

Глава 5 дислипопротеидемии
Дислипопротеидемии это изменения содержания липопротеидов в крови, характеризующееся их повышением, снижением или полным отсутствием. Более частым явлением является гиперлипопротеидемии (ГЛП ), отражающая увеличение какого либо класса ЛП в крови. Классификация типов ГЛП была разработана и предложена еще в 1970 г. D. Fredrikson. В настоящее время она дополнена тем, что выявлена антиатерогенная роль ЛПВП. Установлено существование дисальфа – липопротеидемий, расширены представления о нарушениях обмена этого класса липопротеидемий. Различают следующие типы: тип 1 – гиперхиломикронемия; тип II α – гипербета-липопротеидемия; тип II б – гипербета - и гиперпребета - липопротеидемия; тип III – дисбета - липопротеидемия; тип 1V- гиперпребета - липопротеидемия; тип V – гиперпребета - липопротеидемия и гиперхиломикронемия.
Классификация выделяет связь нарушений обмена ЛП с развитием атеросклероза.
Различают первичные ДЛП, имеющие генетическую природу - эти заболевания носят семейный характер. Семейная предрасположенность имеется у небольшого числа людей, большую часть составляют нарушения обмена ЛП, связанные с воздействием факторов внешней среды.
ДЛП может встречаться также как сопутствующий синдром при некоторых заболеваниях внутренних органов - это – вторичные ДЛП.
Правильная диагностика типа нарушений липидного и ЛП – обмена первичных и вторичных гиперлипидемий необходима для назначения адекватного лечения и прогнозирования исходов. Фенотипирование ДЛП позволяет определить степень риска развития ИБС и наметить пути профилактики и лечения соответствующих нарушений обмена ЛП.
5.1. Первичные дислипопротеидемии
Среди первичных ДЛП различают варианты, связанные с нарушением метаболизма апо В – содержащих ЛП (I, II α, II б, III, IV, V типы ГЛП, а также абета и гипобета - липопротеидемии) и апо А - содержащих ЛП (α – альфа -, гипоальфа - и гиперальфа - липопротеидемии) (табл. 5).
Семейная гиперхиломикронемия – характерно резкое нарастание уровня ТГ (от 4000 до 10 000 мг/ дл), при этом уровень ХС в плазме также нарастает, но не столь значительно (соотношение ТГ: ХС около 9), ТГ возрастают за счет фракции ХМ. Они появляются в плазме при стоянии ее в холодильнике. В некоторых случаях повышено содержание ЛПОНП, а содержание ЛПНП и ЛПВП снижено.
Таблица 5
Генетические варианты первичных гиперлипопротеидемий
Тривиальное название |
Тип по D. Frederekson |
Причина ГЛП |
Связь с атеросклерозом |
Распространенность в популяциях |
Семейная ГХС гомозиготная гетерозиготная |
IIa |
Дефицит ЛПННП рецепторов |
++++ |
1:104 |
++ |
1:500 |
|||
Семейная ГХС обусловленная структурным дефектом апо В-100 |
IIa |
Нарушение катаболизма ЛПНП: дефект апо В (мутация 3500) |
+++ |
1:500-1.600 |
Семейная диабета-липопротеидемия (ремнатная гиперлипедемия) |
III |
Гомозиготнй тип Е2/Е2, усиление образование ЛПОНП, нарушение катаболизма ремиантных частиц |
++ |
1:5000 |
Семейная комбинированная гиперлипедемия |
IIa, или IIб, или IV |
Повышенный синтез апо В-100 и ЛПОНП |
++ |
1:100-1:300 |
Полигенная ГХС |
IIa |
? |
+ |
? |
Семейная ГТГ |
IV |
Усиленное образование и замедленный клирекс ТГ ЛПОНП |
+ |
1:500 |
Семейная гиперхило-микроанемия |
I и V |
Дефицит ЛПЛ или апо С-II |
- |
1^104 |
Семейная гиперЛП(а)емия |
Повышенный уровень ЛП(а) |
Усиленное образование ЛП(а) |
+++ |
? |
Семейная гиперальфа-липопротеидемия |
Повышенный уровень ЛПВП |
Усиленное образование апо А-t; дефицит ЭХС-ПБ; замедление катаболизма ЛПВП |
_ |
? |
При гиперхиломикронемии микроциркуляция крови в различных органах замедляется из-за трудности прохода большого числа крупных ХМ через капилляры. При секреции липазы в панкреатические капилляры образуется избыточное количество медленно перемещающихся продуктов расщепления ТГ, в частности НЭЖК, которые вызывают местное раздражение и воспаление. Этому способствует повышенная активность свободных радикалов, ведущая к образованию продуктов ПОЛ, проявляющих цитотоксическое и провоспалительное действие.
