

1) Металлы 8 группы: Ni, Co, Pt, Pd, Fe. 2)Окислы и сульфиды: Mo, w, Cr, MoS3
Кроме названных кат-ров имеются носители с развитой удельной поверхностью, высокой механической прочностью. Они инертные или с кислотными свойствами.
Ni, Co, Pt, Pd – кат-ры гидрирующего и дегидрирующего действия. Они не обладают устойчивостью к действию контактных ядов и не могут использоваться отдельно в процессах гидроочистки. Наибольшее распространение в гидрогенизирующих процессах получили следующие катализаторы: АКМ, АНМ, АНКМ, АНМС, АНВ, АКВ. Получают развитие цеолитсодержащие катализаторы. Предпологается, что гидрогенолиз гетероорганических соединений протекает многостадийно через хемосорбцию участников реакции на активных центрах Ni или Co, Mo или W. При этом на Co и Ni происходит активация водорода и спилловер атомарного активного водорода. А на Mo и Wo происходит осернение, азотирование и окисление с образованием соединений Mo(S), Mo(N), Mo(O) , к-рые подвергаются обессериванию, диазотированию и восстановлению.
Параметры процессов гидрогенизации.
1)Сырьё- бензиновые, керосиновые и дизельные фракции, а также вакуумный газойль и смазочные масла, содержащие S, N и непредельные УВ. Содержание гетероатомных соединений в сырье колеблется весьма значительно. По мере утяжеления сырья увеличивается не только общее содержание, но и доля наиболее устойчивых к гидрогенолизу гетероатомных соединений. В то же время требование к содержанию примесей в гидрогенизатах снижается по мере утяжеления сырья.
2) Температура, объемная скорость сырья и давление- они определяются кинетикой процесса. Так для ДТ требуемая глубина обессеривания 90-93% достигается при объемной скорости 4 ч-1, давлении 4 МПа и 350-380ºС. При Т=420ºС из-за реакций гидрокрекинга возрастает выход газов и легких УВ, увеличивается коксообразование и расход водорода..
Сырье, выкипающее выше 350ºС, находится в условиях гидроочистки в жидкой фазе и здесь повышение давления увеличивает скорость реакции более значительно, чем когда сырье в газовой фазе.
3) Парциальное давление водорода и краткость циркуляции ВСГ. При повышении общего давления растет Рпарц водорода. На Рпарц водорода влияет кратность циркуляции ВСГ и концентрация водорода в ВСГ. Последняя находится в пределах 60-90%. Чем выше содержание водорода, тем ниже кратность циркуляции ВСГ. Кратность циркуляции возрастает с утяжелением сырья. Процессы гидроочистки проводят в адиабатических реакторах без отвода тепла реакции. Повышение Т по высоте реактора не превышает 10ºС. При гидроочистке высококипящих и высокосернистых фракций предусматривается отвод тепла за счет подачи холодного ВСГ между слоями ВСГ.
4) Регенерация кат-ра. Кат-р теряет свою активность за счет закоксовывания и отложения на его поверхности тяжелых металлов, регенерацию проводят паро-воздушной смесью при Т= 530ºС.
Промышленные установки гидроочистки включают след. блоки: реакторный, сепарации газопродуктовой смеси с выделением ВСГ, очистки ВСГ от сероводорода, компрессорный, стабилизации гидрогенизата.
Принципиальная тех. схема уст-ки гидроочистки ДТ ЛЧ-24-2000.
Циркуляционный ВСГ смешивается с сырьем, смесь нагревается в сырьевых теплообменниках и в трубчатой печи П-1 до Т реакции и поступает в реактор Р-1. После реактора газо-продуктовая смесь частично охлаждается в сырьевых теплообменниках до Т= 210-230ºС и поступает на сепарацию ВСГ в сепараторы С-1 и С-2. ВСГ после С-2 поступает на очистку МЭА в абсорбер К-2 и далее на смешение со свежим ВСГ в качестве циркуляционного газа. Гидрогенезаты горячего из С-1 и холодного из С-2 сепараторов смешиваются и направляются в стабилизационную колонну К-1, где подачей подогретого в печи П-1 отдувочного ВСГ из гидроочищенного продукта удаляются УВ-ные газы и бензин. Расход водорода составляет 0,4% масс. на сырье. Выход гидроочищенного топлива 96,9%, бензина–1,3%, УВ-ных газов–0,6%, сероводорода–1,2% и потери 0,4%.
Параметры уст-ки ЛЧ24-2000:
Мощность 2 млн. т/год по сырью. Расход Н2 0,4% масс
Р=5 МПа. W=4,5 ч-1. Т=360-400 ºС.
В-22.Каталитический риформинг.
Кат.риформинг предназначен для повышения детонационной стойкости бензинов, получения индивидуальных аренов (бензола, толуола, ксилолов), для получения дешевого водородсодержащего газа.
Химизм и термодинамика процесса. Целевыми в каталитическом риформинге являются реакции образования ароматических углеводородов за счет: дегидрирование шестичленных цикланов; дегидроизомеризации циклопентана; дегидроциклизации парафиновых УВ (С6 - С7) и нежелательные реакции- гидрокрекинга, изомеризации парафинов и гидроизомеризации нафтенов. В результате побочных реакций образуются как низкомолекулярные, так и высокомолекулярные УВ, а также продукты уплотнения - кокс, к-рый откладывается на поверхности кат-ра.
Промышленные процессы риформинга проводятся при повышенных давлениях с целью подавления реакции коксообразования. Снижение равновесной глубины ароматизации при повышении давления компенсируют повышением Т.
Катализаторы и механизм их действия. Кат.риформинг осуществляется на бифункциональных кат-рах, состоящих из металлов и оксидов металлов. Основным компонентом катализатора являются металлы: Pt; Pt промотированная добавками рения и иридия, олова, галия и германия, тонко диспергированные на носителе. Вторым компонентом катализатора является оксид алюминия Al2O3, одновременно является носителем для Pt и ее промоторов. Для усиления и регулирования кислотной функции носителя в состав кат-ра вводят галогены: F или Сl. Совместное использование компонентов кат-ра резко усиливает их эффективность и позволяет повышать ОЧ бензинов на 40 - 50 Применение биметаллических катализаторов позволяет снизить давление риформинга от 3,5 до 2 - 1,5 МПа и увеличить выход бензина с ОЧ = 95 по ОЧИМ на 6 %. Полиметаллические кластерные кат-ры обладают стабильностью биметаллических, но характеризуются повышенной активностью, лучшей селективностью и обеспечивают более высокий выход реформата. Срок их службы составляет 6 - 7 лет. Другое преимущество этих катализаторов - хорошая регенерируемость при пониженном содержании Pt.
Параметры процесса.
1). Качество сырья риформинга - определяется фракционным и химическим составом бензина. Качество сырья влияет на выход риформата, октановое число (выход аренов), выход водорода, закоксовывание катализатора.
