (атомов) в одно-или многокомпонентных газовых либо конденсир. средах. Такой перенос осуществляется при наличии градиента концентрации частиц или при его отсутствии; в последнем случае процесс наз. самодиффузией (см. ниже). Различают диффузию коллоидных частиц (т. наз. броуновская диффузия), в твердых телах, молекулярную, нейтронов, носителей заряда в полупроводниках и др.; о переносе частиц в движущейся с определенной скоростью среде (конвективная диффузия) см. Массообмен.Переноса процессы, о диффузии частиц в турбулентных потоках см. Турбулентная диффузия. Все указанные виды диффузии описываются одними и теми же феноменологич. соотношениями.
Применение диффузии. Диффузия используется при выплавке стали. Для придания стальным деталям прочности их помещают в специальные печи, где, находясь в разогретом состоянии, они насыщаются кислородом. Атомы углерода проникают в поверхностный слой металла и повышают его прочность. С ее помощью изготавливают многие полупроводниковые приборы.
40.
Первый закон Рауля
Первый закон Рауля связывает давление насыщенного пара над раствором с его составом; он формулируется следующим образом:
Парциальное давление насыщенного пара компонента раствора прямо пропорционально его мольной доле в растворе, причём коэффициент пропорциональности равен давлению насыщенного пара над чистым компонентом.
![]()
Для бинарного раствора, состоящего из компонентов А и В (компонент А считаем растворителем) удобнее использовать другую формулировку:
Относительное понижение парциального давления пара растворителя над раствором не зависит от природы растворённого вещества и равно его мольной доле в растворе.
![]()
Растворы, для которых выполняется закон Рауля, называются идеальными. Идеальными при любых концентрациях являются растворы, компоненты которых очень близки по физическим и химическим свойствам (оптические изомеры, гомологи и т. п.), и образование которых не сопровождается изменением объёма и выделением либо поглощением теплоты. В этом случае силымежмолекулярного взаимодействия между однородными и разнородными частицами примерно одинаковы, и образование раствора обусловлено лишь энтропийным фактором.
Отклонения от закона Рауля
Растворы, компоненты которых существенно различаются по физическим и химическим свойствам, подчиняются закону Рауля лишь в области очень малых концентраций; при больших концентрациях наблюдаются отклонения от закона Рауля. Случай, когда истинные парциальные давления паров над смесью больше, чем вычисленные по закону Рауля, называют положительными отклонениями. Противоположный случай, когда парциальные давления паров компонентов оказываются меньше вычисленных — отрицательные отклонения.
Причиной отклонений от закона Рауля является то обстоятельство, что однородные частицы взаимодействуют друг с другом иначе, чем разнородные (сильнее в случае положительных и слабее в случае отрицательных отклонений).
Реальные растворы с положительными отклонениями от закона Рауля образуются из чистых компонентов с поглощением теплоты (ΔНраств > 0); объём раствора оказывается больше, чем сумма исходных объёмов компонентов (ΔV > 0). Растворы с отрицательными отклонениями от закона Рауля образуются с выделением теплоты (ΔНраств < 0); объём раствора в этом случае будет меньше, чем сумма исходных объёмов компонентов (ΔV < 0).
Второй закон Рауля
Тот факт, что давление паров над раствором отличается от давления паров над чистым растворителем, существенно влияет на процессы кристаллизации и кипения. Из первого закона Рауля выводятся два следствия, касающиеся понижения температуры замерзания и повышения температуры кипения растворов, которые в объединённом виде известны как второй закон Рауля.
Вант-Гоффа закон осмотического давления, определяет давление молекул растворённого вещества на полупроницаемую перепонку, отделяющую раствор от чистого растворителя и непроницаемую для растворённого вещества. Открыт Я. Х. Вант-Гоффом.
Антифриз (от греч. ἀντι- — против и англ. freeze — замерзать) — общее название для жидкостей, не замерзающих при низких температурах. Применяются в установках, работающих при низких температурах, для охлаждения двигателей внутреннего сгорания, в качестве авиационных противообледенительных жидкостей. В качестве базовых жидкостей антифризов используются смеси этиленгликоля, пропиленгликоля, глицерина, одноатомных спиртов и других веществ с водой.
41. РАСТВОРЫ НЕЭЛЕКТРОЛИТОВ, бинарные или многокомпонентные мол. системы, состав к-рых может изменяться непрерывным образом (по крайней мере, в нек-рых пределах). В отличие от растворов электролитов, в растворах неэлектролитов (мол. р-рах) заряженные частицы в сколько-нибудь заметных концентрациях отсутствуют. Растворы неэлектролитов могут быть твердыми, жидкими и газообразными. В данной статье рассматриваются жидкие р-ры; см. также Твердые растворы.
Взаимная р-римость двух жидкостей при заданных т-ре Т и давлении р м. б. полной (неограниченной) или ограниченной. В последнем случае р-ры в нек-рой области составов расслаиваются, т. е. разделяются на две жидкие фазы, отличающиеся по концентрации. В многокомпонентных расслаивающихся р-рах число сосуществующих жидких фаз м. б. более двух. Если один (или более) из компонентовраствора неэлектролитов в чистом состоянии при заданных Т и р является газом или твердым телом, область существования растворанеэлектролитов простирается от чистой жидкости (смеси жидкостей), выступающей в роли р-рителя, до состава, отвечающего насыщ. р-ру.
Растворы неэлектролитов служат средой, в к-рой протекают многие прир. и пром. процессы. Изучение и прогнозирование св-в этих систем тесно связаны с такими практич. проблемами, как подбор р-рителей для реализации технол. процессов, получение систем с заданными св-вами, разделение прир. и пром. смесей (включая газы и нефти), глубокая очистка в-в.
Физ. химия изучает широкий диапазон св-в р-ров. Наиб. разработана и имеет практически важные применения равновеснаятермодинамика р-ров; дальнейший материал посвящен в осн. этому разделу физ. химии р-ров. Кроме того, изучаются транспортные св-ва р-ров-диффузия, теплопроводность, вязкость (см. Физико-химическая гидродинамика), а также спектроскопич., электрич., акустич. и др. физ. св-ва. Методы исследования макроскопич. св-в растворов неэлектролитов и их структурных характеристик во многом аналогичны методам исследования индивидуальных жидкостей, но осн. внимание уделяется рассмотрению концентрац. зависимостей св-в. Важнейшая задача физ.-хим. исследований - установление связи между наблюдаемыми на опыте св-вами, структурой р-ров и характеристиками межмолекулярных взаимодействий. Эксперим. информацию о структуре р-ров и межмолекулярных взаимод. в них дают методы оптической и радиоспектроскопии, дифракционные, электрич. и др. Важную роль в изучении растворов неэлектролитов играет физико-химический анализ, основанный на построении и исследовании фазовых диаграмм, концентрац. зависимостей термодинамич. и др. физ. св-в (показателя преломления, вязкости, теплопроводности, акустич. характеристик и др.). При этом одна из главных задач состоит в том, чтобы на основании анализа диаграмм состав - свойство устанавливать факт образования хим. соединений между компонентами растворов неэлектролитов и находить их характеристики.
Сильные электролиты при растворении в воде практически полностью диссоциируют на ионы независимо от их концентрации в растворе.
Поэтому в уравнениях диссоциации сильных электролитов ставят знак равенства (=).
К сильным электролитам относятся:
- растворимые соли;
- многие неорганические кислоты: HNO3, H2SO4, HCl, HBr, HI;
- основания, образованные щелочными металлами (LiOH, NaOH, KOH и т.д.) и щелочно-земельными металлами (Ca(OH)2, Sr(OH)2, Ba(OH)2).
Слабые электролиты в водных растворах лишь частично (обратимо) диссоциируют на ионы.
Поэтому в уравнениях диссоциации слабых электролитов ставят знак обратимости (⇄).
К слабым электролитам относятся:
- почти все органические кислоты и вода;
- некоторые неорганические кислоты: H2S, H3PO4, H2CO3, HNO2, H2SiO3 и др.;
- нерастворимые гидроксиды металлов: Mg(OH)2, Fe(OH)2, Zn(OH)2 и др.
Количественная характеристика диссоциации электролитов называется степенью диссоциации(обозначение α); для веществ-электролитов всегда больше нуля (α = 0 в случае неэлектролитов). Степень диссоциации может выражаться как в долях от 1, так и в процентах.
42.
Частным случаем диссоциации (процесса распада более крупных частиц вещества — молекул ионов или радикалов — на частицы меньшего размера) является электролитическая диссоциация, при которой нейтральные молекулы вещества, называемого электролитом, в растворе (в результате воздействия молекул полярного растворителя) распадаются на заряженные частицы: катионы и анионы. Этим объясняется способность растворов электролитов проводить ток.
Принято делить все электролиты на две группы: слабые и сильные. Вода относится к слабым электролитам, диссоциация воды характеризуется небольшим количеством диссоциированных молекул, так как они достаточно стойкие и практически не распадаются на ионы. Чистая (без примесей) вода слабо проводит электрический ток. Это обусловлено химической природой самой молекулы, когда положительно поляризованные атомы водорода внедрены в электронную оболочку сравнительно небольшого атома кислорода, который поляризован отрицательно.
Ио́нное произведе́ние воды́ — произведение концентраций ионов водорода Н+ и ионов гидроксида OH− в воде или в водныхрастворах, константа автопротолиза воды.
Водоро́дный показа́тель (pH)
количественная мера активной кислотности или щелочности среды, численно равная отрицательному десятичному логарифму концентрации водородных ионов; определение pH биологических жидкостей широко применяется при диагностических и экспериментальных исследованиях.
Гидролиз солей – это взаимодействие ионов соли с водой с образованием малодиссоциирующих частиц.
Гидролиз, дословно, - это разложение водой. Давая такое определение реакции гидролиза солей, мы подчеркиваем, что соли в растворе находятся в виде ионов, и что движущей силой реакции является образование малодиссоциирующих частиц (общее правило для многих реакций в растворах).
Всегда ли ионы способны образовывать с водой малодиссоциирующие частицы? Разбирая этот вопрос с учениками, отмечаем, что катионы сильного основания и анионы сильной кислоты таких частиц образовать не могут, следовательно, в реакцию гидролиза не вступают.
Какие типы гидролиза возможны? Поскольку соль состоит из катиона и аниона, то возможно три типа гидролиза:
гидролиз по катиону (в реакцию с водой вступает только катион);
гидролиз по аниону (в реакцию с водой вступает только анион);
совместный гидролиз (в реакцию с водой вступает и катион, и анион);
Растворимость, способность вещества образовывать с другим веществом однородную, термодинамически устойчивую систему переменного состава, состоящую из двух или большего числа компонентов. Такие системы возникают при взаимодействии газов с жидкостями, жидкостей с жидкостями и т.д. (см. Растворы).Соотношение компонентов может быть либо произвольным, либо ограниченным некоторыми пределами. В последнем случае Р. называют ограниченной. Мерой Р. вещества при данных условиях служит концентрация его насыщенного раствора. Р. различных веществ в определённом растворителе зависит от внешних условий, прежде всего — от температуры и давления. Давление наиболее сильно сказывается на Р. газов. Изменение внешних условий влияет на Р. в соответствии с принципом смещения равновесий (см. Ле Шателье — Брауна принцип).Для наиболее важных растворителей составлены таблицы Р. различных веществ в зависимости от внешних условий или только для стандартных условий.
Произведение растворимости (ПР, Ksp) — произведение концентраций ионов малорастворимого электролита в его насыщенном растворе при постоянной температуре и давлении. Произведение растворимости — величина постоянная.
При постоянной температуре в насыщенных водных растворах малорастворимых электролитов устанавливается равновесие между твердым веществом и ионами, образующими это вещество. Например, в случае для CaCO3 это равновесие можно записать в виде:
![]()
Константа этого равновесия рассчитывается по уравнению:
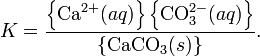
В приближении идеального раствора с учётом того, что активность чистого компонента равна единице, уравнение упрощается до выражения:
![]()
Константа равновесия такого процесса называется произведением растворимости.
В общем виде, произведение растворимости для вещества с формулой AmBn, которое диссоциирует на m катионов An+ и n анионов Bm-, рассчитывается по уравнению:
![]()
где [An+] и [Bm-] — равновесные молярные концентрации ионов, образующихся при электролитической диссоциации.
Из произведений растворимости можно рассчитать концентрации катионов и анионов в растворе малорастворимого электролита. Значения произведений растворимости приведены в справочниках.
43.
Природная вода, это скорее раствор, который имеет в своем составе и растворенные газы, и разнообразные химические соединения.
Как известно ученым всего мира – состав природных вод на всей поверхности планеты, имеет некоторые отличия в зависимости от территории, на которой они находятся. На самом деле – природная вода, это скорее раствор, который имеет в своем составе и растворенные газы, и разнообразные химические соединения.
Чтобы определить качество воды, необходимо принять во внимание всю совокупность концентраций примесей, которые в ней имеются. Можно выделить несколько показателей.
Во-первых, это физические свойства воды – цвет, температура, вкус и запах, прозрачность – наличие взвеси.
Во-вторых, анализируются химические свойства воды, такие как жесткость, окисляемость, активная реакция и сухой остаток.
В-третьих, анализу подвергаются ее биологические свойства. Необходимо определить то, сколько бактерий живет в водоеме и всех гидробионтов (живые существа, населяющие конкретный водоем).
Одним из самых важных и главных признаков любой природной воды является ее прозрачность. В зависимости от того, каков состав природных вод, меняется их прозрачность, а значит и глубина проникновения в глубину солнечного света. Чтобы определить, насколько прозрачна вода, в нее опускают белый диск или диск Секи (именно этот исследователь в свое время изобрел очень простой и удобный способ для определения проницаемости солнечного света). Интересно, что главным местом, где трудился преподобный Секи была обыкновенная католическая церковь, где он занимал должность священнослужителя. И в гидрологической науке данный способ исследования непременно называют божественным даром или вкладом Ватикана в процедуру изучения экосистем водоемов.
То, насколько будет прозрачной вода, зависит от того, какие вещества в ней растворены, присутствуют ли в ней белковые вещества, жиры, органические кислоты, органика, гумусовые отложения. При этом цветность вод, чаще всего определяется именно наличием гумусовых отложений. Интересно, что гумусовые вещества, которые не могут раствориться в воде, встречаются в ней только в состоянии взвеси, в коллоидном истинном виде растворения находятся только гуминовая кислота и некоторые другие соединения. Особенно часто они встречаются в виде солей металлов щелчеземельных или щелочных.
При этом цветность вод несколько изменяется в зависимости от увеличения жесткости (для поверхностных вод) или щелочности (для гидрокарбонатных вод). Если же вода мягкая и имеет большой показатель щелочности, то, скорее всего, у нее будет более высокая цветность. Кроме того, зачастую вода оказывается окрашена соединениями железа, которые образуются в период «цветения» рек и озер или обуславливаются стоками промышленных отходов.
Есть и показатель мутности, который выражается, прежде всего, в том, что в воду попадают взвешенный частицы, такие как планктон, песок, глина или ил. Мутность обычно определяют при помощи значений мг/л, ее определяют при помощи колориметра.
Состав природных вод, кроме того, влияет на такой показатель как рН. Природные воды можно разделить на несколько групп. В первой группе оказались природные воды, которые имеют рН от 1 до 3, это кислые воды. Слабокислые воды имеют рН от 4 до 6. Есть и нейтральный воды, которые имеют показатель в районе 7 единиц. Следующая группа от 8 до 10 – слабощелочная вода. А щелочные воды имеют рН свыше 11. Чаще всего природная вода имеет рН, который колеблется в небольших пределах от 6,6 до 8,8. Но иногда, из-за того, что стали обыденностью кислотные дожди, в некоторых водоемах кислотность может достигать значений в 4 или чуть ниже.
В сухом остатке можно увидеть все примеси, которые имеют не органическое происхождение. Этот остаток выявляется при выпаривании необходимого объема вод при температуре, которая составляет 110 градусов. Это производится до того момента, пока масса остатка не будет составлять постоянную величину. Если же в водах содержится большое число хлоридов, то можно ожидать то, что в результате выпаривания мы получим довольно большое количество сухого остатка.
Марганец и железо можно встретить в поверхностной воде только в виде минеральных или органических соединениях комплексного типа. Также, они встречаются и в виде коллоидного раствора, а также мелкодисперсной смеси. Это содержание марганца и железа может составлять порядка нескольких десятков мг/л
Для природной воды, кроме того, можно определить и такой показатель, как окисляемость. Окисляемость, это свойство воды, связанное с тем, что в ее составе имеется двухвалентное железо, сероводород или сульфиты. Если в воде определена повышенная окисляемость, это может свидетельствовать только о том, что она подвержена повышенному уровню загрязнения. Окисляемость обычно является главной характеристикой для того, чтобы определить ее гигиеническое состояние.
Кроме того, в природной воде могут оказываться нитриты и нитраты – азотосодержащие вещества, которые образуются при взаимодействие гумусовых элементов и сероводорода. Эти реакции носят окислительно-восстановительный характер. Также, они могут образовать при разложении элементов, имеющих в своей основе белок. К примеру, нитраты железа могут восстанавливаться сероводородом, в результате чего образуется нитрит. По форме азотосодержащих соединений, можно производить интересные исследования, такие как определение того, в какое конкретно время произошло поступление в водоем вод из стока. К примеру, если в составе воды определены ионы аммония, это может свидетельствовать о том, что загрязнение было произведено совсем недавно.
Надо отметить, что огромное воздействие на состав воды, и изменение его химических элементов влияет то, какое именно у этой воды питание. К примеру, весной, когда реки питаются преимущественно грунтовыми водами и подземными источниками, в ней наблюдается наименьшая минерализация. В итоге, можно сделать вывод о том, что природная вода имеет в своем составе множество элементов, которые склонны к периодическому изменению.
Жёсткость воды и способы её устранения.
Растворимые соли кальция и магния обуславливают общую жёсткость воды. Если они присутствуют в воде в небольших количествах, то вода называется мягкой. При большом содержании этих солей (100 – 200 мг. солей кальция – в 1 л. в пересчёте на ионы) вода считается жёсткой. В такой воде мыло плохо пенится, так как соли кальция и магния образуют с ним нерастворимые соединения. В жёсткой воде плохо развариваются пищевые продукты, и при кипячении она даёт на стенках паровых котлов накипь. Накипь плохо проводит теплоту, вызывает увеличение расхода топлива и ускоряет изнашивание стенок котла. Образование накипи – сложный процесс. При нагревании кислые соли угольной кислоты кальция и магния разлагаются и переходят в нерастворимые карбонаты:
Са + 2НСО3 = Н2О + СО2 + СаСО3↓
Растворимость сульфата кальция СаSO4 при нагревании также снижается, поэтому он входит в состав накипи.
Жёсткость вызванная присутствием в воде гидрокарбонатов кальция и магния, называется карбонатной или временной, так как она устраняется при кипячении. Помимо карбонатной жёсткости, различают ещё некарбонатную жёсткость, которая зависит от содержания в воде сульфатов и хлоридов кальция и магния. Эти соли не удаляются при кипячении, и поэтому некарбонатную жёсткость называют также постоянной жёсткостью. Карбонатная и некарбонатная жёсткость в сумме даёт общую жёсткость.
Для полного устранения жёсткости воду иногда перегоняют. Для устранения карбонатной жёсткости воду кипятят. Общую жёсткость устраняют или добавлением химических веществ, или при помощи так называемых катионитов. При использовании химического метода растворимые соли кальция и магния переводят в нерастворимые карбонаты, например добавляют известковое молоко и соду:
Са + 2НСО3 + Са + 2ОН = 2Н2О + 2СаСО3↓
Са + SO4 + 2Na + CO3 = 2Na + SO4 + CaCO3↓
Устранение жёсткости при помощи катионитов – процесс более совершенный. Катиониты – сложные вещества (природные соединения кремния и алюминия, высокомалекулярные органические соединения), состав которых можно выразить формулой Na2R, где R – сложный кислотный остаток. При фильтровании воды через слой катионита происходит обмен ионов (катионов) Na на ионы Са и Mg:
44.
Водоподготовкой (водоочисткой) называется комплекс мер, направленных на повышение качества жидкости из природных источников путем ее освобождения от солей, примесей и биологических агентов.
Цели водоподготовки
Основное назначение обработки жидкости – получение воды, пригодной для бытовых и промышленных нужд и безопасной для конечного и промежуточного потребителя. При этом подразумевается использование только экономически оправданных и наиболее эффективных способов очистки.
Оптимальные технологические методики существенно различаются в каждом конкретном случае, однако, всех их можно отнести к той или иной стадии процесса подготовки воды.
Этапы водоочистки
Осветление воды – ряд процедур, направленных на снижение ее мутности. Механические примеси затрудняют любую дальнейшую очистку жидкости, поэтому операция осветления обычно проводится в первую очередь.
Обесцвечивание – устранение окрашивания жидкости из-за присутствия в ее составе некоторых коллоидов и растворенных веществ.
Деминерализация включает обессоливание, обезжелезивание и умягчение жидкости. Очистка может осуществляться различными методами в зависимости от состава примесей.
Обеззараживание или дезинфекция используется при обработке технической и питьевой воды для подавления жизнеспособности микроорганизмов и их спор.
Коррекция щелочности/кислотности не входит в список обязательных этапов водоподготовки. Тем не менее, ее проведение зачастую необходимо и для технической, и для питьевой воды. Жидкость с ненормализованным уровнем pH способствует коррозии оборудования, а ее употребление оказывает негативное влияние на здоровье человека.
Бытовая водоподготовка
Контроль над соответствием требованиям безопасности параметров подаваемой населению воды давно не является приоритетом для муниципальных служб. Проблему отчасти решает установка дополнительных систем очистки силами граждан. Наиболее доступное решение в этой сфере – бытовые фильтры для воды. Стоимость фильтрующего оборудования обычно невысока, а сложность их монтажа и регулярного обслуживания минимальна.
При отсутствии централизованного водоснабжения возникает однозначная необходимость в более совершенных системах очистки. В загородном доме или на даче источником воды обычно служит закрытый колодец. Даже если качество жидкости устраивает владельцев большую часть года, при сезонных повышениях уровня грунтовых вод оно может резко ухудшаться.
Оптимальным вариантом в этом случае станет установка комплексной системы фильтров, обеспечивающей достаточный уровень очистки от механических загрязнений с последующим обеззараживанием.
Особенно значимую роль играет водоподготовка для котельной частного дома или коттеджа. Возможности по очистке в этом случае ограничены соображениями безопасности: в частности, для умягчения воды и связывания солей жесткости не используются многие эффективные технологии из-за возможного пагубного влияния на здоровье людей. Стоит помнить, что установка любого дополнительного оборудования для бытового газового котла требует обязательного согласования проекта в соответствующих службах.
Промышленная водоподготовка
На сегодняшний день водоподготовка и водоочистка для нужд производства и коммунальных сетей является обязательной мерой, без которой работа системы водоснабжения попросту невозможна.
Наиболее важна обработка воды в энергетической промышленности, где жидкость выступает в роли теплоносителя. Если при простом перекачивании многие соли жесткости и другие примеси остаются в виде ионов, то нагревание и тем более испарение воды многократно повышает их активность и приводит к оседанию в виде накипи. Недостаточная очистка воды в теплоэнергетике становится причиной порчи дорогостоящего оборудования: котлов, трубопроводов, градирен.
Собственная станция очистки есть и на крупных предприятиях пищевой промышленности. Здесь требования к кондициям воды еще более жесткие, ведь она входит в состав продукции таких комбинатов.
В наименьшей степени водоподготовка значима при использовании жидкости в системах охлаждения и опрыскивания. Тем не менее, даже в этом случае необходимо проведение большинства процедур по ее очистке и обеззараживанию.
Следует учитывать тот факт, что промышленная водоподготовка включает не только методики, применяемые на производстве, но и всякую обработку жидкости с использованием оборудования соответствующего класса. К примеру, очистка воды в коммунальных сетях или хлорирование бассейнов также относятся к этой категории водоподготовки. Сюда же входит водоподготовка для коттеджа или частного дома, если они подключены к станции очистки воды дачного поселка или города.
45.
Мельчайшие частички почвы, заряженные ионами водорода H+ действуют как слабая кислота, обуславливая кислую реакцию почвы, низкий pH. Напротив, частички почвы удерживающие кальций, магний, калий и натрий обуславливают щелочную реакцию, высокий pH. Почвы становятся кислыми вследствие вытеснения ионами водорода H+ катионов кальция, магния, натрия и калия. Процесс этот обратимый, pH почвы можно повысить внесением этих элементов. Но наиболее экономично использовать кальций. Кальций наиболее экономичен для повышения pH почвы, кроме того, он является очень важным элементом питания растений, улучшает структуру почвы, делает ее рассыпчатой, гранулированной, стимулирует развитие полезных почвенных микроорганизмов, особенно бактерий обогащающих почву азотом. Подобными свойствами обладает и магний, часто эти элементы используют вместе. Внесение кальциевомагниевых соединений приводит к значительному улучшению роста растений. Внесение кальция или кальциевомагниевых соединений с целью снижения кислотности называется известкованием. Хотя термин "известь" относится к CaO (негашеная известь), известью называют и другие соединения кальция или кальция и магния.
pH - это водородный показатель, характеризующий концентрацию свободных ионов водорода в воде. Для удобства отображения был введен специальный показатель, названный рН и представляющий собой логарифм концентрации ионов водорода, взятый с обратным знаком, т.е pH = -log[H+]. Если говорить проще, то величина рН определяется количественным соотношением в воде ионов Н+ и ОН-, образующихся при диссоциации воды. Если в воде пониженное содержание свободных ионов водорода (рН>7) по сравнению с ионами ОН-, то вода будет иметь щелочную реакцию, а при повышенном содержании ионов Н+ (рН<7)- кислую. В идеально чистой дистиллированной воде эти ионы будут уравновешивать друг друга. В таких случаях вода нейтральна и рН=7. При растворении в воде различных химических веществ этот баланс может быть нарушен, что приводит к изменению уровня рН. Очень часто показатель рН путают с такими параметрами, как кислотность и щелочность воды. Важно понимать разницу между ними. Главное заключается в том, что рН - это показатель интенсивности, но не количества. То есть, рН отражает степень кислотности или щелочности среды, в то время как определение кислотности и щелочности характеризует количественное содержание в воде веществ, способных нейтрализовывать соответственно щелочи и кислоты. В качестве аналогии можно привести пример с температурой, которая характеризует степень нагрева вещества, но не количество тепла. Например, опустив руку в воду, мы можем сказать какая вода - прохладная или теплая, но при этом не сможем определить сколько в ней тепла (т.е. условно говоря, как долго эта вода будет остывать). Для чего нужно измерять pH? pH воды - один из важнейших рабочих показателей качества воды, во многом определяющих характер химических и биологических процессов, происходящих в воде. В зависимости от величины pH может изменяться скорость протекания химических реакций, степень коррозионной агрессивности воды, токсичность загрязняющих веществ и т.д. Для наглядности приведу таблицу доступности питиательных вещ-в для усвоения растениями при различных значениях рН: Оптимальная требуемая величина рН варьируется для различных систем водоочистки в соответствии с составом воды, характером материалов, применяемых в системе распределения, а также в зависимости от применяемых методов водообработки. Обычно уровень рН находится в пределах, при которых он непосредственно не влияет на потребительские качества воды. Так, в речных водах pH обычно находится в пределах 6.5-8.5, в атмосферных осадках 4.6-6.1, в болотах 5.5-6.0, в морских водах 7.9-8.3. Известно, что при низком рН вода обладает высокой коррозионной активностью, а при высоких уровнях (рН>11) вода приобретает характерную мылкость, неприятный запах, способна вызывать раздражение глаз и кожи. Именно поэтому для питьевой и хозяйственно-бытовой воды оптимальным считается уровень рН в диапазоне от 6 до 9. Для растений - от 5.5 до 6.5
46.
Качественный анализ — совокупность химических, физико-химических и физических методов, применяемых для обнаружения элементов, радикалов и соединений, входящих в состав анализируемого вещества или смеси веществ. В качественном анализе используют легко выполнимые, характерные химические реакции, при которых наблюдается появление или исчезновение окрашивания, выделение или растворение осадка, образование газа и др. Реакции должны быть как можно более селективны и высокочувствительны. Качественный анализ в водных растворах основан на ионных реакциях и позволяет обнаружить катионы или анионы. Основоположником качественного анализа считается Р.Бойль, который ввёл представление о химических элементах как о неразлагаемых основных частях сложных веществ и систематизировал все известные в его время качественные реакции.
47.
Количественный анализ — совокупность методов аналитической химии для определения количества (содержания) элементов (ионов), радикалов, функциональных групп, соединений или фаз в анализируемом объекте.
Цели количественного анализа
Количественный анализ позволяет установить элементный и молекулярный состав исследуемого объекта или содержание отдельных его компонентов.