2). Температурный режим процесса и распределение объема катализатора по реакторам. Т.к. процесс риформирования сильно эндотермичен его проводят в каскаде из 3 - 4 реакторов с промежуточным подогревом сырья. В первом реакторе протекает высоко эндотермическая реакция дегидрирования нафтенов, в последнем реакторе протекают преимущественно эндотермические реакции дегидроциклизации и экзотермические реакции гидрокрекинга парафинов.
Т на входе в реактор находится в пределах 480-500ºС. По мере закоксовывания кат-ра и потери его активности Т на входе в первый реактор постепенно повышают,
Давление. Оно оказывает влияние на выход и качество риформата. при прочих равных параметрах с понижением парциального давления водорода возрастает глубина ароматизации сырья, при этом повышается селективность превращения парафинов и тормозит реакции гидрокрекинга. Однако при снижении давления возрастает скорость дезактивации кат-ра за счет его закоксовывания.
4). Кратность циркуляции ВСГ.. С увеличением мольного соотношения водород - сырье скорость дезактивации катализатора сокращается, а межрегенерационный цикл удлиняется. Однако увеличение Квсг приводит к увеличению энергозатрат, росту гидравлических сопротивлений и объема аппаратов.
5). Объемная скорость подачи сырья (W)- оказывает влияние на риформинг как параметр, обратный времени контакта сырья с кат-ром. С увеличением W снижается глубина реакции ароматизации и более значительно реакций гидрокрекинга парафинов. В результате повышение W приводит к: 1)к увеличению выхода риформата, но с пониженным октановым числом и меньшим содержанием аренов; 2)снижению выхода ВСГ, но с более высокой концентрацией водорода; 3)повышению селективности процесса и удлинению межрегенерационного цикла
6). Содержание Cl в катализаторе. Cl обеспечивает активность кислотных центров кат-ра, но т.к. Сl летучий, то кислотность центров понижается.
Тех. схема. Гидроочищенное и осушенное сырье смешивается с циркулирующим ВСГ, подогревается в теплообменнике, затем в секции печи П-1 и поступает в реактор первой ступени Р-1. На установке имеются 3-4 адиабатических реактора и соответствующее число секций в печи П-1 для межступенчатого подогрева реакционной смеси. На выходе из последнего реактора смесь охлаждается в теплообменнике и холодильнике до 20-40ºС и поступает в сепаратор высокого давления С-1 для отделения циркулирующего ВСГ от катализата. Часть ВСГ после осушки цеолитами в адсорбере Р-4 поступает на прием циркуляционного компрессора, а балансовый избыток отводится в блок гидроочистки бензола и передается другим потребителям водорода. Нестабильный катализат из С-1 поступает в сепаратор низкого давления С-2, где от него отделяют легкие УВ. После С-2 газовая и жидкая фазы поступают в абсорбер К-1. Абсорбентом служит стабильный катализат– бензин. Низ абсорбера подогревается горячей струей через печь П-2. В абсорбере при P=1,4 МПа и Тниза=165 и Тверха=40ºС отделяется сухой газ (IV). Нестабильный катализат с низа К-1 после подогрева в теплообменнике поступает в колонну стабилизации К-2. Тепло в низ К-2 подводится за счет циркуляции стабильного катализата через печь П-2. Головная фракция стабилизации после конденсации и охлаждения поступает в приемник С-3, откуда частично идет на орошение К-2. Балансовый избыток выводится с установки. Часть стабильного катализата с низа К-2 после охлаждения в теплообменнике подается во фракционирующий абсорбер К-1. Балансовый избыток выводится с установки.
В-23 Сущность реакций алкилирования их класс-ция.
Алкилированием наз-ют процесс введения алкильных групп в молекулы органических и нек-ых неорганических веществ. Эти процессы имеют большое практическое значение прежде всего для синтеза алкилароматических УВ, к-ые исп-ют в производстве синтетических каучуков, ПАВов, пластмасс, синт.волокон. Ведущее место занимает алкил-ие бензола олефинами. В больших масштабах производят продукты алкилирования меркаптанов, парафинов, сульфидов, аминов, спиртов.
Классификация процессов алк-ния:
1) По атому углерода (С-алкилирование). Такое замещение происходит в парафинах и аренах. Kat
ArH + RCl → ArR+HCl
AlCl3
2) По атомам кислорода и серы (O- и S- алк.).
ArOH + RCl +NaOH→ ArOR+NaCl+H2O
NaSH + RCl → RSH + NaCl
3)По атому азота (N-алк). В аммиаке и аминах Н2 замещается на Алк.
NH3 + RCl → RNH2 + HCl
4) По атомам других элементов (Si,Pb,Al). это важнейший путь получения металлорганических соединений.
2RCl + Si→R2SiCl2
Реа-ции алк-ния различают по строению алкильной группы, вводимой в соединение:
- циклоалкилирование циклогексилбензол
![]()
-арилирование С6H5Cl +NH3 → C6H5NH2 + HCl
- винилирование ROH + CH≡CH → ROCH═CH2
Алкильные группы могут содержать различные заместители – Cl, -COOH, -NO2,-OH.
Например, β- оксиалкилирование C2H4-O +HOH→ HO-CH2-CH2-OH.
Алкилирующие агенты и катализаторы.
1) Олефины. Чаще прим.этилен, пропилен, бутилен и высшие жидкие олефины. Их прим. в основном для С-алк-ния парафинов и аренов. Они не прим-ся для N-алк-ния и не всегда эффективны при S-и О- алк. Алкилирование при этом протекает по ионному механизму и катализируется протонными и апротонными кислотами.
R-CH=CH2 +H+→ R-C+H-CH3, карб-катион
В соответствии с устойчивостью карб-катиона реакционная способность олефинов изменяется в ряду:
CH2=C(CH3)2 > i-CH2=CH-CH2-CH3 > CH3-CH=CH2 > CH2=CH2
Удлинение и разветвление цепи углеродных атомов в олефине значительно повышает его реакционную способность.
2) Алкилгалогениды, в основном прим.хлорпроизводные. Они пригодны для всех типов реакций алк-ния. Механизм электрофильный и нуклеофильный, иногда радикальный.
Реакционная способность зависит от устойчивости карб-катиона и повышается при удлинении и разветвлении алкильной группы:
(CH3)3 –C-Cl > (CH3)2 –CH-Cl >CH3 –CH2-Cl
Реакционная способность алкилгалогенидов в реаях нуклеофильного замещения изменяется в ряду:
ArCH2Cl > CH2=CH-CH2Cl > AlkCl > ArCl; Перв. AlkCl > втор.AlkCl > трет.AlkCl
3) Спирты и простые эфиры. Спирты прим.для реакций O-,N-алк. В тех случаях когда они дешевле и доступнее хлорпроизводных. Процесс идет под действием кислотных Kat.
..
R-:O:H + H+ ↔ R-O δ+-H→ H2O + R+
H
Из простых эфиров исп. только оксиды олефинов. Они исп.для O-,N,-,S-алкил. Реакции идут под действием основных кат-ров или без катал-ра.
Энергетическая характеристика основных реакций алкилирования.