В зависимости от объекта исследования различают неорганический и органический анализ. В свою очередь их разделяют на элементный анализ, задача которого — установить, в каком количестве содержатся элементы (ионы) в анализируемом объекте, на молекулярный и функциональный анализы, дающие ответ о количественном содержании радикалов, соединений, а также функциональных групп атомов в анализируемом объекте.
Методы количественного анализа
Классическими методами количественного анализа являются гравиметрический (весовой) анализ и титриметрический (объемный) анализ.
Физико-химический анализ — комплекс методов анализа физико-химических систем путем построения и геометрического анализа диаграмм состояния и диаграмм состав-свойство. Этот метод позволяет обнаружить существование соединений (например, медистого золота CuAu), существование которых невозможно подтвердить другими методами анализа. Первоначально исследования в области физико-химического анализа были сосредоточены на изучении зависимостей температур фазовых переходов от состава. Однако на рубеже XIX—XX веков Н. С. Курнаков показал, что любое физическое свойство системы является функцией состава, а для изучения фазового состояния можно использовать электропроводность, вязкость, поверхностное натяжение, теплоёмкость, коэффициент рефракции, упругость и другие физические свойства[1].
В основе теории физико-химического анализа лежат сформулированные Н. С. Курнаковым принципы соответствия и непрерывности. Принцип непрерывности утверждает, что если в системе не образуются новые фазы или не исчезают существующие, то при непрерывном изменении параметров системы свойства отдельных фаз и свойствасистемы в целом изменяются непрерывно. Принцип соответствия утверждает, что каждому комплексу фаз соответствует определённый геометрический образ надиаграмме состав-свойство.
Анализ химический
имеет задачей исследовать состав тел. Он разделяется на качественный и количественный А. При помощи первого убеждаются в присутствии тех элементов или соединений, которые входят в состав исследуемого вещества; с помощью второго определяетсяколичество этих составных частей в весовой единице анализируемого тела. Прежде чем приступить к количественному А., необходимо знать природу составных частей исследуемого тела, что достигается посредством качественного А. Таким образом, этот последний должен всегда предшествовать количественному А.
Качественный анализ основан на том, что при известного рода обработке в каждом химическом соединении или элементе можно вызвать своеобразные явления, реакции, свойственные только одному какому-либо телу и указывающие, таким образом, на присутствие этого тела. Известные реакции протекают одинаково для целых групп тел, тогда как для других вовсе не наступают. Таким образом, возможно выработать систематический ход А. и разбить отдельные тела на группы, а затем уже доискиваться в известной группе отдельных членов. Только придерживаясь систематического порядка при употреблении вызывающих известные реакции реактивов, возможно легко и верно достигнуть цели и притом тем легче, если не пренебрегать предварительными пробами, дающими указание на присутствие или отсутствие целых групп элементов. Поясним на примере ход качественного А. Для исследования пусть будет дано белое кристаллическое вещество. Предварительные А. пробы следующие: нагревают слегка небольшую порцию вещества в стеклянной трубочке; оно при этом плавится, не образуя налета от влажности; нет также возгона и нет выделения угля; отсюда надо вывести заключение, что мы имеем дело с безводным неорганическим соединением, не содержащим аммиачных и ртутных солей. Другую пробу нагревают на куске угля в пламени паяльной трубки: она плавится, всасывается углем и, попадая на раскаленные места его, производит вспышку, что указывает на присутствие азотнокислых, хлорновато— и бромноватокислых солей. Третью пробу вещества смешивают с безводной содой и нагревают в небольшом углублении на угле в пламени паяльной трубки: вещество при плавлении пенится и оставляет небольшой металлический королек, не образуя на угле налета, откуда, во-первых, можно заключить, что мы имеем дело с металлом, отсутствие же налета указывает на отсутствие свинца, кадмия, цинка, висмута, сурьмы и олова, кроме того, вещество легко растворимо в воде. Теперь приступают собственно к исследованию: водный раствор вещества смешивают с сернистоводородной водой; появляется обильный черный осадок. Возможно присутствие: свинца, серебра, меди, висмута, кадмия, ртути, мышьяка, сурьмы, олова, золота, платины, молибдена, вольфрама; предварительными пробами уже показано отсутствие ртути и других названных с нею металлов. В жидкость пропускают сероводород до тех пор, пока она не будет явственно им пахнуть; черный осадок собирают на фильтр и промывают водой; часть жидкости выпаривают досуха в фарфоровой чашке; при этом не остается никакого остатка, что указывает на то, что, кроме осаждающихся сероводородом металлов, в анализируемом веществе нет никаких нелетучих тел. Полученный при действии сероводорода черный осадок обрабатывают сернистым аммонием, в котором растворимы сернистые соединения мышьяка, сурьмы, олова, золота, платины, вольфрама и молибдена; так как осадок не изменяет своего наружного вида, а отфильтрованная от него жидкость не дает никакого осадка при нейтрализовании соляной кислотой, то это указывает на отсутствие всех только что упомянутых элементов. Могут быть, следовательно, даны: свинец, серебро, медь, висмут, кадмий и ртуть, но все они, за исключением серебра и меди, могли бы быть открыты при предварительных пробах, а потому мы имеем дело с серебром или с медью или же со смесью этих обоих металлов. Черный осадок промывают водой и нагревают с крепкой азотной кислотой, при чем он растворяется с выделением серы. Последнюю удаляют фильтрованием, а избыток азотной кислоты — выпариванием досуха и получают таким образом совершенно белый сухой остаток, цвет которого уже указывает на отсутствие меди, потому что малейшие количества ее окрасили бы остаток в голубовато-зеленый цвет. Так как таким образом доказано отсутствие всех прочих металлов, то в данном веществе может находиться только серебро. Чтобы доказать присутствие его, растворяют белый остаток в нескольких каплях воды и прибавляют в раствор каплю соляной кислоты, при чем образуется обильный белый, творожистый осадок хлористого серебра, легко растворимый в аммиаке. Для анализа, таким образом, была дана серебряная соль, и остается еще определить содержащуюся в ней кислоту, при чем поступают подобным же образом, как только что было описано.
48.
ВАЛЕНТНОСТЬ (от лат. valentia - сила), способность атома присоединять или замещать определенное число др. атомов или атомных групп с образованием хим. связи. Количеств. мерой валентности атома элемента Э служит число атомов водорода или кислорода (эти элементы принято считать соотв. одно- и двухвалентными), к-рые Э присоединяет, образуя гидрид ЭНx или оксид ЭnОm. Валентность элемента м.б. определена и по др. атомам с известной валентностью. В разл. соединениях атомы одного и того же элемента могут проявлять разл. валентности. Так, сера двухвалентна в H2S и CuS, четырехвалентна в SO2 и SF4, шестивалентна в SO3 и SF6. До развития электронных представлений о строении в-ва валентность трактовалась формально. В рамках электронной теории химической связи валентность атома определяется числом его неспаренных электронов в основном или возбужденном состоянии, участвующих в образовании общих электронных пар с электронами др. атомов. Поскольку электроны внутр. оболочек атома не участвуют в образовании хим. связей, макс. валентность элемента считают равной числу электронов во внеш. электронной оболочке атома. Макс. валентность элементов одной и той же группы периодич. системы обычно соответствует ее порядковому номеру. Напр., макс. валентность атома С должна быть равной 4, С1 - 7. Электростатич. теория хим. связи привела к формулировке близкого к валентности и дополняющего ее понятия степени окисления (окислит. числа), соответствующей заряду, к-рый приобрел бы атом, если бы все электронные пары его хим. связей сместились в сторону более электроотрицат. атомов. При этом электронные пары, обобщенные одинаковыми атомами, делятся пополам. По знаку степень окисления, как правило, совпадает с экспериментально определяемымэффективным зарядом атома, но численно намного превышает его. Напр., степень окисления серы в SO3 равна +6, а ее эффективный заряд - ок. + 2.
Развитие
представлений о донорно-акцепторных
связях (см. Координационная
связь), водородной
связи,
обнаружение новых классов хим. соед.
(мостиковых электронодефицитных,
металлоорг., сэндвичевых,
металлич. кластеров,![]() комплексов),
а также исследования кристаллич. структур
показали ограниченность правил
определения формальной валентности.
Было найдено, что координац. числаатомов могут
превышать их формальную валентность.
Так, атом С
в карбонильном кластере [CFe5(CO)15]
связан с 5 атомами Fe,
ванионе [ССо8(СО)18]2- -
с 8 атомами Со.
Хим. связь в подобных соед. не м. б. описана
с помощью представлений о двух-электронных
двухцентровых связях и обусловлена
образованием много центровой связи.
комплексов),
а также исследования кристаллич. структур
показали ограниченность правил
определения формальной валентности.
Было найдено, что координац. числаатомов могут
превышать их формальную валентность.
Так, атом С
в карбонильном кластере [CFe5(CO)15]
связан с 5 атомами Fe,
ванионе [ССо8(СО)18]2- -
с 8 атомами Со.
Хим. связь в подобных соед. не м. б. описана
с помощью представлений о двух-электронных
двухцентровых связях и обусловлена
образованием много центровой связи.
Термин "валентность" введен в 60-х гг. 19 в. На представлениях о валентности была основана классич. теория хим. строения A.M. Бутлерова. В совр. теории хим. строения представления о валентности часто отождествляют с общим учением о хим. связи.
Степенью окисления атома называется фактическая либо кажущаяся величина электростатического заряда атома в простом веществе, в молекуле химического соединения, в ионе.
Примеры степеней окисления атома кислорода (O) в молекулах H2O, H2O2, KO2, KO3, O2, O3F2, O2F2 и OF2
Q(O in H2O) = −2, то есть кажущаяся величина электростатического заряда атома кислорода в молекуле воды равна −2.
Q(O in H2O2) = −1, то есть кажущаяся величина электростатического заряда каждого атома кислорода в молекуле пероксида водорода равна −1.
Q(O in KO2) = −1/2, то есть кажущаяся величина электростатического заряда каждого атома кислорода в молекуле надпероксида калия равна −1/2.
Q(O in KO3) = −1/3, то есть кажущаяся величина электростатического заряда каждого атома кислорода в молекуле озонида калия равна −1/3.
Q(O in O2) = 0, то есть фактическая величина электростатического заряда каждого атома кислорода в молекуле кислорода равна 0.
Q(O in O3F2) = 2/3, то есть кажущаяся величина электростатического заряда каждого атома кислорода в молекуле триоксидифторида равна 2/3.
Q(O in O2F2) = 1, то есть кажущаяся величина электростатического заряда каждого атома кислорода в молекуле диоксида фтора равна 1.
Q(O in OF2) = 2, то есть кажущаяся величина электростатического заряда атома кислорода в молекуле оксида фтора равна 2.
ТИПЫ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ РЕАКЦИЙ
В реакциях межмолекулярного окисления-восстано
вления степени окисления изменяют атомы, входящие в состав различных соединений:
![]()
![]()
К реакциям внутримолекулярного окисления-восстановления относят те, в которых степени окисления изменяют атомы различных элементов, входящих в состав молекул одного и того же вещества:
![]()
![]()
Для реакций диспропорционирования (самоокисления-самовосстановления) характерно одновременное повышение и понижение степеней окисления атомов одного и того же элемента:
![]()
![]()
возможно и обратное диспропорционирование:
![]()
49.
ГЕТЕРОГЕННЫЕ РЕАКЦИИ, хим. р-ции с участием в-в, находящихся в разл. фазах и составляющих в совокупности гетерог. систему. Типичные гетерогенные реакции: термич. разложение солей с образованием газообразных и твердых продуктов (напр., СаСО3 -> СаО + СО2), восстановление ожсидов металлов водородом или углеродом (напр., РЬО + С -> Рb + СО), растворение металлов в к-тах (напр., Zn + + H2SO4 -> ZnSO4 + Н2), взаимод. твердых реагентов (А12О3 + NiO -> NiAl2O4). В особый класс выделяют гетерогенно-каталитич. р-ции, протекающие на пов-сти катализатора; при этом реагенты и продукты могут и не находиться в разных фазах. Напр., при р-ции N2 + + ЗН2 -> 2NH3, протекающей на пов-сти железного кат., реагенты и продукт р-ции находятся в газовой фазе и образуют гомог. систему.
Особенности гетерогенных реакций обусловлены участием в них конденсированных фаз. Это затрудняет перемешивание и транспортреагентов и продуктов; возможна активация молекул реагентов на пов-сти раздела фаз. Кинетика любой гетерогенной реакцииопределяется как скоростью самого хим. превращения, так и процессами переноса (диффузией), необходимыми для восполнения расхода реагирующих в-в и удаления из реакц. зоны продуктов р-ции. В отсутствие диффузионных затруднений скорость гетерогеннойреакции пропорциональна размерам реакц. зоны; т. наз. удельная скорость р-ции, рассчитанная на единицу пов-сти (или объема) реакц. зоны, не изменяется во времени; для простых (одностадийных) р-ций она м.б. определена на основе действующих масс закона. Этот закон не выполняется, если диффузия в-в протекает медленнее, чем хим. р-ция; в этом случае наблюдаемая скорость гетерогенной реакции описывается ур-ниями диффузионной кинетики (см. Макрокинетика).
При гетерогенных реакциях с участием одного или неск. твердых реагентов часто образуются твердофазные продукты. Такие р-ции, как правило, локализованы на пов-сти раздела фаз или в поверхностном слое и обычно протекают нестационарно. Они характеризуютсяпериодом индукции, в течение к-рого возникают зародыши (ядра) новой фазы. Их образование связано с перестройкой атомной структуры твердого реагента и требует затраты энергии. Поэтому такие гетерогенные реакции чувствительны ко всем нарушениям структуры, облегчающим образование зародышей, и м. б. активированы термич., радиац., мех. и др. воздействиями, увеличивающимиконцентрацию дефектов, в первую очередь плотность дислокаций (см. Дефекты в кристаллах). Кинетич. ур-ние р-ции в этом случае отражает изменение во времени не только концентраций реагирующих в-в, но и пов-сти раздела твердых фаз реагента и продукта: по мере роста зародышей пов-сть раздела увеличивается и скорость р-ции сначала возрастает, затем проходит через максимум и снижается вследствие соприкосновения растущих зародышей и образования сплошного слоя твердого продукта (подробнее см.Топохимические реакции).
В природе гетерогенные реакции входят в комплекс процессов, приводящих к образованию осадочных пород и выветриванию. В хим. технологии гетерогенные реакции газа с жидкостью (окисление воздухом, кислородом, озоном; хлорирование и др.) обычно проводят при интенсивном перемешивании спец. мех. устройствами или самим газом (в т. наз. барботажном реакторе). Р-ции термич. разложения составляют основу фотографич. процесса, р-ции между газами или жидкостями и твердыми в-вами-основу обжига,восстановления и окисления металлов, горения, произ-ва твердых катализаторов, выщелачивания, экстракции и др. Часто сочетаются гетерогенные реакции в трех-и многофазных системах, напр. хлорирование твердых оксидов металлов газообразным хлором в присут. твердого углеродсодержащего восстановителя. Важная область использования гетерогенных реакций-получение тонких поверхностных слоев и покрытий при взаимод. твердого тела с жидкостью. При низких т-рах диффузия в глубь твердого материала протекает медленно, что позволяет получать стабильные тонкие поверхностные слои, а в отдельных случаях-двухмерные фазы, толщина к-рых по порядку величины близка к параметру кристаллич. решетки. Иногда стабильные поверхностные слои образуются самопроизвольно; таковы защитные оксидные пленки на металлах, препятствующие дальнейшему окислению (см. Газовая коррозия).
ЭЛЕКТРОДНЫЙ
ПОТЕНЦИАЛ,
разность электростатич. потенциалов
между электродом и
находящимся с ним в контакте электролитом.
Возникновение электродного потенциала
обусловлено пространств. разделением
зарядов противоположного знака на
границе раздела фаз и образованием
двойного электрического слоя.
На границе между металлич. электродом и
р-ром электролита пространств.
разделение зарядов связано со след.
явлениями: переносом ионов из металла в
р-р в ходе установления электрохим. равновесия,
кулоновской адсорбцией ионов из
р-ра на пов-сть металла,
смещением электронного газа за
пределы положительно заряженного
ионного остова кристаллич. решетки,
специфич. (некулоновской) адсорбцией ионов или полярных
молекул р-рителя
на электроде и
др. Последние два явления приводят к
тому, что электродный потенциал не равен
нулю даже при условиях, когда заряд
пов-сти металларавен
нулю (см. Потенциал
нулевого заряда).
Абс.
величину электродного потенциала
отдельного электрода определить
невозможно, поэтому измеряют всегда
разность потенциалов исследуемого электрода и
нек-рого стандартного электрода
сравнения.
Электродный потенциал равен эдс
электрохим. цепи, составленной из
исследуемого и стандартного электродов (диффузионный
потенциал между
разными электролитами,
обусловленный различием скоростей
движения ионов,
при этом должен быть устранен). Для
водных р-ров в качестве
стандартногоэлектрода обычно
используют водородный
электрод (Pt,
Н2[0,101
МПа] | Н+[a=
1]), потенциал к-рого при давлении водорода 0,101
МПа и термодинамич. активности а ионов Н+ в
р-ре, равной 1, принимают условно равным
нулю (водородная шкала электродных
потенциалов). При схематич. изображении
цепи водородный
электрод всегда
записывают слева; напр., потенциал
медного электрода в
р-ре соли меди равен
эдс цепи Pt, H2|HCl![]() CuCl2|Cu|Pt
(две штриховые черты означают,
что диффузионный
потенциал на
фанице НС1 и СuС12 устранен).
Если
исследуемый электрод находится
в стандартных условиях,
когда активности всех ионов,
определяющих электродный потенциал,
равны 1, а давление газа (для
газовых электродов)
равно 0,101 МПа, значение электродного
потенциала наз. стандартным (обозначение
E°).
Оно связано со стандартным
изменением энергии
Гиббса
CuCl2|Cu|Pt
(две штриховые черты означают,
что диффузионный
потенциал на
фанице НС1 и СuС12 устранен).
Если
исследуемый электрод находится
в стандартных условиях,
когда активности всех ионов,
определяющих электродный потенциал,
равны 1, а давление газа (для
газовых электродов)
равно 0,101 МПа, значение электродного
потенциала наз. стандартным (обозначение
E°).
Оно связано со стандартным
изменением энергии
Гиббса![]() и константой
равновесия Кр электрохим.
р-ции ур-нием:
и константой
равновесия Кр электрохим.
р-ции ур-нием:![]() ,
где F - число Фарадея; п - число электронов,
участвующих в р-ции; R - газовая
постоянная;
Т - абс. т-ра. Значения E° электрохим.
систем по отношению к водородному
электроду и
протекающие на электродах р-ции
сведены в спец. таблицы (подробнее
см. Стандартный
потенциал).
Зависимость
электродного потенциала от
термодинамич. активностей ai участников
электрохим. р-ции выражается Нернста
уравнением:
,
где F - число Фарадея; п - число электронов,
участвующих в р-ции; R - газовая
постоянная;
Т - абс. т-ра. Значения E° электрохим.
систем по отношению к водородному
электроду и
протекающие на электродах р-ции
сведены в спец. таблицы (подробнее
см. Стандартный
потенциал).
Зависимость
электродного потенциала от
термодинамич. активностей ai участников
электрохим. р-ции выражается Нернста
уравнением:
![]()
где vi - стехиометрич. коэф. участника р-ции, причем для исходных в-в это отрицат. величина, а для продуктов р-ции -положительная. Если через электрод протекает электрич. ток, электродный потенциал отклоняется от равновесного значения из-за конечной скорости процессов, происходящих непосредственно на границе электрод - электролит (см. Поляризация).
50.
Химические источники тока, устройства, вырабатывающие электрическую энергию за счёт прямого преобразования химической энергии окислительно-восстановительных реакций. Первые химические источники тока созданы в 19 в. (Вольтов столб, 1800; элемент Даниела — Якоби, 1836; Лекланше элемент, 1865, и др.). До 60-х гг. 19 в. химические источники тока были единственными источниками электроэнергии для питания электрических приборов и для лабораторных исследований. Основу химических источников тока составляют два электрода (один — содержащий окислитель, другой — восстановитель), контактирующие с электролитом. Междуэлектродами устанавливается разность потенциалов — электродвижущая сила (эдс), соответствующая свободной энергииокислительно-восстановительной реакции. Действие химических источников тока основано на протекании при замкнутой внешней цепи пространственно разделённых процессов: на отрицательном электроде восстановитель окисляется, образующиеся свободныеэлектроны переходят по внешней цепи (создавая разрядный ток) к положительному электроду, где участвуют в реакции восстановленияокислителя.
В зависимости от эксплуатационных особенностей и от электрохимической системы (совокупности реагентов и электролита) химические источники тока делятся на гальванические элементы (обычно называются просто элементами), которые, как правило, после израсходования реагентов (после разрядки) становятся неработоспособными, и аккумуляторы, в которых реагенты регенерируются при зарядке — пропускании тока от внешнего источника (см. Зарядное устройство). Такое деление условно, т.к. некоторые элементы могут быть частично заряжены. К важным и перспективным химическим источникам тока относятся топливные элементы (электрохимические генераторы), способные длительно непрерывно работать за счёт постоянного подвода к электродам новых порций реагентов и отвода продуктов реакции. Конструкция резервных химических источников тока позволяет сохранять их в неактивном состоянии 10—15 лет (см. также Источники тока).
С начала 20 в. производство химических источников тока непрерывно расширяется в связи с развитием автомобильного транспорта, электротехники, растущим использованием радиоэлектронной и др. аппаратуры с автономным питанием. Промышленность выпускает химические источники тока, в которых преимущественно используются окислители PbO2, NiOOH, MnO2 и др., восстановителями служат Pb, Cd. Zn и др. металлы, а электролитами — водные растворы щелочей, кислот или солей (см., например, Свинцовый аккумулятор).
Основные характеристики ряда химических источников тока приведены в табл. Лучшие характеристики имеют разрабатываемые химические источники тока на основе более активных электрохимических систем. Так, в неводных электролитах (органическихрастворителях, расплавах солей или твёрдых соединениях с ионной проводимостью) в качестве восстановителей можно применятьщелочные металлы (см. также Расплавные источники тока). Топливные элементы позволяют использовать энергоёмкие жидкие или газообразные реагенты.
51.
Электро́лиз — физико-химический процесс, состоящий в выделении на электродах составных частей растворённых веществ или других веществ, являющихся результатом вторичных реакций на электродах, который возникает при прохождении электрического тока через раствор либо расплав электролита.
Электролиз - это ещё один способ получения чистых металлов и неметаллов. Кроме того, электролиз можно провести и в домашних условиях. Нужен источник тока, два электрода (какие электроды бывают и какой в каком случае брать - расссказано дальше) и, конечно, электролит. Электролит - это раствор, который проводит электрический ток.
Различают электролиз растворов и электролиз расплавов. Оба эти процесса существенно отливчаются друг от друга. Отличие - в наличии растворителя. При электрролизе растворов кроме ионов самого вещества в процессе учавствуют ионы растворителя. При электролизе расплавов - только ионы самого вещества. Для того, чтобы получить нужный продукт (газ, металл или неметалл), нужно правильно выбрать электрод и раствор электролита. Электродами могут служить любые материалы, проводящие электрический ток. В основном применяют металлы и сплавы, из неметаллов электродами могут служить, например, графитовые стержни (или углерод). Реже в качестве электрода используют жидкости. Электрод, заряженный положительно - анод. Электрод, заряженный отрицательно - катод. При электролизе происходт окисление анода (он растворяется) и восстановление катода. Именно поэтому анод следует брать таким, чтобы его растворение не повлияло на химический процесс, протекающий в растворе или расплаве. Такой анод называют инертным электродом. В качестве инертного анода можно взять графит (углерод) или платину
Применение электролиза для обработки поверхностей включает как катодные процессы гальванотехники (в машиностроении, приборостроении, авиационной, электротехнической, электронной промышленности), так и анодные процессы полировки, травления, размерной анодно-механической обработки, оксидирования (анодирования) металлических изделий (см. также Электрофизические и электрохимические методы обработки). Путём электролиза в контролируемых условиях осуществляют защиту от коррозии металлических сооружений и конструкций (анодная и катодная защита).
52.
ФАРАДЕЯ
ЗАКОНЫ,
основные законы электролиза,
отражающие общий закон сохранения в-ва
в условиях протекания злектрохим. р-ции.
Установлены M. Фарадеем в 1833-34. Согласно
1-му закону, масса в-ва т, прореагировавшего
в процессе электролиза,
прямо пропорциональна силе тока I и
времени электролиза t,
т. е. кол-ву пропущенного электричества
Q = It (предполагается, что I не зависит от
t; в противном случае масса т
пропорциональна![]() где
t1 и
t2 -
моменты включения и выключения тока).
Согласно 2-му закону, для разных электродных
процессов при
одинаковом кол-ве пропущенного
электричества Q массы прореагировавших
в-в относятся друг к другу так же,
как эквиваленты
химические этих
в-в. Оба Фарадея закона объединяются
одним ур-нием:
где
t1 и
t2 -
моменты включения и выключения тока).
Согласно 2-му закону, для разных электродных
процессов при
одинаковом кол-ве пропущенного
электричества Q массы прореагировавших
в-в относятся друг к другу так же,
как эквиваленты
химические этих
в-в. Оба Фарадея закона объединяются
одним ур-нием:
![]()
где M - мол. м. в-ва, участвующего в электролизе, z - число элементарных зарядов, соответствующее превращению одной молекулыэтого в-ва, 1/F- коэф. пропорциональности, общий для всех в-в, F - Фарадея постоянная, равная 96484,56 Кл/моль.
Фарадея
законы относятся к числу строгих законов,
но в ряде случаев могут наблюдаться
кажущиеся отклонения от них, вызываемые
след. причинами: 1) в нестационарных
условиях электролиза часть
электричества затрачивается на
заряжение двойного
электрического слоя;
2) если электролит обладает
электронной проводимостью (напр., р-р
металлич. Na в жидком аммиаке),
то часть тока черезэлектролит переносят электроны,
а не ионы,
и соответствующее кол-во электричества
не участвует в процессе электролиза;
3) наряду с основным процессом электролиза,
напр, образованием металлич. Zn по р-ции
Zn2+ +
2е![]() Zn,
часть тока может затрачиваться на
протекание параллельных электрохим.
р-ций, напр.: 2H3O+ +
2е = H2 +
2H2O;
O2 +
4е + 4H3O+ =
6H2O.
Системы, в к-рых полностью исключены
указанные причины кажущихся отклонений
от Фарадея законов, получили назв.
кулонометров; их использование позволяет
по кол-ву образовавшихся
продуктов электролиза точно
определить кол-во пропущенного
электричества. В кулонометрах обычно
применяют электрохим. р-ции Ag+ +
е = Ag или 3I- =
I3- +
2е.
Zn,
часть тока может затрачиваться на
протекание параллельных электрохим.
р-ций, напр.: 2H3O+ +
2е = H2 +
2H2O;
O2 +
4е + 4H3O+ =
6H2O.
Системы, в к-рых полностью исключены
указанные причины кажущихся отклонений
от Фарадея законов, получили назв.
кулонометров; их использование позволяет
по кол-ву образовавшихся
продуктов электролиза точно
определить кол-во пропущенного
электричества. В кулонометрах обычно
применяют электрохим. р-ции Ag+ +
е = Ag или 3I- =
I3- +
2е.
Фарадея законы сыграли важную роль в понимании природы хим. связи и развития атомно-молекулярной теории. Их используют при выводе всех ур-ний, описывающих электрохим. превращения B-B на границах раздела проводников 1-го и 2-го рода (см.Электрохимическая кинетика). Практич. применение Фарадея законы находят в кулонометрии, а также при определении выхода р-ции по току, т.е. отношения теоретич. кол-ва электричества, рассчитанного на основе Фарадея законов, к кол-ву электричества, реально затраченному на получение данного в-ва в процессе электролиза.
Поляризация электрохимическая, отклонение электродного потенциала Е от стационарного потенциала Ест, который электродприобретает в отсутствие внешнего тока. Поляризация электрохимическая измеряется в вольтах (милливольтах). Если отклонение отрицательно (вызвано подводом электронов, которые должны расходоваться в реакциях, идущих в катодном направлении), тополяризацию электрохимическую называют катодной; при противоположном направлении тока — анодной. Графики функциональной связи между поляризацией электрохимической и плотностью тока i называют соответственно катодными и анодными поляризационными кривыми и широко используют при описании и исследовании электрохимических и коррозионных процессов.