Все р-ции алкирирования экзотермичны, но значения тепловых эффектов сильно различаются в зависимости от типа разрывающейся связи и алкилирующего агента. При одном и том же алкилирующем агенте ∆Н реакции уменьшается в след.порядке:
Саром.> Солиф. > N > O
Для разных алкил.агентов существует след.зависимость ∆Н:
СН≡СН > С2Н4О > R-CH=CH2 > ROH > RCl
В-24 Алкилирование ароматических УВ на хлориде алюминия.
Алкил-щие агенты и катализаторы.Олефины, алкилгалогениды, спирты, но лучше олефины. При алк-нии хлорпроизводными в качестве кат-ра исп.только апротонные кислоты (AlCl3):
С6Н6 + С2Н5Cl → C6H5C2H5 +HCl
При алк-нии олефинами в качестве кат-ра прим.протонные кислоты: HF> H2SO4 > H3PO4 и апротонные кислоты : AlCl3, FeCl3, SnCl4, BF3, алюмосиликаты и цеолиты.
Наиболее активны HF, H2SO4. Процесс жидкофазный , Т=10-40; Р=0,1-1 МПа. HF не разлагает и не осмоляет УВ, кат-ор легко отделяется, хорошо регенерируется, нет промывных вод. «-» : большая летучесть и токсичность, сильное коронирующее действие; катализирует сильно изомеризацию и образование полимеров. H2SO4 «+» низкая Т. «-»коррозия аппаратуры, большой расход реагентов и нейтрализующих веществ,много промывных вод и как побочные продукты образуются сульфоэфиры, сложная технология. H3PO4 используют нанесением на твердый носитель. Т=250. «+ » процесс парофазный Р=2-6 МПа. «-» нужен большой избыток бензола и небольшой срок службы (25-30 суток), продукты содержат примесь олефинов.
Процесс с применением цеолитов, Т=400, Р=0,2- 0,6. «+» нет сточных вод, низкий расход кат-ра, в качестве сырья можно прим-ть разбавленные фракции олефинов. «-» создание прочного катализатора. Этот процесс конкурирует с жидкофазным процессом алкил-ния олефинами в присутствии AlCl3: «+» высокая активность, мягкие условия Т=100-130, Р=3-5 атм., высокая селективность. «-» коррозия аппаратуры, много промывных вод, высокие требования к качеству сырья.
Химизм и механизм процесса. СН2=СН2 +Н+ → СН3-С+Н2
Хлорид алюминия в твердом виде практически не растворим в УВ-дах и слабо катализирует реакцию, но в присутствии HCl, AlCl3 растворяется с образованием темного жидкого вещ-ва к-ое не растворяется в избытке УВ-ов- комплекс Густавсона
nC6H6
Бензол + HCl + Al2Cl6
↔
![]() [ *(n-1)C6H6
]+ Al2Cl7-
; n =1-6
[ *(n-1)C6H6
]+ Al2Cl7-
; n =1-6
При алк-нии высшими олефинами наблюдается изомеризация алк-ных групп:
СН3-СН2-СН2-СН=СН2 + Н+ → СН3-СН2-СН2-С+Н-СН3 ↔ СН3-СН2-С+Н-СН2-СН3
В более жестких условиях у гомологов бензола происходит изомеризация с внутримолекулярной миграцией алкильных групп с образованием более устойчивого изомера:
. Способность алкильных групп к миграции изменяется в ряду: (СН3)3С > (СН3)2СН > СН2-СН3 >> СН3

Последовательность алк-ния, селективность процесса. При алк-нии аром.соединений в присутствии любых кат-ров происходит последовательное замещение атомов водорода с образованием смеси продуктов разной степени алк-ния:
k1 k2
С6Н6+ С2Н4 →С6Н5-С2Н5 +С2Н4→ С6Н4(С2Н5)2 + С2Н4 → С6Н3(С2Н5)3 и т.д. до гексаэтилбензола.
Состав реакц-ной массы зависит от соотношения исходных реагентов.
В прис-вии кат-ров имеет место реакция переалкилирования:
С6Н4(С2Н5)2 + С6Н6↔2 С6Н5-С2Н5
Максимальное содержание моноалкилбензолов в равновесной смеси =50%, т.е. эта реакция явл-ся более выгодной для образования С6Н5-С2Н5.
При этом избыток бензола уменьшают до 2-3 кратного.
На селективность процесса влияют также побочные реакции :
1) Смолообразование
2) Деструкция алк.групп происходит на стадии образования карб-катиона
R-C+H-CH2-R → R-CH=CH2 +R+ +2C6H6→ C6H5-CHR-CH3 + C6H5-R
3) полимеризация происходит в рез-те последовательного взаимодействия карб-кат. с олефинами
СН3-C+H2 + С2Н4→ СН3-СН2-СН2-C+H2 …..
Эти реакции увеличиваются при повышении Т. Их подавляют избытком бензола, и при более низких концентрациях кат-ра ведут процесс.
Технологическая схема получения этил- и изопропилбензола. Состоит из нескольких узлов:
- узел осушки бензола
- приготовления кат-кого комплекса
-проведения реакции алк-ния
-обработка и разделение продуктов реакции.
Свежий бензол вместе с возвратным со стадии разделения идет в к-ну 3,в к-ой происходит осушка бензола азеотропной ректификацией. С верха к-ны выходит низкокипящая азеотропная смесь бензола с водой. Она конденсируется в конд-ре 4 и разделяется в сеп-ре 5 на два слоя. Воду с растворенным в ней бензолом отводят или исп-ют для промывки реакционной массы. Бензольный слой из С-5 стекает на верхнюю тарелку к-ны 3 создавая орошение. Осушенный бензол из куба К-3 подогревается в теплообменнике 2 и поступает в сборник 7,откуданасосом закачивается в алкилатор 9. Каталитический комплекс готовится в аппарате 8 с мешалкой и рубашкой для обогрева паром.В него загружают смесь бензола и ПАБов =1:1 и AlCl3 в количестве 1 моль на 2-3 моля ароматических УВ. После этого при Т и перемешивание подают этилхлорид или небольшое количество воды.
Бензол с этилхлоридом в прис-вии кат-ра образуют этилбензол. А в производстве ИПБ добавляют воду:
AlCl3 +Н2О = AlОНCl2 + НCl
Приготовленный кат-кий комплекс при температуре 60-65 периодически поступает в алкилатор 9(адиаб. колонного типа),где идет реакция алк-ния. В низ 9 поступает газообразный олефин, бензол из 7, рециркулирующий кат.комплекс из С-16 и клнденсат из С-11. Мольное соотношение бензола к олефину =3-3,5:1, Т=100-130, Р=3-4 атм. в алк-ре 9. Парогазовая смесь с верха колонны проходит конд.-холод. 10 где конденсируется бензол. В С-11 конденсат бензола отделяется и возвращается в алкилатор. Газы из С-11 содержат еще много паров бензола. Для его улавливания они идут в абсорбер 12, к-ый орошается ПАБами. Раствор бензола в ПАБах идет в реактор для переалкилирования. Газы после абсорбера 12 промывают щелочью в аб-ре 13 для удаления НCl ,затем водой аб-ре14 для удаления солей и остатков щелочи. После этого абгазы выводят на сжигание- они содержат метан, этан, пропан, олефинов практически нет. Реакционная масса выводится через боковой перелив вверху к-ны, охлаждается в хол-ке 15 и в С-16 отделяется от кат.комплекса. Состав рек-ной смеси (алкилат): бензол 45-55%, моноалкилбензол 35-40%. Диалкилбензол 8-12%; ПАБ,смолы, побочные продукты до 3%.