В общем случае связь между i и поляризацией электрохимической криволинейна, однако в интервале отклонений ± 10—15 мв от Естона, как правило, прямолинейна. Угловой коэффициент этого участка (т. е. отношение приращения поляризации электрохимической к приращению i) имеет размерность сопротивления единицы поверхности (омсм2) и называется поляризационным сопротивлениемэлектрода Rп. Электроды с большим Rп называются сильнополяризуемыми, т.к. уже при очень малых i их потенциалы сильно отклоняются от Ест. Электроды с малым Rп — слабополяризуемые. Существует обратная пропорциональность между Rп и интенсивностью того обмена электрическими зарядами, который происходит между электродом и электролитом при Ест. На коррелирующем электроде эта интенсивность обычно совпадает с плотностью коррозионного тока, и потому измерение Rп иногда используют для определения скорости электрохимической коррозии. Если на электроде возможна лишь одна электродная реакция, то Ест совпадает с равновесным потенциалом Ер этой реакции, поляризация электрохимическая — с её перенапряжением, a Rпоказывается обратно пропорциональным равновесному току обмена.
Термином «концентрационная поляризация» обозначают те изменения Е, которые связаны с замедленным переносом исходных или конечных компонентов протекающей на электроде реакции. В зоне реакции концентрация первых (сисх) понижается, а вторых (скон) — увеличивается. Это повышает тенденцию реакции протекать в обратном направлении, что и должно компенсироваться приложением дополнительной разности потенциалов. Последняя особенно резко растет, когда скорость реакции достигает предельно возможной скорости диффузионных потоков, так что либо сисх снижается практически до 0, либо конечные продукты кристаллизуются, закрывая электродную поверхность. Эту предельную диффузионную плотность тока можно повысить, улучшив массоперенос, например, путёмперемешивания. Вместо термина «концентрационная поляризация» также пользуются термином «концентрационноеперенапряжение», т.к. обозначаемое им отклонение Е должно фактически отсчитываться не от Ест, а от Ер соответствующей индивидуальной реакции.
Явления поляризации электрохимической могут быть и вредны, и полезны. Например, при электролизе они повышают расход электроэнергии, а при работе гальванического элемента понижают отдачу электроэнергии; зато при коррозии могут вести к торможению нежелательных процессов. См. также ст. Пассивирование.
Перенапряжение электрохимическое, отклонение электродного потенциала от его равновесного (по отношению к приэлектродному составу раствора) термодинамического значения при поляризации электрода внешним током. При заметном удалении от равновесияперенапряжение () и плотность поляризующего тока (i) обычно связаны соотношением = а + b lg i (уравнение Тафеля), где а и b — эмпирические постоянные. Перенапряжение зависит от температуры, природы электродного материала и состава раствора. Перенапряжение необходимо для ускорения нужной электродной реакции. Если скорость электродной реакции в целом определяется скоростью собственно электрохимической стадии, связанной с переносом заряда, то перенапряжение усиливает электрическое поле, действующее на разряжающиеся частицы, благодаря чему снижается энергия активации разряда. Поскольку электрическое поле в значительной степени обусловлено строением двойного электрического слоя, перенапряжение оказывается зависящим отконцентрации постороннего электролита и адсорбирующихся веществ, влияющих на распределение потенциала в двойном слое. На повышении перенапряжения основано действие многих ингибиторов коррозии металлов (см. Ингибиторы химические), что является одной из положительных сторон перенапряжения. В то же время перенапряжение в промышленном электролизе, неизбежно связанное с дополнительным расходом энергии, приводит к увеличению себестоимости продукции.
53.
Коррозия металлов
Коррозия металлов — разрушение металлов вследствие химического или электрохимического взаимодействия их с коррозионной средой.[2] Для процесса коррозии следует применять термин «коррозионный процесс», а для результата процесса — «коррозионное разрушение». Образование гальванических пар с пользой применяют для создания батарей и аккумуляторов. С другой стороны, образование такой пары приводит к неблагоприятному процессу, жертвой которого становится целый ряд металлов, — коррозии. Под коррозией понимают происходящее на поверхности электрохимическое или химическое разрушение металлического материала. Наиболее часто при коррозии металл окисляется с образованием ионов металла, которые при дальнейших превращениях дают различные продукты коррозии. Коррозия может быть вызвана как химическим, так и электрохимическим процессом. Соответственно, различают химическую и электрохимическую коррозию металлов.
[править]Типы коррозии
[править]Электрохимическая коррозия
Разрушение металла под воздействием возникающих в коррозионной среде гальванических элементов называют электрохимической коррозией. Не следует путать с электрохимической коррозией коррозию однородного материала, например, ржавление железа или т. п. При электрохимической коррозии (наиболее частая форма коррозии) всегда требуется наличие электролита (Конденсат, дождевая вода и т. д.), с которым соприкасаются электроды — либо различные элементы структуры материала, либо два различных соприкасающихся материала с различающимися окислительно-восстановительными потенциалами. Если в воде растворены ионы солей, кислот, или т. п., электропроводность её повышается, и скорость процесса увеличивается.
![]()
Коррозионный элемент
При соприкосновении двух металлов с различными окислительно-восстановительными потенциалами и погружении их в раствор электролита, например, дождевой воды с растворенным углекислым газом CO2, образуется гальванический элемент, так называемый коррозионный элемент. Он представляет собой не что иное, как замкнутую гальваническую ячейку. В ней происходит медленное растворение металлического материала с более низким окислительно-восстановительным потенциалом; второй электрод в паре, как правило, не корродирует. Этот вид коррозии особо присущ металлам с высокими отрицательными потенциалами. Так, совсем небольшого количества примеси на поверхности металла с большим редокспотенциалом уже достаточно для возникновения коррозионного элемента. Особо подвержены риску места соприкосновения металлов с различными потенциалами, например, сварочные швы или заклёпки.
Если растворяющийся электрод коррозионно-стоек, процесс коррозии замедляется. На этом основана, например, защита железных изделий от коррозии путём оцинковки — цинк имеет более отрицательный потенциал, чем железо, поэтому в такой паре железо восстанавливается, а цинк должен корродировать. Однако в связи с образованием на поверхности цинка оксидной плёнки процесс коррозии сильно замедляется.
Водородная и кислородная коррозия
Если происходит восстановление ионов H3O+ или молекул воды H2O, говорят о водородной коррозии или коррозии с водородной деполяризацией. Восстановление ионов происходит по следующей схеме:
2H3O+ + 2e− → 2H2O + H2
или
2H2O + 2e− → 2OH− + H2
Если водород не выделяется, что часто происходит в нейтральной или сильно щелочной среде, происходит восстановление кислорода и здесь говорят о кислородной коррозии или коррозии с кислородной деполяризацией:
O2 + 2H2O + 4e− → 4OH−
Коррозионный элемент может образовываться не только при соприкосновении двух различных металлов. Коррозионный элемент образуется и в случае одного металла, если, например, структура поверхности неоднородна.
[править]Химическая коррозия
Химическая коррозия — взаимодействие поверхности металла с коррозионно-активной средой, не сопровождающееся возникновением электрохимических процессов на границе фаз. В этом случае взаимодействия окисление металла и восстановление окислительного компонента коррозионной среды протекают в одном акте. Например, образование окалины при взаимодействии материалов на основе железа при высокой температуре с кислородом:
4Fe + 3O2 → 2Fe2O3
При электрохимической коррозии ионизация атомов металла и восстановление окислительного компонента коррозионной среды протекают не в одном акте и их скорости зависят от электродного потенциала металла (например, ржавление стали в морской воде).
Коррозионные процессы отличаются широким распространением и разнообразием условий и сред, в которых они протекают. Поэтому пока нет единой и всеобъемлющей классификации встречающихся случаев коррозии.
По типу агрессивных сред, в которых протекает процесс разрушения, коррозия может быть следующих видов:
газовая коррозия;
атмосферная коррозия;
коррозия в неэлектролитах;
коррозия в электролитах;
подземная коррозия;
биокоррозия;
коррозия под воздействием блуждающих токов.
По условиям протекания коррозионного процесса различаются следующие виды:
контактная коррозия;
щелевая коррозия;
коррозия при неполном погружении;
коррозия при полном погружении;
коррозия при переменном погружении;
коррозия при трении;
межкристаллитная коррозия;
коррозия под напряжением.
По характеру разрушения:
сплошная коррозия, охватывающая всю поверхность:
равномерная;
неравномерная;
избирательная[1];
локальная (местная) коррозия, охватывающая отдельные участки:
пятнами;
язвенная;
точечная (или питтинг);
сквозная;
межкристаллитная (расслаивающая в деформированных заготовках и ножевая в сварных соединениях).
Главная классификация производится по механизму протекания процесса. Различают два вида:
химическую коррозию;
электрохимическую коррозию.
54.
Электрохимическая коррозия
Разрушение металла под воздействием возникающих в коррозионной среде гальванических элементов называют электрохимической коррозией. Не следует путать с электрохимической коррозией коррозию однородного материала, например, ржавление железа или т. п. При электрохимической коррозии (наиболее частая форма коррозии) всегда требуется наличие электролита (Конденсат, дождевая вода и т. д.), с которым соприкасаются электроды — либо различные элементы структуры материала, либо два различных соприкасающихся материала с различающимися окислительно-восстановительными потенциалами. Если в воде растворены ионы солей, кислот, или т. п., электропроводность её повышается, и скорость процесса увеличивается.
При соприкосновении двух металлов с различными окислительно-восстановительными потенциалами и погружении их в раствор электролита, например, дождевой воды с растворенным углекислым газом CO2, образуется гальванический элемент, так называемый коррозионный элемент. Он представляет собой не что иное, как замкнутую гальваническую ячейку. В ней происходит медленное растворение металлического материала с более низким окислительно-восстановительным потенциалом; второй электрод в паре, как правило, не корродирует. Этот вид коррозии особо присущ металлам с высокими отрицательными потенциалами. Так, совсем небольшого количества примеси на поверхности металла с большим редокспотенциалом уже достаточно для возникновения коррозионного элемента. Особо подвержены риску места соприкосновения металлов с различными потенциалами, например, сварочные швы или заклёпки.
Если растворяющийся электрод коррозионно-стоек, процесс коррозии замедляется. На этом основана, например, защита железных изделий от коррозии путём оцинковки — цинк имеет более отрицательный потенциал, чем железо, поэтому в такой паре железо восстанавливается, а цинк должен корродировать. Однако в связи с образованием на поверхности цинка оксидной плёнки процесс коррозии сильно замедляется.
Водородная и кислородная коррозия
Если происходит восстановление ионов H3O+ или молекул воды H2O, говорят о водородной коррозии или коррозии с водородной деполяризацией. Восстановление ионов происходит по следующей схеме:
2H3O+ + 2e− → 2H2O + H2
или
2H2O + 2e− → 2OH− + H2
Если водород не выделяется, что часто происходит в нейтральной или сильно щелочной среде, происходит восстановление кислорода и здесь говорят о кислородной коррозии или коррозии с кислородной деполяризацией:
O2 + 2H2O + 4e− → 4OH−
Коррозионный элемент может образовываться не только при соприкосновении двух различных металлов. Коррозионный элемент образуется и в случае одного металла, если, например, структура поверхности неоднородна.
Химическая коррозия
Химическая коррозия — взаимодействие поверхности металла с коррозионно-активной средой, не сопровождающееся возникновением электрохимических процессов на границе фаз. В этом случае взаимодействия окисление металла и восстановление окислительного компонента коррозионной среды протекают в одном акте. Например, образование окалины при взаимодействии материалов на основе железа при высокой температуре с кислородом:
4Fe + 3O2 → 2Fe2O3
При электрохимической коррозии ионизация атомов металла и восстановление окислительного компонента коррозионной среды протекают не в одном акте и их скорости зависят от электродного потенциала металла (например, ржавление стали в морской воде).
55.
Коррозия приводит ежегодно к миллиардным убыткам, и разрешение этой проблемы является важной задачей. Основной ущерб, причиняемый коррозией, заключается не в потере металла как такового, а в огромной стоимости изделий, разрушаемых коррозией. Вот почему ежегодные потери от неё в промышленно развитых странах столь велики. Истинные убытки от неё нельзя определить, оценив только прямые потери, к которым относятся стоимость разрушившейся конструкции, стоимость замены оборудования, затраты на мероприятия по защите от коррозии. Ещё больший ущерб составляют косвенные потери. Это простои оборудования при замене прокорродировавших деталей и узлов, утечка продуктов, нарушение технологических процессов.
Идеальная защита от коррозии на 80 % обеспечивается правильной подготовкой поверхности, и только на 20 % качеством используемых лакокрасочных материалов и способом их нанесения.[3]. Наиболее производительным и эффективным методом подготовки поверхности перед дальнейшей защитой субстрата являетсяабразивоструйная очистка[4].
Обычно выделяют три направления методов защиты от коррозии:
Конструкционный
Активный
Пассивный
Для предотвращения коррозии в качестве конструкционных материалов применяют нержавеющие стали, кортеновские стали, цветные металлы. При проектировании конструкции стараются максимально изолировать от попадания коррозионной среды, применяя клеи, герметики, резиновые прокладки.
Активные методы борьбы с коррозией направлены на изменение структуры двойного электрического слоя. Применяется наложение постоянного электрического поля с помощью источника постоянного тока, напряжение выбирается с целью повышения электродного потенциала защищаемого металла. Другой метод — использование жертвенного анода, более активного материала, который будет разрушаться, предохраняя защищаемое изделие.
В качестве защиты от коррозии может применяться нанесение какого-либо покрытия, которое препятствует образованию коррозионного элемента (пассивный метод).
Красочное покрытие, полимерное покрытие и эмалирование должны, прежде всего, предотвратить доступ кислорода и влаги. Часто также применяется покрытие, например, стали другими металлами, такими как цинк, олово, хром, никель. Цинковое покрытие защищает сталь даже когда покрытие частично разрушено. Цинк имеет более отрицательный потенциал и корродирует первым. Ионы Zn2+ токсичны. При изготовлении консервных банок применяют жесть, покрытую слоем олова. В отличие от оцинкованной жести, при разрушении слоя олова корродировать, притом усиленно, начинает железо, так как олово имеет более положительный потенциал. Другая возможность защитить металл от коррозии — применение защитного электрода с большим отрицательным потенциалом, например, из цинка или магния. Для этого специально создаётся коррозионный элемент. Защищаемый металл выступает в роли катода, и этот вид защиты называют катодной защитой. Растворяемый электрод, называют, соответственно, анодом протекторной защиты. Этот метод применяют для защиты от коррозии морских судов, мостов, котельных установок, расположенных под землей труб. Для защиты корпуса судна на наружную сторону корпуса крепят цинковые пластинки.
Если сравнить потенциалы цинка и магния с железом, они имеют более отрицательные потенциалы. Но тем не менее корродируют они медленнее вследствие образования на поверхности защитной оксидной плёнки, которая защищает металл от дальнейшей коррозии. Образование такой плёнки называют пассивацией металла. У алюминия её усиливают анодным окислением (анодирование). При добавлении небольшого количества хрома в сталь на поверхности металла образуется оксидная плёнка. Содержание хрома в нержавеющей стали — более 12 процентов.
Система холодного цинкования
Система холодного цинкования предназначена для усиления антикоррозионных свойств комплексного многослойного покрытия. Система обеспечивает полную катодную (или гальваническую) защиту железных поверхностей от коррозии в различных агрессивных средах
Система холодной оцинковки бывает одно-, двух- или трехупаковочной и включает:
связующее — известны составы на хлоркаучуковой, этилсиликатной, полистирольной, эпоксидной, уретановой, алкидной (модифицированной) основе;
антикоррозионный наполнитель — цинковый порошок («цинковая пыль»), с содержанием более 95 % металлического цинка, имеющего размер частиц менее 10 мкм и минимальную степень окисления.;
отвердитель (в двух- и трех- упаковочных системах)
Одноупаковочные системы холодного цинкования поставляются готовыми к применению и требуют лишь тщательного перемешивания состава перед нанесением. Двух- и трехупаковочные системы могут поставляться в нескольких упаковках и требуют дополнительных операций по приготовлению состава перед нанесением (смешивание связующего, наполнителя, отвердителя).
После приготовления (двух- и трёхупаковочные системы), нанесения состава на защищаемую поверхность металла кистью, валиком, методом пневматического илибезвоздушного распыления и высыхания на поверхности металла образуется цинкнаполненное противокоррозионное покрытие — полимерно-цинковая плёнка, сохраняющая все свойства полимерного покрытия, которое использовалось в качестве связующего, и одновременно обладающая всеми защитными достоинствами обычного цинкового покрытия.
Преимущества системы холодной оцинковки по сравнению со способом горячей гальванизации:
Простота и меньшая трудоёмкость технологии нанесения защитного цинкового покрытия. Для нанесения покрытия не требуется специальное оборудование.
Возможность антикоррозионной защиты металлоконструкций любых размеров, как в заводских так и в полевых условиях.
Возможность исправления непосредственно на месте абразивных повреждений покрытия и дефектов, возникающих при сварке металлоконструкций.
Экологически чистый процесс нанесения покрытия: нет необходимости производить работы в горячем цеху.
Создание на поверхности железа гибкого слоя цинка (не образующего микротрещин при изгибании металлоизделия).
Система холодного цинкования применяется во всех видах промышленности и в быту, где требуется надёжная и долговечная защита железных поверхностей от коррозии.
Помимо использования в качестве грунтовочного слоя в комплексном многослойном покрытии система холодной оцинковки может применяться как самостоятельное антикоррозийное покрытие металлических поверхностей.
[править]Газотермическое напыление
Для борьбы с коррозией используют также методы газотермического напыления. С помощью газотермического напыления на поверхности металла создается слой из другого металла/сплава, обладающий более высокой стойкостью к коррозии (изолирующий) или наоборот менее стойкий (протекторный). Такой слой позволяет остановить коррозию защищаемого металла. Суть метода такова: газовой струей на поверхность изделия на огромной скорости наносят частицы металлической смеси, в результате чего образуется защитный слой толщиной от десятков до сотен микрон. Газотермическое напыление также применяется для продления жизни изношенных узлов оборудования: от восстановления рулевой рейки в автосервисе до нефтедобывающих компаний[5].
[править]Термодиффузионное цинковое покрытие
(ГОСТ 9.316-2006). Для эксплуатации металлоизделий в агрессивных средах, необходима более стойкая антикоррозионная защита поверхности металлоизделий.Термодиффузионное цинковое покрытие является анодным по отношению к чёрным металлам и электрохимически защищает сталь от коррозии. Оно обладает прочным сцеплением (адгезией) с основным металлом за счет взаимной диффузии железа и цинка в поверхностных интерметаллитных фазах, поэтому не происходит отслаивания и скалывания покрытий при ударах, механических нагрузках и деформациях обработанных изделий.
Диффузионное цинкование, осуществляемое из паровой или газовой фазы при высоких температурах (375—850 °C), или с использованием разрежения (вакуума) — при температуре от 250 °C, применяется для покрытия крепёжных изделий, труб, деталей арматуры и др. конструкций. Значительно повышает стойкость стальных, чугунных изделий в средах, содержащих сероводород (в том числе против сероводородного коррозионного растрескивания), промышленной атмосфере, морской воде и др. Толщина диффузионного слоя зависит от температуры, времени, способа цинкования и может составлять 0,01—1,5 мм. Современный процесс диффузионного цинкования позволяет образовывать покрытие на резьбовых поверхностях крепёжных изделий, без затруднения их последующего свинчивания. Микротвёрдость слоя покрытия Hμ = 4000 — 5000 МПа. Диффузионное цинковое покрытие также значительно повышает жаростойкость стальных и чугунных изделий, при температуре до 700 °C. Возможно получение легированных диффузионных цинковых покрытий, применяемое для повышения их служебных характеристик.
Цинкование
Цинкование — это процесс нанесения цинка или его сплава на металлическое изделие для придания его поверхности определённых физико-химических свойств, в первую очередь высокого сопротивления коррозии. Цинкование — наиболее распространённый и экономичный процесс металлизации, применяемый для защиты железа и его сплавов от атмосферной коррозии. На эти цели расходуется примерно 40 % мировой добычи цинка. Толщина покрытия должна быть тем больше, чем агрессивнее окружающая среда и чем длительнее предполагаемый срок эксплуатации. Цинкованию подвергаются стальные листы, лента, проволока, крепёжные детали, детали машин и приборов, трубопроводы и др. металлоконструкции. Декоративного назначения цинковое покрытие обычно не имеет; некоторое улучшение товарный вид приобретает после пассивирования оцинкованных изделий в хроматных, или фосфатных растворах, придающих покрытиям радужную окраску. Наиболее широко используется оцинкованная полоса, изготовляемая на автоматизированных линиях горячего цинкования, то есть методом погружения в расплавленный цинк. Методы распыления и металлизация позволяют покрывать изделия любого размера (например, мачты электропередач, резервуары, мостовые металлоконструкции, дорожные ограждения). Электролитическое цинкование ведётся в основном из кислых и щёлочно-цианистых электролитов; специальные добавки позволяют получать блестящие покрытия.
56.
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ, химические СОЕДИНЕНИЯ, содержащие химический элемент УГЛЕРОД. Их насчитывается приблизительно в сто раз больше, чем неорганических соединений. К органическим соединениям относятся УГЛЕВОДОРОДЫ, базовая структура, которая, соединяясь с атомами других элементов (например, кислородом или азотом), образует большое количество органических соединений, включая и те, что являются жизненно необходимыми.
Виды гибридизации
sp-гибридизация
Происходит при смешивании одной s- и одной p-орбиталей. Образуется две равноценные sp-атомные орбитали, расположенные линейно под углом 180 градусов и направленные в разные стороны от ядра атома углерода. Две оставшиеся негибридные p-орбитали располагаются во взаимно перпендикулярных плоскостях и участвуют в образовании π-связей, либо занимаются неподелёнными парами электронов.
sp2-гибридизация
Происходит при смешивании одной s- и двух p-орбиталей. Образуется три гибридные орбитали с осями, расположенными в одной плоскости и направленными к вершинам треугольника под углом 120 градусов. Негибридная p-атомная орбиталь перпендикулярна плоскости и, как правило, участвует в образовании π-связей
sp3-гибридизация
Происходит при смешивании одной s- и трех p-орбиталей, образуя четыре равноценные по форме и энергии sp3-гибридные орбитали. Могут образовывать четыре σ-связи с другими атомами или заполняться неподеленными парами электронов.
Оси sp3-гибридных орбиталей направлены к вершинам правильного тетраэдра. Тетраэдрический угол между ними равен 109°28', что соответствует наименьшей энергии отталкивания электронов. Так же sp3-орбитали могут образовывать четыре σ-связи с другими атомами или заполняться неподеленными парами электронов.
57.
Классификация
Основные классы органических соединений биологического происхождения — белки, липиды, углеводы, нуклеиновые кислоты — содержат, помимо углерода, преимущественно водород, азот, кислород, серу и фосфор. Именно поэтому «классические» органические соединения содержат прежде всего водород, кислород, азот исеру — несмотря на то, что элементами, составляющими органические соединения, помимо углерода могут быть практически любые элементы.
Соединения углерода с другими элементами составляют особый класс органических соединений — элементоорганические соединения. Металлоорганические соединения содержат связь металл-углерод и составляют обширный подкласс элементоорганических соединений.
[править]Характерные свойства
Существует несколько важных свойств, которые выделяют органические соединения в отдельный, ни на что не похожий класс химических соединений.
Различная топология образования связей между атомами, образующими органические соединения (прежде всего, атомами углерода), приводит к появлению изомеров — соединений, имеющих один и тот же состав и молекулярную массу, но обладающих различными физико-химическими свойствами. Данное явление носит название изомерии.
Явление гомологии — существование рядов органических соединений, в которых формула любых двух соседей ряда (гомологов) отличается на одну и ту же группу — гомологическую разницу CH2. Целый ряд физико-химических свойств в первом приближении изменяется симбатно[неизвестный термин] по ходу гомологического ряда. Это важное свойство используется в материаловедении при поиске веществ с заранее заданными свойствами.
Изотермия — постоянство температуры тела— в процессе развития организма развивается постепенно. У новорожденного ребенка способность поддерживать постоянство температуры тела слабая. Вследствие этого может наступить охлаждение (гипотермия) или перегревание (гипертермия) организма при таких температурах окружающей среды, которые не оказывают влияния на взрослого человека. Кроме того, даже небольшая мышечная работа, например связанная с длительным криком ребенка, может повысить температуру тела.
58.
В свою очередь, углеводороды достаточно широко распространены в природе и могут быть выделены из различных природных источников: нефти, попутного нефтяного и природного газа, каменного угля. Рассмотрим их подробнее.
Основные современные виды топлива
[править]Твёрдые топлива
Древесина, древесная щепа, древесные пеллеты
Горючий сланец
Сапропель
Торф
Уголь
Битуминозные пески
Порох
Соединения азота
Твёрдое ракетное топливо
[править]Жидкие топлива
Просты в транспортировке, но при этом велики потери при испарении, разливах и утечках.
Нефтяные топлива
Дизельное топливо (газойль, соляровое масло)
топливо печное бытовое
Керосин
Лигроин
Бензин, газолин
Масла
Сланцевое масло
Отработавшее машинное масло
Растительные (рапсовое, арахисовое) или животные масла (жиры)
Спирты
Этанол
Метанол
Пропанол
Жидкое ракетное топливо
Эфиры
(Изомеры) спиртов
Метил-трет-бутиловый эфир (МТБЭ)
Диметиловый эфир (ДМЭ)
жирных кислот
Этерифицированные растительные масла (биодизель)
Эмульсии
Водотопливная эмульсия
Этиловый спирт в бензинах
Масла в бензинах
Синтетические топлива, производимые на основе процесса Фишера-Тропша
Из угля (CTL)
Из биомассы (BTL)
Из природного газа (GTL)
[править]Газообразные топлива
Ещё более транспортабельны, при этом ещё большие потери, а также при нормальных условиях ниже энергетическая плотность.
Пропан
Бутан
Метан, природный газ, метан угольных пластов,сланцевый газ, рудничный газ, болотный газ, биогаз, лэндфилл-газ, гидрат метана
Водород
Сжатый (компримированный) природный газ (CNG)
Продукты газификации твёрдого топлива
Угля — (синтез-, генераторный, коксовый) газы, возможна подземная газификация углей
Древесины
Смеси
Пропан-бутановая смесь (LPG)
Смесь водорода и природного газа (HCNG)
[править]Дисперсные системы, растворы
Аэрозоли
Угольная пыль
Алюминиевая, магниевая пыль
Пены
Газодизель (смесь природного газа с дизельным топливом)
Смесь водорода с бензином
Суспензии
Водоугольное топливо
Водонитратное топливо («жидкий порох»)
59.
Нефть (из тур. neft, от персидск. нефт[1]) — природная маслянистая горючая жидкость, состоящая из сложной смесиуглеводородов и некоторых других органических соединений. По цвету, нефть бывает красно-коричневого, иногда почти чёрного цвета, хотя иногда встречается и слабо окрашенная в жёлто-зелёный цвет и даже бесцветная нефть; имеет специфический запах, распространена в осадочных породах Земли. Сегодня нефть является одним из важнейших длячеловечества полезных ископаемых.
Цель переработки нефти (нефтепереработки) — производство нефтепродуктов, прежде всего различных видов топлива (автомобильного, авиационного, котельного и т. д.) и сырья для последующей химической переработки.
Первичные процессы
Первичные процессы переработки не предполагают химических изменений нефти и представляют собой ее физическое разделение на фракции. Сначала промысловая нефть проходит первичный технологический процесс очистки добытой нефти от нефтяного газа, воды и механических примесей - этот процесс называется первичнойсепарацией нефти[1].
[править]Подготовка нефти
Нефть поступает на НПЗ в подготовленном для транспортировки виде. На заводе она подвергается дополнительной очистке от механических примесей, удалению растворённых лёгких углеводородов (С1-С4) и обезвоживанию на электрообессоливающих установках (ЭЛОУ).
[править]Атмосферная перегонка
Нефть поступает в ректификационные колонны на атмосферную перегонку (перегонку при атмосферном давлении), где разделяется на несколько фракций: легкую и тяжёлую бензиновые фракции, керосиновую фракцию, дизельную фракцию и остаток атмосферной перегонки — мазут. Качество получаемых фракций не соответствует требованиям, предъявляемым к товарным нефтепродуктам, поэтому фракции подвергают дальнейшей (вторичной) переработке.
Материальный баланс атмосферной перегонки западно-сибирской нефти
ПРЕДЕЛЫ ВЫКИПАНИЯ, °С |
ВЫХОД ФРАКЦИИ, % (МАСС.) |
Газ |
1,1 % |
Бензиновые фракции |
|
<62°С |
4,1% |
62—85°С |
2,4% |
85—120°С |
4,5% |
120—140°С |
3,0% |
140—180°С |
6,0% |
Керосин |
|
180—240°С |
9,5% |
Дизельное топливо |
|
240—350°С |
19,0% |
Мазут |
49,4% |
Потери |
1,0% |
[править]Вакуумная дистилляция
Основная статья: Вакуум-дистилляция
Вакуумная дистилляция — процесс отгонки из мазута (остатка атмосферной перегонки) фракций, пригодных для переработки в моторные топлива, масла, парафины и церезины и другую продукцию нефтепереработки и нефтехимического синтеза. Остающийся после этого тяжелый остаток называется гудроном. Может служить сырьем для получения битумов.