Алкилат идет на очистку от растворенной НCl и AlCl3.для этого его промывают водой а затем щелочью, а потом снова водой. Далее алкилат идет на ректификацию в систему ректиф. колонн. В первое к-не отгоняют бензол с водой; в след.к-не в вакууме отгоняют целевой продукт – моноалкилбензол. Далее в след. к-не отгоняют диалкилбензол и ПАБы. Остаток из последней кол-ны- это смолы, их сжигают. Выход целевого продукта достигает 94-95% при расходе AlCl3 от 5 до 10 кг на тонну моноалкилбензола.
В-25. Производство изопарафинового алкилата.МТБЭ.
Алкилат- высокооктановый изокомпонент бензина. Производится С-алкилированием изобутана бутиленами и пропиленом. Алкилат- состоит практически нацело из изопарафинов, имеет высокое ОЧММ=90.Основным компонентом явл-ся изооктан (ОЧ=100). В общем виде: CnH2n+2 + CmH2m↔Cn+mH2(n+m)+2
Механизм карбоний ионный: CH3CH=CHCH3 + H+→ CH3CH2C*HCH3
CH3CH2C*HCH3 + (CH3)3CH → CH3CH2CH2CH3 + (CH3)3C+
RH + CH2=CHR*↔RR*CHCH3
Катализаторы: AlCl3, HF, H2SO4.
Алкилируются только изопарафины.
Побочные реакции: получение высших УВ, деструкция карб-катиона, полимеризация олефина. Их подавляют избытком изопарафинов.
Сырье - бутан-бутеновая фракция крекинг газов. Реакционная масса – двухфазная система, которую эмульгируют при помощи мешалок. Большое значение имеет концентрация H2SO4 (лучшей явл-ся 98-100 %). Объемное отношение H2SO4 и УВ-дов = 1:1.
Реакционные узлы: двух видов, отличаются способом отвода выделяющегося тепла- охлаждением хладоагентом (аммиаком или пропаном) через теплообменную поверхность и охлаждением за счет испарения избыточного изобутана. В первом случае валкилаторе – контакторе вертикального или горизонтального типа, снабженном мощной мешалкой, имеются охлаждающие трубы, в к-рых хладоагент испаряется, пары которого направляются затем в холодильную установку, где они снова превращаются в жидкость. Реакторы второго типа – горизонтальные каскадные, в к-рых охлаждение реакционной смеси осуществляется за счет частичного испарения изобутана, что облегчает регулирование Т. Реактор имеет несколько зон смешения, разделенных перегородками, и двухсекционную зону отстоя. Бутилен подводится отдельно в каждую секцию, вследствие чего концентрация олефинов очень мала, это позволяет подавить побочные реакции. Применение каскадных реакторов упрощает и удешевляет установки.
Тех.схема установки производства алкилата. Т=5-15; Р=0,6-1 МПа. Исходная УВ-ная смесь поступает пятью параллельными потоками в смесительные секции реактора-алкилатора Р, в первую секцию вводятся H2SO4 и жидкий изобутан. Из отстойной секции Р выводятся продукты алкилирования, к-рые после нейтрализации щелочью и промывки водой идут в К-2 для отделения циркулируемого изобутана. Испарившиеся в реакторе изобутан и пропан через сепаратор Р-рессивер компрессором через холодильник подаются в колонну-депропанизатор К-1. Нижний продукт К-1-изобутан через кипятильник и теплообменник присоединяется к циркулирующему потоку изобутана из К-2. Нижний продукт К-2 поступает вколонну дебутанизатор К-3, а остаток К-3 – в К-4 для перегонки суммарного алкилата. С верха этой колонны отбирается целевой продукт – легкий алкилат, а с низа – тяжелый алкилат (компонент ДТ).
МТБЭ. МТБЭ явл-ся высокооктановым кислородсодер-жащим компонентом автобензина. По сравнению с другими компонентами МТБЭ обладает более высоким ОЧ и низкой температурой кипения (+ 55,2 С), что в совокупности позволяет повысить ОЧ преимущественно головных фракций базового бензина, тем самым и равномерность распределения детонационной стойкости по его фракциям. В товарные автобензины МТБЭ добавляют в количестве 5-15 %. Эфирсодержа-щие бензины характеризуются большой полнотой сгорания и меньшей токсичность выхлопных газов.
Процесс получения МТБЭ основан на реакции взаимодействия метанола с изобутиленом на ионообменном катализаторе кислотного типа : СН2 = С(СН3)(СН3) + СН3ОН ↔ СН3—С(СН3)(СН3)—ОСН3
Реакция синтеза МТБЭ из изобутилена и метанола протекает по цепному карбоний ионному механизму с выделением 66 кДж/моль тепла ,а её равновесие смещается вправо при повышении давления и снижения Т.
1. Первой стадией О- алкилирования метанола изобутиленом является протониро-вание последнего гидрид ионом кислотного катализатора : СН3—С(СН3)=СН2 + Н А ↔ СН3—С+(СН3)—СН3 + А+
2. Образовавшийся третичный бутеновый карбониевый ион вступает в реакцию с метанолом (при его избытке):
СН3—С(СН3)(СН3) —СН3 + СН3 ОН ↔ СН3–С(СН3)(СН3) –ОН–СН3 ↔ СН3–(СН3)(СН3)С–О–СН3 + Н-
3. Образовавшийся протон далее реагирует с изобутеном, как и в стадии 1.
4. Причиной обрыва цепи может стать возврат протона к катализатору Н- + А+ ↔ НА
Помимо основной целевой реакции О-алкилирования, при синтезе МТБЭ протекают побочные реакции:
- димеризация изобутена с образованием диизобутилена. Эта реакция идет с выделением большого количества тепла.
- гидратация изобутилена водой, содержащейся в исходном сырье с образованием изобутилового спирта;
- гидроконденсация метанола с образованием диметилового эфира.
Обычно реакцию образования МТБЭ ведут с небольшим избытком метанола чтобы ограничить побочные реакции. Получение МТБЭ- каталитический процесс. Применяют гомогенные (серная, фосфорная, соляная, борная кислоты) и гетерогенные катализаторы (цеолиты, оксиды алюминия, железа, никеля ,ионообменные смолы) кислотно-основного типа. Все существующие технологии синтеза МТБЭ обычно включают в себя три основных узла: реакторный узел, фракционирование и очистку отходящей фракции. Качество товарного МТБЭ колеблется в пределах 97 – 99% масс. В качестве промышленных реакторов используются трубчатые изотермические реакторы со съемом тепла циркулирующей водой, каскад адиабатических реакторов с промежуточным отводом тепла реакции или сочетание разных типов реакторных систем, реакторы реакционно-ректификационного типа, в которых отвод тепла осуществляется испарением потока флегмы. Сырьё: ББф кат.крекинга и пиролиза, и метанол.