[править]Вторичные процессы
Целью вторичных процессов является увеличение количества производимых моторных топлив, они связаны с химической модификацией молекул углеводородов, входящих в состав нефти, как правило, с их преобразованием в более удобные для окисления формы.
По своим направлениям, все вторичные процессы можно разделить на 3 вида:
Углубляющие: каталитический крекинг, термический крекинг, висбрекинг, замедленное коксование, гидрокрекинг, производство битумов и т.д.
Облагораживающие: риформинг, гидроочистка, изомеризация и т.д.
Прочие: процессы по производству масел, МТБЭ, алкилирования, производство ароматических углеводородов и т.д.
[править]Риформинг
Основная статья: Каталитический риформинг
Каталитический риформинг - каталитическая ароматизация нефтепродуктов (повышение содержания аренов в результате прохождения реакций образования ароматических углеводородов). Риформингу подвергаются бензиновые фракции с пределами выкипания 85-180°С[2]. В результате риформинга бензиновая фракция обогащается ароматическими соединениями и его октановое число повышается примерно до 85. Полученный продукт (риформат) используется как компонент для производства автобензинов и как сырье для извлечения ароматических углеводородов.
[править]Гидроочистка
Основная статья: Гидроочистка
[править]Каталитический крекинг
Основная статья: Каталитический крекинг
Каталитический крекинг - процесс термокаталитической переработки нефтяных фракций с целью получения компонента высокооктанового бензина и непредельных жирных газов. Сырьем для каталитического крекинга служат атмосферный и легкий вакуумный газойль, задачей процесса является расщепление молекул тяжелых углеводородов, что позволило бы использовать их для выпуска топлива. В процессе крекинга выделяется большое количество жирных (пропан-бутан) газов, которые разделяются на отдельные фракции и по большей части используются в третичных технологических процессах на самом НПЗ. Основными продуктами крекинга являются пентан-гексановая фракция (т. н. газовый бензин) и нафта крекинга, которые используются как компоненты автобензина. Остаток крекинга является компонентом мазута.
[править]Гидрокрекинг
Основная статья: Гидрокрекинг
Гидрокрекинг — процесс расщепления молекул углеводородов в избытке водорода. Сырьем гидрокрекинга является тяжелый вакуумный газойль (средняя фракция вакуумной дистилляции). Главным источником водорода служит газ риформинга. Основными продуктами гидрокрекинга являются дизельное топливо и т. н. бензин гидрокрекинга (компонент автобензина).
[править]Коксование
Основная статья: Коксование
Процесс получения нефтяного кокса из тяжелых фракций и остатков вторичных процессов.
[править]Изомеризация
Основная статья: Изомеризация
Процесс получения изоуглеводородов (изобутан, изопентан, изогексан, изогептан) из углеводородов нормального строения. Целью процесса является получение сырья для нефтехимического производства (изопрен из изопентана, МТБЭ и изобутилен из изобутана) и высокооктановых компонентов автомобильных бензинов.
[править]Алкилирование
Основная статья: Алкилирование
Алкилирование — введение алкила в молекулу органического соединения. Алкилирующими агентами обычно являются алкилгалогениды, алкены, эпоксисоединения,спирты, реже альдегиды, кетоны, эфиры, сульфиды, диазоалканы.
[править]Экстракция ароматики
60.
Горюче-смазочные материалы (сокращённо ГСМ) — нефтепродукты, к которым относят различные виды горючего и смазки, в основном в применении к автотранспорту:топливо (бензин, дизельное топливо, сжиженный нефтяной газ, сжатый природный газ), смазочные материалы (моторные, трансмиссионные и специальные масла, пластичные смазки), специальные жидкости (тормозные и охлаждающие).
Кре́кинг (англ. cracking, расщепление) — высокотемпературная переработка нефти и её фракций с целью получения, как правило, продуктов меньшей молекулярной массы — моторных топлив, смазочных масел и т. п., а также сырья для химической и нефтехимической промышленности. Крекинг протекает с разрывом связей С—С и образованием свободных радикалов иликарбанионов. Одновременно с разрывом связей С—С происходит дегидрирование, изомеризация, полимеризация и конденсация как промежуточных, так и исходных веществ. В результате последних двух процессов образуются т. н. крекинг-остаток (фракция стемпературой кипения более 350 °C) и нефтяной кокс.
Первая в мире промышленная установка непрерывного термического крекинга нефти была создана и запатентована инженером В. Г. Шуховым и его помощником С. П. Гавриловым в 1891 году (патент Российской империи № 12926 от 27 ноября 1891 года). Была сделана экспериментальная установка. Научные и инженерные решения В. Г. Шухова повторены У. Бартоном при сооружении первой промышленной установки в США в 1915—1918 годах. Первые отечественные промышленные установки крекинга построеныВ. Г. Шуховым в 1934 году на заводе «Советский крекинг» в Баку.
Крекинг проводят нагреванием нефтяного сырья или одновременным воздействием на него высокой температуры и катализаторов.
В первом случае процесс применяют для получения бензинов (низкооктановые компоненты автомобильных топлив) и газойлевых (компоненты флотских мазутов, газотурбинных и печных топлив) фракций, высокоароматизированного нефтяного сырья в производстве технического углерода (сажи), а также альфа-олефинов (термический крекинг); котельных, а также автомобильных и дизельных топлив (висбрекинг); нефтяного кокса, а также углеводородных газов, бензинов и керосино-газойлевых фракций; этилена, пропилена, а также ароматических углеводородов (пиролиз нефтяного сырья).
Во втором случае процесс используют для получения базовых компонентов высокооктановых бензинов, газойлей, углеводородных газов (каталитический крекинг); бензиновых фракций, реактивных и дизельных топлив, нефтяных масел, а также сырья для процессов пиролиза нефтяных фракций и каталитического риформинга(гидрокрекинг).
Используют также др. виды пиролитического расщепления сырья, например процесс получения этилена и ацетилена действием электрического разряда в метане(электрокрекинг), осуществляемый при 1000—1300 °C и 0,14 МПа в течение 0,01—0,1 с.
Крекинг используют для повышения октанового числа бензина (увеличения массовой доли C8H18). В ходе каталитического крекинга протекают также процессы изомеризации алканов.
61.
Бензи́н — горючая смесь лёгких углеводородов с температурой кипения от 30 до 200 °C. Плотность около 0,75 г/см³. Теплотворная способность примерно 10 500 ккал/кг (46 МДж/кг, 34,5 МДж/литр). Температура замерзания ниже −60 °C в случае использования специальных присадок.
Получение
Бензин получают путем возгонки и отбора фракций нефти, выкипающих в определенных температурных пределах; до 100 °C — бензин I сорта, до 110 °C — бензин специальный, до 130 °C — бензин II сорта, до 265 °C — керосин («метеор»), до 270 °C — керосин обыкновенный, примерно до 300 °C — производится отбор масляных фракций. Остаток считается мазутом.
Повышение качества автомобильного бензина
Повысить качество автомобильных бензинов можно за счет следующих мероприятий:
отказа от применения в составе бензинов соединений свинца;
снижения содержания в бензине серы до 0,05 %, а в перспективе до 0,003 %;
снижения содержания в бензине ароматических углеводородов до 45 %, а в перспективе до 35 %;
нормирования концентрации фактических смол в бензинах на месте применения на уровне не более 5 мг на 100 см³;
деления бензинов по фракционному составу и давлению насыщенных паров на 8 классов с учетом сезона эксплуатации автомобилей и температуры окружающей среды, характерной для конкретной климатической зоны. Наличие классов позволяет выпускать бензин со свойствами, оптимальными для реальных температур окружающего воздуха, что обеспечивает работу двигателей без образования паровых пробок при температурах воздуха до +60 °С, а также гарантирует высокую испаряемость бензинов и легкий пуск двигателя при температурах ниже −35 °С;
введения моющих присадок, не допускающих загрязнения и осмоления деталей топливной аппаратуры. Наиболее массовые отечественные бензины А-76, АИ-93 (ГОСТ 2084-77) и АИ-92 (ТУ 38.001165-97) не отвечают указанным требованиям по содержанию свинца (для этилированных бензинов), массовой доли серы, отсутствию регламентации содержания бензола и моющих присадок.
[править]Применение
В конце XIX века бензин не находил лучшего применения, чем антисептическое средство (продавался в аптеках) и топлива для примусов. Зачастую из нефти отгоняли только керосин, а все остальное, включая бензин, либо сжигали, либо просто выбрасывали. Однако с появлением двигателя внутреннего сгорания, работающего по циклу Отто, бензин стал одним из главных продуктов нефтепереработки, хотя по мере распространения дизельных двигателей благодаря их более высокому КПД на первый план выходит дизельное топливо.
Бензин применяется как топливо для карбюраторных и инжекторных двигателей, высокоимпульсное ракетное топливо (Синтин), при производстве парафина, какрастворитель, как горючий материал, сырье для нефтехимии прямогонный бензин или бензин газовый стабильный (БГС).
[править]Разновидности бензина
[править]Автомобильные бензины
В России автомобильные бензины выпускаются по ГОСТ 2084-77, ГОСТ Р 51105-97 и ГОСТ Р 51866-2002.
Автомобильные бензины подразделяются на летние и зимние (в зимних бензинах содержится больше низкокипящих углеводородов).
Основные марки автомобильных бензинов ГОСТ 2084-77:
А-72 — с октановым числом по моторному методу не менее 72;
А-76 — с октановым числом по моторному методу не менее 76 (позже был наименован АИ-80 в соответствии с исследовательским методом);
АИ-91 — с октановым числом по исследовательскому методу не менее 91;
АИ-92 — с октановым числом по исследовательскому методу не менее 92;
АИ-95 — с октановым числом по исследовательскому методу не менее 95;
АИ-98 — с октановым числом по исследовательскому методу не менее 98;
[править]Авиационные бензины
Авиационный бензин отличается от автомобильного более высокими требованиями к качеству, обычно имеет более высокое октановое число (что характеризует его детонационную стойкость на бедной смеси) и подразделяется по «сортности» (что характеризует его детонационную стойкость на богатой смеси).
Для авиабензина основными показателями качества являются:
детонационная стойкость (определяет пригодность бензина к применению в двигателях с высокой степенью сжатия рабочей смеси без возникновения детонационного сгорания)
фракционный состав (говорит об испаряемости бензина, что необходимо для определения его способности к образованию рабочей топливовоздушной смеси; характеризуется диапазонами температур выкипания (40—180(°)С) и давлений насыщенных паров (29—48 кПа))
химическая стабильность (способность противостоять изменениям химического состава при хранении, транспортировке и применении)
Основной способ производства авиационных бензинов — прямая перегонка нефти, каталитический крекинг или риформинг без добавки или с добавкой высококачественных компонентов, этиловой жидкости и различных присадок.
Классификация авиационных бензинов основывается на их антидетонационных свойствах, выраженных в октановых числах и в единицах сортности. Сорта российских авиационных бензинов маркируются по ГОСТ 1012-72, как правило, дробью: в числителе — октановое число или сортность на бедной смеси, в знаменателе — сортность на богатой смеси, например, Б-91/115 и Б-95/130. Встречается маркировка авиационных бензинов и по одним октановым числам, например, Б-70 (изготовляется по ТУ 38.101913-82) и Б-92 (изготовляется по ТУ 38.401-58-47-92).[1]
Бензины Б-91/115, Б-95/130 и Б-92 этилированные, а бензин Б-70 — нет (он используется в основном как растворитель).
[править]Бензины-растворители
Основная статья: Нефтяной растворитель
Нашли применение узкие легкокипящие продукты каталитического риформинга (БР-2) или прямой перегонки малосернистых нефтей (БР-1) (ГОСТ 443-76) в качестве растворителя для приготовления резиновых клеев при производстве печатных красок, мастик; для обезжиривания электрооборудования, тканей, кожи, поверхностей металлов перед нанесением металлических покрытий; для промывки подшипников, арматуры перед консервацией, в производстве искусственных мехов; для изготовления быстросохнущих масляных красок и электроизоляционных лаков; для извлечения канифоли из древесины, приготовления спирто-бензиновой смеси для промывки печатных плат в электротехническом производстве.
Экстракционные бензины (температура кипения 70-95 °C) прямой перегонки малосернистых нефтей применяются для экстракции растительных масел, извлечения жира из костей, никотина из махорочного листа, как растворитель в резиновой и лакокрасочной промышленности.
Малосернистый деароматизированный экстракционный бензин (температура кипения 70-85 °C) применяется для выработки масел в районах с жарким климатом (высокой испаряемостью).
Получаемый из рафината каталитического риформинга (температура кипения 105—125 °C) растворитель БЛХ, содержащий в основном парафиновые углеводороды линейного и изомерного строения, производится специально для лесохимической промышленности и применяется для извлечения канифоли из древесной щепы, иногда при приготовлении резиновых клеев и лаковых рецептур типографских красок.
Узкую фракцию прямой перегонки (температура кипения 110—185 °C) (озокеритовый растворитель) применяют для экстракции озокерита из руд.
Широкое применение получил Нефрас С 50/170 (ГОСТ 8505-80) (широкая фракция прямой перегонки малосернистых нефтей или рафината каталитического риформинга) в качестве растворителя при производстве искусственных кож, для химической чистки тканей, промывки деталей перед ремонтом, для смывания с деталей противокоррозийных покрытий и др.
Ксилольный рафинат каталитического риформинга и толуола с содержанием ароматики до 30 % — Нефрас САР применяется при производстве монолитных конденсаторов.
Особенно распространён бензин-растворитель для лакокрасочной промышленности — Уайт-спирит.
Нефрас С 150/200 узкой фракции прямой перегонки сернистых нефтей, близок по свойствам и применяется так же, как и уайт-спирит, однако содержит больше серы и имеет более резкий запах.
В народе легкокипящие бензины-растворители бытового применения часто называют «Калоша», кроме того, на российском рынке встречается продукт Нефрас С2 80/120 схожий по составу с БР1 и товарным наименованием «Калоша».