Тех.схема. Исходная бутан-бутиленовая фракция через емкость Е поступает в верхнюю часть реактора форконтактной очистки. Очищенная смесь после нагрева в теплообменнике до 60ºС поступает в зону синтеза под каждый слой кат-ра реактора Р-1(2). В верхнюю часть реакционной зоны во избежание перегрева кат-ра подается свежий метанол с Т=50-60 С. Жидкие продукты, состоящие из МТБЭ с примесью метанола и УВ, выводятся из куба реактора и идут в отпарную колонну К-2 с паровым кипятильником на отпарку примесей. МТБЭ с куба К-2 после теплообменников и холодильников идет в товарный парк. Паровая фаза, состоящая из отработанной ББФ, метанола и следов МТБЭ, поступает на конденсацию МТБЭ в К-1. Конденсированный МТБЭ возвращается на верхнюю тарелку Р-1(2) в качестве холодного орошения. С верха К-1 отводятся пары ББФ и метанола, к-рые после конденсации идут в С-1, далее в К-3 разделяются экстракцией водой. Отработанная ББФ с верха К-3 после охлаждения в холодильниках идет в товарный парк. Отгонка циркуляциооного метанола от воды проводится в ректификационной колонне К-4, охлаждается и собирается в рефлюксной емкости С-3. Часть метанола исп-ся в качестве холодного орошения К-4, а остальная часть идет в Е. Вода из куба К-4 после охлаждения подается в экстрактор К-3 для отмывки метанола от ББФ.
В-26. Синтезы на основе α-оксидов.
Реакции идут с разрушением цикла. Этилен оксид
химически активен, взаимодействует с веществами, содержащими подвижный атом водорода.
СН2СН2О + HA↔ HOCH2CH2A
Примеры синтезов:
1. Взаимодействие с водой СН2СН2О + НОН→ НОСН2СН2ОН
2. Вз-вие со спиртами и фенолами СН2СН2О + RОН→ НОСН2СН2ОR
3. Вз-вие с H2S и RSH: СН2СН2О+ H2S → НОСН2СН2SH
СН2СН2О+ RSH → НОСН2СН2SR
4. Вз-ие с карбоновыми и неорганическими кислотами: СН2СН2О + RCOOH→ HOCH2CH2OCOR
5. Вз-ие с аммиаком, аминами и амидами: СН2СН2О + =NH→ HOCH2CH2N=
6. Реакции с расширением цикла: с СО2 и СН3СООН.
Механизм: эти реакции часто идут без кат-ра, но сильно ускоряются кислотами или щелочами. При взаимодействии с О-сод. соед-ми, H2S, RSH или амидами наиболее типичными кат-ми явл-ся основания (NaOH, Na2CO3, NR3)- катализ основный или нуклеофильный. NaOH + НА → Н2О + Na+ + А-
![]()
По такому же механизму протекает и не каталитическая реакция. Нуклеофилом явл-ся сама молекула реагента, но реакция идет медленнее. С достаточной скоростью она протекает при Т=180-220ºС, при нуклеофильной реакции 100-150ºС. Кислотный катализ тех же реакций протонными кислотами эффективен в сильнополярных средах (Н2О, низшие спирты).
![]()
В малополярной среде протонные кислоты присоединяются к α-оксидам и становятся неактивными – применяют гетерогенные кат-ры кислотного типа (BF3, SnCl4, Al2O3, HF). Реакции кислотного катализа протекают при Т от 20-40 до 100-150.
Продукты:
1) Гликоли- прим-ся для производства антифризов, взрывчатых веществ, растворителей, полимерных материалов.
2) Этаноламины- прим-ся для произ-ва взрывчатых веществ и для очистки газов от кислотных примесей.
3) Неионогенные ПАВы- применяют как компоненты моющих средств.
Технология и реакционные узлы. Все производства на основе α-оксидов по технологическим признакам делятся на три типа :
1) Реакции осуществляющиеся при большом избытке реагента – произ-во гликолей и эфиров. Теплота реакции воспринимается избыточным реагентом. Процесс осущ-ся в адиабатических и полностью гомогенных условиях.
(А) (Б) (В) (Г)
![]()
![]()
![]()
![]()
Рис.А: Непрерывно действующая реакционная колонна, не имеющая поверхности теплообмена. Исходная смесь, предварительно подогретая паром, подается с верху и поступает в низ колонны по центральной трубе, в к-ой она еще подогревается реакционной массой. «-»: снижение селективности за счет наличия продольного перемешивания продукта.
2)Для непрерывного осуществления таких реакций в более интенсивном режиме, больше подходят кожухотрубчатые реакторы (Рис.Б). Применяется для реакций, в к-рых мольное отношение α-оксида к реагенту поддерживают от 1-(4-5) до (2-3)-1: синтез этаноламинов, тиогликолей, тиоэфиров. Применяется реакционный узел с циркуляцией реакционной массы через выносной холодильник (Рис.В). «-»: аппарат применим только для тех реакций, в к-рых последовательное превращение не играет существенной роли: получение этиленкарбоната, тиоспиртов и тиоэфиров. 3) Реакции, в к-рых мольное отношение этиленоксида к реагенту превышает 3:1 – синтез полигликолей и неионогенных ПАВ. Процесс осуществляется периодическим способом в аппарате с разбрызгиванием жидкой реакционной массы в атмосферу газообразного этиленоксида (Рис.Г). Охлажденную в кубе жидкость непрерывно прокачивают через спец.форсунки и впрыскивают в газообразное пространство реактора, куда вводят этиленоксид. Образуются капельки ПАВ, к-ые оседают в жидкую фазу.
В-12. Процессы этерификации.
- это обратимое взаимодействие орг. и неорг кислот со спиртами с образованием эфиров и воды.
R-СООН + R`ОН ↔ R-СОО-R` + Н2О ;
HNO3 + R`OH ↔ R`ONO2 + H2O
Если спирт многоатомный, то:
HOH2C-СН2ОН + RСООН ↔ HOH2C-CH2OCOR ↔ ↔ROCO-H2C-CH2-OCOR
Если спирт трехатомный, то реакция пойдет в три ступени. Если и к-та и спирт, как минимум бифункциональны , то образуются полимеры:
nHO-R-OH + n HO-CО-R`-CО-OH→
→ [-O-R-O-CО-R`-CО-]n
Обратные реакции называются гидролизом сложных эфиров или омылением их.
Р-ции могут идти без кат-ра, но медленно. Для увеличения скорости процесса нужно увеличить Т до 200-300°С, в том случае если нельзя использовать кат-р: кислотного типа: а) гомогенные (неорг к-ты:H2SO4, HCl, H3PO4 б) гетерогенные ( Al2O3, алюмосиликаты MgO, Cr2O3, ZrO], фосфаты Ме [МеРО4; Са3(РО4)2; Мg(РО4)2 ], ионообменные смолы).
Механизм р-ции этерификации с кат-ом.
O OH
║ │
R-C-OH ↔R-C-O+H ↔ R-C+-OH ↔ R-C-OH ↔
\ \ │
OH OH R`-O+-H
OH
│
↔ R-C─O+H2 ↔ R-C+-OH ↔ R-C=O
│ \ \
R`-O O-R` O-R`
Карбоновые к-ты, их ангидриды и хлорангидриды – это этерифицирующие агенты.
По активности располагаются в ряду : RCOOH< (RCO)2O< RCOCl
К реакциям этерификации относятся:
1). Р-ия алкоголиза ( спирт со сложным эфиром):
R-CОOR` + R``OH ↔ R-CОOR`` + R`OH
2). Р-ий ацидолиза ( карбоновая к-та со сложным эфиром):
R-CОOR` + R``-CОOH ↔ R-CОOH + R``-CОOR`
3) Р-ия переэтерификации ( р-ия 2-х сложных эфиров):
R-COOR` + R``-COOR``` ↔ R-COOR``` + R``-COOR`
Эти р-ции лучше катализируются сильными протонными кислотами. Это р-ции обменного разложения.
Термодинамика. Р-ции спиртов с карбоновыми к-ми протекают без теплового эффекта. Р-ции спиртов с неорг. кислотами идут с выделением тепла за счет разбавления неорг к-ты образующейся водой. С выделением тепла идут р-ции с хлорангидридами к-т и первая стадия с ангидридами кислот.
Чем длиннее и разветвленнее молекула спирта, тем ниже Кр. В этом случае в качестве этерифицирующих агентов лучше брать ангидрид или хлорангидрид. С удлинением и разветвлением молекулы к-ты Кр немного повышается
С ↑ t , при проведении р-ции в газовой фазе на гетерогенном кат-ре, Кр снижается. Чем длиннее и разветвленнее молекулы спирта и кислоты, тем скорость р-ции будет ниже.
Технология процессов этерификации. Процесс ведут в жидкой и газовой фазе в зависимости от используемого кат-ра. Основная задача технологии – наиболее полно сдвинуть равновесие р-ции вправо. Самый простой путь постоянно отгонять один из продуктов р-ции из реакционной массы, снижая тем самым его концентрацию. Равновесная Si стремится восполнить эту потерю и р-ция идет до полного превращения исх в-в.
К + С ↔ Э + В
К-кислота, С- спирт, Э –эфир, В- вода.
![]()
![]()
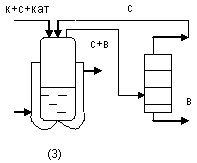

1). К,С,Э – высококипящие компоненты (ВКК); В- НКК. Процесс ведут при постоянной отгонке воды, поэтому реакторе образуется эфир . Для обеспечения отгонки воды используют вакуум или продувку инертным газом. В качестве к-ты – фталевою, адипиновую, себациновую, спирта- высшие спирты (глицерин, гликоли).
2). К,Э – ВКК; С+В – НКК( азеотроп). Из реакционной массы отгоняют азеотроп, разделяют и возвращают спирт в реактор. Азеотроп отгоняют, охлаждают, конденсирут и подвергают расслоению в отстойнике Если азеотроп не расслаивается, то в реактор вводят азеотропную добавку
( бензол или дихлорэтан) и вода отгоняется с этой добавкой, а затем эта добавка легко отслаивается от воды и возвращается в реактор. Но если нельзя использовать азеотропную добавку, то разделение С+В проходит в ректификационной колонне.
3). К- ВКК, Э+В или Э+С+В – азеотроп с большим содержанием воды. Из реактора отгоняют азеотроп, охлаждают, конденсируют и расслаивают на 2 слоя
( органический и водный).
4). К – ВКК, С+В+Э – азеотроп с большим содержанием эфира. Азеотроп отгоняется из реактора и подвергается расслоению. Чтобы вести процесс непрерывно необходимо использовать несколько реакторов кубового типа, в к-ых стадия загрузки и выгрузки чередуется. Концентрация кат-ра ~ 0,1 %, время р-ции 2-6 ч. Если с азеотропом отгоняется спирт, то берут его избыток. «-» жидкофазной этерификации - необходимость нейтрализации и промывки получаемого эфира от к-ты, поэтому чаще используют гетерогенный катализ. Процесс ведут при Т=150-160° в реакторе 1 со сплошным слоем кат-ра, продукты р-ции поступают в экстракционную колонну 3, где водой извлекают непревращенный спирт и кислоту, а в отпарной колонне 2 отгоняют спирт с кислотой от воды.
Основные продукты процессов этерификации. 1)растворители и экстрагенты- этилацетат, изопропилацетат, алилацетат, моноацетат этиленгликоля, а также ди- и триацетаты этиленгликоля. 2)пластификаторы и синтетические смазочные масла – полные эфиры двухосновных карбоновых кислот высшими одноатомными спиртами, полные эфиры ди- , три и полиэтиленгликолей или пентаэритрита с высшими одноосновными карбоновыми к-ми С6-С9. 3)мономеры – полиметилметакрилат, пентафталевые смолы, поликарбонаты.
В-13. Прямая гидратация олефинов.
-это присоединение воды по двойной связи с образованием спиртов
R-CH=CH2 + H2O ↔ R-CH(ОН)-CH3
Р-ция обратима, с выделением тепла и уменьшением реакционного объема. Для смещения равновесия вправо необходимо ↓ Т и ↑ Р, но при низких Т скорость р-ции будет невысокая. На практике стараются поддерживать высокую t и высокое Р. Р-ция относится к каталитическим процессам. Используется любой кат-р кислотного типа ( протонные и апротонные к-ты).
R-CH-CH2 ↔ R-CH = CH2 ↔ R-C+H-CH3 ↔ R-CH-CH3
↓ │ │
H+ +OH2 OH
↔R-CH-CH3
Р-ция идет через образование иона карбония, Чем длиннее и разветвленнее молекула олефина, тем устойчивее ион карбония. Ряд активности:
i-C4H8> н-С4Н8>С3Н6>С2Н4.
Побочные р-ции: 1.образование простых эфиров
2ROH ↔ ROR +H2O ; 2.полимеризация
n R-CH=CH2 -[ CH(R)-CH2-]n ; 3.дегидрирование СН3-СН2ОН СН3-СНО + Н2
Технологическая схема производства этанола. Процесс осуществляется прямой гидратацией этилена. Свежий этилен компрессором 1 и рециркулирующий этилен компрессором 2 сжимают до 8 МПа, смешивают с водой, подоваемой насосом 14. Полученную газовую смесь сначала нагревают в теплообменнике 4, затем в печи 3 до 280-330° и подают в реактор 5. Реактор 5 представляет собой полую стальную колонну. Соотношение этилена к водяному пару = 1,5:1. Кат-тор фосфорная к-та на носителе. Пары этилена и воды проходят слой кат-ра сверху вниз. Из-за невысокой степени конверсии ( 4%) тепловой эффект незначителен и отвод тепла не нужен. Продукты р-ции выходят с низа реактора и далее нейтрализуются от паров фосфорной кислоты впрыскиванием раствора NaOH. Образующиеся фосфаты натрия отделяются в сепараторе 6. Далее горячие реакционные газы отдают свое тепло в теплообменнике 4, дополнительно охлаждаются в водяном холодильнике 7 и разделяются в сепараторе 8. Несконденсировавшиеся пары спирта из С-8 поступают в нижнюю часть абсорбера 9, орошаемого водой с насоса 14. Оставшийся газ, содержащий непревращенный этилен, поступает на всас компрессора 2.