[править]Нафта (бензины для нефтехимии)
Основная статья: Лигроин
Нафта представляет собой фракцию нефти с пределами выкипания до 180 градусов Цельсия, состоят преимущественно из нормальных парафинов С5-С9. Получают прямой перегонкой нефти с добавлением небольшого количества вторичных фракций. Применяется как сырьё пиролиза для получения этилена на нефтехимических предприятиях, для блендинга и для экспорта. В РФ известны следующие товарные названия нафты:
Бензин газовый стабильный (БГС)
Бензин для химической промышленности
Бензин прямогонный (БП)
Дистиллят газового конденсата легкий (ДГКл)
Прочие продукты-аналоги
Лучше всего хранить смазочные материалы в помещении при относительно постоянной умеренной температуре. Любое хранилище, открытое или закрытое, необходимо расположить таким образом, чтобы оно удовлетворяло следующим условиям:
1. Удобный подъезд для транспортных средств. 2. Возможность свободного маневрирования транспортных средств при разгрузке. 3. Наличие рядом с хранилищем разгрузочной площадки со всем необходимым оборудованием. 4. Возможность вскрытия емкостей и отлива масел в чистом, не запыленном месте. 5. Легкость доставки смазочных материалов к основным местам использования. 6. Простота инвентаризации, легкость визуального контроля состояния емкостей. 7. Наличие специального места для пустых бочек и возвратной тары.
Открытое хранение
Погодные условия (кроме экстремальных температур и проникновения воды) не влияют на большинство смазочных материалов, поэтому в течение ограниченного времени их можно хранить на открытых площадках.
Однако, если температура может опуститься ниже 0°С, следует обеспечить защиту смазочных материалов, чувствительных к воздействию мороза (например, масло-водяных эмульсий или разбавленных водой жидкостей). Ни в коем случае не следует хранить вне помещений следующие материалы:
1. Электроизоляционные масла 2. Рефрижераторные (холодильные) масла 3. Белые и медицинские масла 4. Масла, смазки и жидкости 5. Пластичные смазки 6. Чистые СОЖ, содержащие жирные масла или соединения, которые при очень низкой температуре могут загустеть и расслоиться 7. Продукты с пищевым допуском
Рекомендуется открывать емкости со смазочными материалами и в последующем хранить их под навесом. Это снижает риск их загрязнения: в неполные бочки легче проникает влага или происходит конденсация. При открытом хранении бочки подвержены температурным колебаниям, которые вызывают соответствующие изменения внутреннего давления. В результате тара, даже имеющая уплотнения, «дышит», что создает условия для попадания влаги внутрь. Такая возможность возрастает, если бочка стоит пробкой вверх, т.к. верхняя часть бочки удерживает дождевую влагу.
Вода, находящаяся на бочке, может также привести к появлению ржавчины и смыть маркировку. Вот почему бочки следует хранить в наклонном положении, на боку или пробкой вниз. Пробки наклоненных и горизонтально расположенных бочек устанавливаются в положение «3 часа» и «9 часов» для того, чтобы сальники бочки соприкасались с маслом.
В любом случае бочки должны стоять не на земле, а располагаться на стеллажах или полках, на значительном расстоянии от поверхностной влаги. Категорически запрещается ставить бочки на поверхность, содержащую коррозионный клинкер.
Емкости следует регулярно осматривать с целью выявления коррозии, течи в швах и уплотнениях и проверки состояния маркировки.
Особое внимание следует обратить на хранение малых емкостей со смазочными материалами (бочонки и ведра). Они не предназначены для хранения в суровых погодных условиях. При вынужденном открытом хранении их следует поместить на стеллажи под навесом или защитить от дождя брезентом, обеспечив тем не менее хорошую циркуляцию воздуха.
Хранение в помещениях
Такое хранение всегда предпочтительнее. Если площадь закрытых хранилищ ограничена, ее нужно использовать для хранения малых емкостей, смазочных материалов, которые не выдерживают мороза, для открытых емкостей, а также для особых категорий смазочных материалов (см. Открытое хранение). В помещениях редко наблюдаются такие низкие температуры, которые могли бы оказать отрицательное влияние на смазочные материалы. Следует избегать чрезмерного местного перегрева от паровых труб, печей и т.п., так как это может вызвать термодеструкцию или испарение продуктов, содержащих растворитель.
Если только одна часть хранилища теплая, там следует разместить масла повышенной вязкости (густые масла). Склад для хранения смазочных материалов должен быть сухим, так как во влажной среде легко возникает коррозия емкостей.
Примечание. Часто условия страховки или противопожарные правила требуют выделения специальных мест для безопасного хранения летучих продуктов.
Хранение в резервуарах
Предпочтительнее располагать резервуары для хранения смазочных материалов в помещениях, однако они могут находиться и на открытых площадках при условии их защиты от дождя, снега и экстремальных температур.
На всех резервуарах, заливных и сливных трубах должны быть таблички с указанием полного наименования содержащегося в них продукта; это позволит избежать случайного смешения сортов при загрузке или сливе. По этому вопросу Вы можете проконсультироваться у сотрудника нашей компании.
Обычные резервуары из низкоуглеродистой стали могут потребовать определенного дооборудования при хранении отдельных видов смазочных материалов. Внутренняя поверхность резервуаров, в которых хранятся электрические и рефрижераторные масла, обычно имеет покрытие из эпоксидной смолы. А их воздухо-приемные отверстия оборудуются силикагелевыми дыхательными клапанами, поглощающими влагу. Для сохранения качества и цвета белых масел их нужно хранить в резервуарах из нержавеющей стали или с внутренним покрытием из эпоксидной смолы.
В резервуарах, не оборудованных силикагелевыми дыхательными клапанами, по мере конденсации влаги на относительно холодных стенках может постепенно накапливаться вода. Это происходит даже тогда, когда резервуары установлены в помещении. Воду следует периодически сливать через запорный (дренажный) вентиль, расположенный в самой низкой точке резервуара. Обычно резервуары устанавливаются с уклоном 1/10 по направлению к дренажному вентилю, что уменьшает вероятность диспергирования загрязненного масла. Некоторые сорта смазочных веществ при попадании в них большого количества воды могут частично или полностью превратиться в эмульсию.
Хранение пластичных смазок
Бочки с пластичной смазкой следует хранить в вертикальном положении. В стандартной 180-килограммовой бочке консистентной смазки имеется большое отверстие, уплотнение которого можно легко повредить при небрежном обращении. Это может привести к утечке мягкой смазки из горизонтально расположенной бочки.
62.
Полиме́ры (греч. πολύ- — много; μέρος — часть) — неорганические и органические, аморфные и кристаллические вещества, состоящие из «мономерных звеньев», соединённых в длинные макромолекулы химическими или координационными связями. Полимер — это высокомолекулярное соединение: количество мономерных звеньев в полимере (степень полимеризации) должно быть достаточно велико. Во многих случаях количество звеньев может считаться достаточным, чтобы отнести молекулу к полимерам, если при добавлении очередного мономерного звена молекулярные свойства не изменяются.[1] Как правило, полимеры — вещества с молекулярной массой от нескольких тысяч до нескольких миллионов.[2]
Поливинилацетат – полимеры жидкого винилацетата, получаемого в результате химического взаимодействия ацетилена (C2H2) и уксусной кислоты:
Полипропилен - термопластичный полимер, получаемый из газа пропилена C3H6. (CH2 = CH - CH3)
Полиизобутилен - продукт полимеризации газа изобутилена.
Полистирол – продукт полимеризации стирола – ненасышенного УВ – винилбензола или фенилэтилена – CH2CHC6H5.
В строении полимера можно выделить мономерное звено — повторяющийся структурный фрагмент, включающий несколько атомов. Полимеры состоят из большого числа повторяющихся группировок (звеньев) одинакового строения, например поливинилхлорид (—СН2—CHCl—)n, каучук натуральный и др. Высокомолекулярные соединения, молекулы которых содержат несколько типов повторяющихся группировок, называют сополимерами или гетерополимерами.
Полимер образуется из мономеров в результате реакций полимеризации или поликонденсации. К полимерам относятся многочисленные природные соединения: белки,нуклеиновые кислоты, полисахариды, каучук и другие органические вещества. В большинстве случаев понятие относят к органическим соединениям, однако существует и множество неорганических полимеров. Большое число полимеров получают синтетическим путём на основе простейших соединений элементов природного происхождения путём реакций полимеризации, поликонденсации и химических превращений. Названия полимеров образуются из названия мономера с приставкой поли-: полиэтилен, полипропилен, поливинилацетат и т. п.
Полимеризация и поликонденсация
Синтетические полимеры получают в результате реакций полимеризации и поликонденсации. Полимеризация — это процесс соединения друг с другом большого числа молекул мономера за счет кратных связей (С = С, С = О и др.) или раскрытия циклов, содержащих гетероатомы (О, N, S). При полимеризации обычно не происходит образования низкомолекулярных побочных продуктов, вследствие чего полимер и мономер имеют один и тот же элементный состав, Поликонденсация — зто процесс соединения друг с другом молекул одного или нескольких мономеров, содержащих две и да более функциональные группы (ОН, СО, СОС, NHS и др.) способные к химическому взаимодействию, при котором происходит отщепление низкомолекулярных продуктов. Полимеры, получаемые поликонденсационным способом, по элементному составу не соответствуют исходным мономерам.
Полимеризация мономеров с кратными связями протекает по законам цепных реакций в результате разрыва непредельных связей. Макромолекула при цепной полимеризации образуется очень быстро и сразу же приобретает конечные размеры, т. е не возрастает при увеличении длительности процесса. Полимеризация мономеров циклического строения происходит за счет раскрытия цикла и в ряде случаев пропекает не по цепному, а по ступенчатому механизму. Макромолекула при ступенчатой полимеризации образуется постепенно, т. е. сначала образуется димер затем тример и т.д., поэтому молекулярная масса полимера растет со временем. Принципиальное отличие ценной полимеризации от ступенчатой и от поликонденсации состоит в том, что на разных стадиях процесса реакционная смесь всегда состоит из мономера и полимера и не содержит ди-, три-, тетрамеров. С увеличением продолжительности реакции растет лишь число макромолекул полимера, а мономер расходуется постепенно. Молекулярная масса полимера не зависит от степени завершенности реакции или, что то же, от конверсии мономера, которая определяет только выход полимера.
63.
Макромолекулы полимеров могут быть линейными, разветвленными и сетчатыми.
Линейные полимеры образуются при полимеризации мономеров или линейной поликонденсации.
Разветвленные полимеры могут образоваться как при полимеризации, так и при поликонденсации. Разветвление полимеров может быть вызвано при росте боковых цепей, передачей цепи на макромолекулу, физическими воздействиями (g-облучение) на смесь полимера и мономеров.
Сетчатые полимеры образуются в результате сшивки цепей при вулканизации.
Форма макромолекул влияет на структуру и свойства полимеров.
В линейных и разветвленных макромолекулах, атомы или группы атомов могут вращаться вокруг ординарных связей, постоянно изменяя свою пространственную форму. Это свойство обеспечивает гибкость макромолекул, и они могут изгибаться, скручиваться, распрямляться. Поэтому для линейных и разветвленных полимеров характерно высокоэластичное состояние, они обладают термопластическими свойствами: размягчаются при нагревании и затвердевают при охлаждении без химических превращений.
При разветвлении эластические термопластические свойства становятся менее выраженными, а при образовании сетчатой структуры термопластичность теряется. Уменьшение длины цепей ведет к уменьшению эластичности полимеров, например, при переходе от каучука к эбониту.
Линейные полимеры могут иметь регулярную и нерегулярную структуру.
В полимерах регулярной структуры отдельные звенья цепи повторяются в определенной последовательности и располагаются в определенном порядке в пространстве, их называют стереорегулярными. Стереорегулярность изменяет тепловые и механические свойства полимеров.
Химические свойства полимеров зависят от их состава, молекулярной массы и структуры, вследствие наличия двойных связей и функциональных групп. Отдельные макромолекулы могут ²сшиваться² поперечными связями. Это процесс вулканизации и перевод линейных макромолекул термореактивных полимеров в сетчатые структуры.
При вулканизации происходит взаимодействие каучука с серой (0.5 - 5% серы) с образованием резины или эбонита (20% и более серы ), например,
К реакциям взаимодействия функциональных групп с низкомолекулярными веществами относятся галогенирование, гидролиз и д.р.
Полимеры могут подвергаться деструкции, т.е. разрушению под действием кислорода, света, теплоты, радиации. В результате деструкции уменьшается молекулярная масса макромолекул, изменяются физические и химические свойства полимеров и он становится непригодным для дальнейшего применения, Этот процесс называется старением полимеров. Чтобы замедлить этот процесс вводят стабилизаторы, чаще всего антиоксиданты.
Механические свойства полимеров определяются элементным составом, молекулярной массой, структурой и физическим состоянием макромолекул.
С ростом молекулярной массы механическая прочность возрастает, а также при переходе от линейных к разветвленным и далее к сетчатым структурам.
Стереорегулярные структуры имеют большую прочность, чем полимеры с разупорядоченной структурой. Самая высокая прочность у полимеров наблюдается в кристаллическом состоянии. Механическую прочность можно повысить добавлением наполнителей - сажи, мела, армированием стекловолокном.
Электрические свойства полимеров. Вещества делятся на диэлектрики, полупроводники и проводники.
Диэлектрики имеют очень низкую проводимость (< 10ˉ8 Омˉ1×смˉ1 ), которая увеличивается с повышением температуры.
Внешнее электрическое поле поляризует диэлектрики, т.е. определенно ориентирует молекулы. Внутри возникает собственное электрическое поле, которое ослабляет воздействие внешнего поля. Характеризуется это диэлектрической проницаемостью. При высоком напряжении внешнего электрического поля диэлектрик теряет свои электроизоляционные свойства. Это напряжение называется напряжением пробоя, а отношение напряжение пробоя к толщине диэлектрика - электрической прочностью.
Большинство полимеров относится к диэлектрикам и определяются эти свойства наличием полярных групп в макромолекулах (Clˉ, OHˉ, COOHˉ, и т.п.) - они ухудшают их диэлектрические свойства. Полимеры, не имеющие этих групп: фторопласт, полиэтилен - хорошие диэлектрики. Увеличение молекулярной массы улучшает диэлектрические свойства. При переходе от стеклообразного к высокоэластичному и вязкотекучему состояниям удельная электрическая проводимость возрастает. Для улучшения диэлектрических свойств необходимо удалять из полимеров ионы и примеси. OHˉ обуславливает гидрофильность полимеров. Они поглощают воду. В результате чего увеличивается электропроводность. OHˉ необходимо связывать между собой или с другими группами.
Диэлектрики применяются в электротехнике и радиотехнике как материалы различных электротехнических изделий, защитных покрытий кабелей, проводов, изоляционных эмалей ионы и лаков.
Некоторые полимеры обладают полупроводниковыми свойствами (проводимость 10ˉ10 - 10ˉ4 Ом–1.см–1), это полимеры с сопряженными двойными связями, у них есть делокализованные p - электроны. К ним относят полиацетилен ( -CH = CH - )n, поливинилен и др.
64.