Конденсат из С-8 и с низа абсорбера 9 проходит дроссель 15, где сбрасывается Р и поступает в С-10 , где отделяются остатки газа. Конденсат ( 15% водный р-р этанола с примесями) из С-10 подвергается очистке в ректификационных колоннах 11 и 12. В колонне 11 отгоняют более летучие побочные продукты – ацетальдегид и диэтиловый эфир, а в колонне 12 от спирта отделяют воду, к-ая поступает на ионообменную очистку в блок 13 и возвращается в процесс. Целевой продукт отбирается с верха колонны 12 в виде 95% этилового спирта.
В-14Гидратация ацетилена.
СН≡СН + Н2О → СН3-СНО ( р-ция Кучерова)
Р-ция идет с выделением тепла, необратимо при низких Т, чтобы р-ция стала обратимой Т нужна >300°С. Процесс может идти и без кат-ра, но с низкой скоростью.
Применяют кат-ры 2 типов;
1.) HgSO4 в 10-20% р-ре H2SO4.
CH≡CH ↔ CH ≡ CH↔ CH = CH↔ Hg+ + CH2 = CH→
¦ ¦ │ │
Hg2+ Hg+ +OH2 +OH2
→CH3- CHO+H++Hg+
Чем выше концентрация H2SO4 и чем выше Т, тем выше скорость р-ции.
Побочные р-ции: 1.образование кротонового альдегида и смол.
CH3-CHO+ CH3-CHO → CH3-СН=СН-СНО →CH3-СН=СН-СН=СН-СНО +….+ →смолы
Р-ция протекает при повышенных Т , поэтому Т нужно ограничивать.
2. CH3-CHO + 2Hg2+ + Н2О → CH3-CООН + 2Hg++ 2Н+ ; Этой р-ции способствуют примеси в ацетилене
Для подавления 1 и 2 р-ций необходимо как можно быстрее удалять ацетальдегид из реакционной массы. Для большего срока службы кат-ра в реакционную массу добавляют Fe2(SO4)3. Железо окисляет ртуть до активной формы: Fe3+ + Hg+→ Fe2+ + Hg2+
(А)

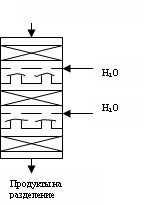
(Б)
Реактор (А) – пустотелая колонна заполненная на 80% водным раствором H2SO4, HgSO4, Fe2(SO4)3. В низ колонны подается ацетилен с парами воды. Ацетилен барбатирует через реакционную массу, выдувает ацетальдегид и вместе с ним походит каплеотбойник 5, охлаждается в холодильнике 2, пары воды конденсируются и отделяются от газов в сепараторе 3. Вода возвращается в реактор, а пары ацетальдегида и ацетилена поступают в низ абсорбера 4, к-ый орошается водой, вода поглощает ацетальдегид и уходит с низа аппарата на ректификацию. А непрореагировавший ацетилен с верха абсорбера 4 возвращается в реактор 1. В реакторе Т= 90-95°, тепло р-ции идет на испарение воды. Выход ацетальдегида до 95%.
«-» токсичность, потери ртути.
2). На нетоксичном кат-ре: Н3РО4, Мg3(РО4)2, Zn3(PO4)2, CdHPO4·Ca3(PO4)2 - менее активны, чем ртутные, поэтому Т↑ до 350-400°. Идет бурное образование кротонового альдегида, поэтому берут 10-кратный избыток водяного пара по отношению к ацетилену.
Реактор (Б) – это колонный аппарат с несколькими слоями гетерогенного кат-ра. Ацетилен с парами воды
предварительно нагревают и вводят в реактор сверху вниз. Тепло р-ции снимается впрыском воды между слоями кат-ра. Чтобы капли воды не попали на кат-р устанавливают колпачковые тарелки. Продукты р-ции идут на конденсацию и разделение, непревращенный ацетилен возвращается в реактор. Выход ацетальдегида до 90%. Срок службы кат-ра до регенерации 5 суток, общий – 100 дней. Это «-».
Ацетальдегид – промежуточный продукт, из него получают уксусную к-ту, уксусный ангидрид, бутиловый спирт, акрилонитрил, молочную к-ту.
В-15.Способы получения ПАВ
Неионогенные Пав. Для их синтеза можно использовать различные соединения, имеющие длинные алкильные цепи и функциональные группы с подвижным атомам водорода.
1.
R
![]() --
OH + n СН2–(О)-CH2→
--
OH + n СН2–(О)-CH2→
→ R - O-[ CH2 – CH2-O-]n-H ; R=C12-C18
оксиэтилирование алкилфенолов.
2. Из высших жирных спиртов:ROH + n СН2 –(О)-CH2→ →R-O--[CH2–CH2-O-]n-H ; R=C12-C18
3. Из высших жирных кислот:RCOOH + n СН2 – (О)-CH2→ → R–C-O--[CH2–CH2-O-]n-H; R=C12-C18
По первым двум р-циям получают около 80% от всех неионогенных ПАВ. Эти ПАВ на ионы в воде не диссоциируют, а растворимость их объясняется образованием водородных связей между атомами водорода в воде и атомами кислорода в полиоксиэтилированнной цепочке.
Ионогенные ПАВ: В зависимости от того какими ионами обусловлена поверхностная активность делятся на 3 группы: анионактивные, катионактивные, амфолитные.
Анионактивные:
1) Алкиларилсульфанаты Na, к-ые получают сульфированием алкилароматики.
2) Алкилсульфаты
-первичные алкилсульфаты Na, к-ые получают сульфатированием первичных спиртов:
ROH + SO3 → ROSO2OH ;
ROSO2OH + NaOH → ROSO2ONa + H2O
-вторичные, к-рые получают сульфатированием вторичных спиртов или сульфатированием олефинов.
3) Алкилсульфанаты натрия, которые получают сульфохлорированием или сульфоокислением парафинов
RH + SO2 + Cl2 → RSO2Cl + HCl ;
RSO2Cl + 2NaOH → RSO2ONa + NaCl + H2O
RH + SO2 + 0.5O2 → RSO2OH ;
RSO2OH + NaOH → RSO2ONa + H2O
4) α- олефинсульфонаты натрия, к-ые получают сульфотированием α- олефинов
R-CH=CH2 + SO3→ R-CH=CH-SO2OH ;
R-CH=CH-SO2OH + NaOH → R-CH=CH-SO2ONa + H2O
5)Алкилкарбонаты натрия R-COONa, их получают омылением либо природных жиров, либо синтетических жирных кислот.
Все 5 анионактивные ПАВ диссоциируют в водном растворе на Na+ и анион-остаток.
Катионактивные: Поверхностной активностью обладает катион. К этим ПАВ относятся различные амины, аммониевые соли и аммониевые основания. Эти ПАВ обладают высокой антибактериальной активностью, применяют в медицине пищевой промышленности. Их широко применяют в медицине, пищевой пром-ти, а также как добавки к СМС.
Амфолитные: Содержат одну или несколько функциональных групп, к-ые в растворе в зависимости от кислотности среды бывают как катионактивные - в кислой, так и анионактивные – в щелочной. Их исп-ют для произв-ва жидких шампуней для волос.
Технологическая схема производства СМС сульфатированием спиртов серным ангидридом.
В реактор 1 пленочного типа непрерывно подают сверху вниз спирт и пары серного ангидрида, разбавленные воздухом до концентрации 4-7%. Снижение концентрации серного ангидрида необходимо для замедления высокой скорости очень экзотермичной реакции, что позволяет избежать перегревов смеси, снижения доли побочных реакций, потемнения продукта. Отвод выделяющегося тепла производится рассолом, проходящим по межтрубному пространству реактора. Продукты реакции поступают в сепаратор 2. где от жидкой фазы отделяется воздух и серный ангидрид. Газы поступают в абсорбер 3. орошаемый водой, для улавливания остатков серного ангидрида. Алкилсульфат из С-2 поступает на нейтрализацию концентрированным раствором щелочи в аппарат 4 с мешалкой и выносным холодильником. В аппарате 4 Т<60°С. Затем в аппарате 6 производится окончательная нейтрализация алкилсульфата до рН = 7. В смесителе 7 к раствору алкилсульфата добавляют ряд компонентов и полученную смесь сушат распылением в токе горячих топочных газов в сушилке 8. Унесенные с газами частицы улавливаются в циклоне 9. Порошок СМС транспортером 10 поступает на расфасовку.
В-17. Сульфатирование спиртов и олефинов.
Сульфатирование спиртов. Эти процессы применяют для получения ПАВ типа алкилсульфатов Nа(ROSO2ONa). ROH→ ROSO2OH; ROSO2OH +NaОН→ROSO2ONa + Н2О
Сульфатирующие агенты- амидосульфоновая к-та, хлорсульфоновая к-та, серный ангидрид и серная к-та.
1). ROH + NH2-SO2OH→ ROSO2ONH4
Р-ция идет необратимо при Т=100-120°, но ее используют редко, т.к к-та дорогая.
2). ROH + Cl-SO2OH → ROSO2OH + HCl
Р-ция иет необратимо при комнатной Т. Эта к-та более дешевая, но образуется НCl и возможно побочное образование хлорпарафинов.
3). ROH + SO3 → ROSO2OH
Р-ция идет очень быстро, необратимо, с большим выделением тепла. Это наиболее перспективный метод.
4). ROH + H2SO4-Н2О↔ ROSO2OH+ ROH ↔
↔ROSO2OR + H2O
Р-ция идет в две стадии, обратимо. На 1-ой стадии образуются моноалкилсульфаты, на второй – диалкилсульфаты. Первая стадия идет с более высокой скоростью. При ограничении времени р-ции содержание МАС больше равновесного.
Серная к-та играет роль также и кат-ра по следующему механизму:
H2SO4 +Н+↔ Н-О+Н─SO2OH+ ROH ↔
↔ R-О+Н─SO2OH + Н2О ↔ ROSO2OH +Н+
Т.к разрыв происходит по связи O-S в молекуле серной к-ты, то строение радикала в алкилсульфате будет точно такое же, что и в исходном спирте. Реакционная способность спиртов лежит в ряду: перв-> втор-> трет.
Р-ция идет с большим выделением тепла за счет разбавления к-ты водой.
Побочные реакции: 1. ROSO2OH + ROH→ ROR + H2SO4
2. R-CH2-CH2-OSO2OH→R-CH=CH2 + H2SO4
Для подавления побочных реакции поддерживают
20-40°С.
Сульфатирование олефинов. В качестве сульфатирующего агента используется только H2SO4. Р-ция идет обратимо в 2 стадии.
СН2 = СНR+ H2SO4↔ СН3-СНR-OSO2OH+ СН2 = СНR ↔ СН3-СНR-OSO2-О-СНR-СН3
Первая стадия идет быстрее второй, поэтому при ограничении времени р-ции содержание МАС больше равновесного. При низких Т равновесие р-ции почти полностью сдвинуто вправо.
Механизм: электрофильное присоединение по правилу Марковникова с образованием ионов карбония.
R – CH = CH2+Н+ ↔R – C+H-CH3 + H2SO4→
→R – CH(OSO2OH)- CH3 + H+
Чем длиннее и разветвленнее молекула исходного олефина, тем устойчивее ион карбония, тем выше реакционная способность олефина. «+» заряд свободно мигрирует по всей цепи молекулы, то в итоге образуется смесь вторичных алкилсульфатов. R – C+H-CH3 ↔
↔R`-C+H-CH2-CH2-CH3 ↔ R`-C+H-R``
Получается смесь продуктов R` R``-CH- OSO2OH.
Технология сульфатирования спиртов и олефинов. 1).Сульфатирование серной к-той. Используется реактор в виде куба с мешалкой и рубашкой адиабатического типа, т.к необходимо интенсивное охлаждение, к-ое осуществляют рассолом. Постепенно добавляют к кислоте спирт и олефин. Если необходимо процесс вести непрерывно, то берут каскад реакторов.
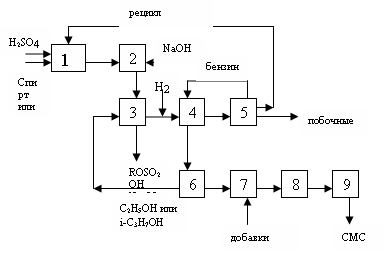
В блоке 1 происходит сульфатирование, далее реакционная масса в блоке 2 нейтрализуется щелочью и там происходят р-ции:
ROSO2OH + NaOH → ROSO2ONa + H2O
ROSO2OR + NaOH → ROSO2ONa + ROH
H2SO4 + 2NaOH → Na2SO4 + 2 H2O
Далее в блоке 3 реакционная масса экстрагируется этиловым и изопропиловым спиртом. При этом в спирте растворяются все орг в-ва, кроме сульфата натрия, к-ый отделяется как осадок. Далее спиртовый р-р разбавляют водой и в блоке 4 реакционную массу экстрагируют бензином. При этом в бензине растворяются побочные и непревращенные орг в-ва и этот экстракт в блоке 5 разделяется. Бензин возвращаетсяв блок 4, непревращенные продукты в блок 1, побочные отводят с установки. Водно-спиртовой р-р алкилсульфата натрия из блока 4 идет в блок 6, где отгоняется спирт, к-ый возвращается в блок 3, а водный р-р алкилсульфата натрия в блоке 7 смешивается с добавками, осушается в блоке 8 и в блоке 9 происходит измельчение и упаковка.