Семейная гипертриглицеридемия (ГТГ) - она соответствует IV типу ГЛП, отличается от гиперхиломикронемии тем, что повышение уровня ТГ в плазме крови при этом варианте ДЛП происходит за счет фракции ЛПОНП, аккумуляции ХМ при этом не наблюдается. Считается, что причиной развития ГТГ является или повышенное образование ЛПОНП в печени, или замедленный их катаболизм, или и то и другое вместе. ЛПОНП, обнаруживаемые у пациентов с семейной ГТГ, увеличены в размерах за счет обогащения триглицеридами. Выраженная ГТГ сочетается с гипоальфа - липопротеидемией, которая может возникнуть из - за ускоренного катаболизма апо А–I и А-II.
При семейной ГТГ отмечается аккумуляция мелких, плотных ЛПНП, содержащих больше белка и меньше ХС по сравнению с нормальными частицами. С их наличием связывают сдвиг липопротеидного спектра в атерогенную сторону у пациентов с ГТГ. Такие частицы отличаются более низкой аффинностью к специфическим ЛПНП - рецепторам на фибробластах и большим сродством к протеогликанам аорты. Поэтому при этой гипертриглицеридемии наблюдаются признаки поражения как коронарных, так и периферических сосудов, но атеросклеротические изменения развиваются медленно.
Семейная комбинированная гиперлипидемия отличается от других видов гиперлипидемий обнаружением различных типов ГЛП в одной и той же семье или у ближайших родственников. Среди них встречаются лица только с ГХС, или только с ГТГ, или с тем и другим одновременно. Определенные изменения выявлены и в ЛП плазмы крови: повышено содержание ЛПОНП, ЛППП, редко ЛПНП. При этом наблюдается гипоальфа - липопротеидемия. Клинически прослеживается связь с атеросклерозом.
Семейная дисбета-липопротеидемия (ремнантная гиперлипидемия).
Это III тип ГЛП, отличается от всех других вариантов присутствием в плазме крови β - ЛПОНП, иногда и ХМ. Эти липид - белковые комплексы обогащены ХС и апо Е. При этом типе ГЛП нарушен катаболизм ХМ и ЛПОНП, что связано с появлением в их составе изоформы апо Е 2, которая слабо взаимодействует с этими рецепторами. Слабое взаимодействие проявляет и гомозиготный тип Е2 : Е2 и гетерозиготный тип Е2 : Е 3 и Е2 : Е4, которые встречаются при данной патологии. Кроме дефектных изоформ апо Е проявлению данной патологии способствуют метаболические аномалии (диабет, гипотиреоз и др.), т. е. те состояния, которые способствуют повышенной секреции богатых триглицеридами ЛП, или ведут к снижению экспрессии рецепторов, через которые осуществляется катаболизм ремнантных частиц.
Изменения в липидах и ЛП спектре крови при III типе ГЛП: повышение ХС, ТГ, накопление β – ЛПОНП - развиваются у человека при избыточном употреблении пищи, богатой жирами. При ремнантной гиперхолестеринемии отмечаются поражения всего сосудистого русла и ксантоматоз.
Семейная ГХС соответствует II а типу ГЛП. Это наиболее серьезная патология в обмене ЛП, так как степень риска развития ИБС в 10- 20 раз выше по сравнению со здоровыми лицами. При этой патологии известно более 300 мутаций в гене апо В, Е рецептора. Делеции в гене белка - рецептора сопровождаются нарушением катаболизма ЛПНП. Их деградация осуществляется неспецифическим путем с помощью скэвенджер - рецепторов клеток РЭС. При этом происходит накопление ХС в макрофагах селезенки, купферовских клетках печени, гистиоцитах костного мозга и клетках других тканей. Часть макрофагов накапливает значительные количества ХС и превращается в пенистые клетки. Нарушение нормального рецепторного катаболизма ЛПНП приводит к резкому повышению уровня ХС в крови. Оно особенно выражено у гомозигот, для которых характерно полное отсутствие апо – В, Е рецепторов. Содержание ХС у них превышает 700 мг/дл (более 18 ммоль/л), а иногда может достигать 1000 мг/дл (более 25 ммоль/л). Клинически семейная ГХС выражается появлением признаков ИБС и инфаркта миокарда в сравнительно молодом возрасте и бугорчатых ксантом в детском возрасте.
Семейная ГХС, обусловленная структурным дефектом апо В-100. Повышенная концентрация ЛПНП обусловлена дефектом апо В-100, приводящая к ослаблению его взаимодействия со специфическими рецепторами. Мутантная форма апо В обнаружена среди людей с ГХС, ее распространенность и клинические проявления близки таковым при гетерозиготной форме семейной ГХС, обусловленной дефицитом апо В, Е–рецепторов.
Полигенная ГХС - является наиболее частой причиной повышенного уровня ХС крови среди лиц, у которых не выявляется патология в фуккционировании рецепторной системы удаления из крови ЛПНП. Эта форма ГХС обусловлена сочетанным действием ряда генов, дефектные белковые продукты которых способствуют умеренному нарастанию уровня ХС крови, а также дополнительному влиянию некоторых внешних факторов, вызывающих метаболические сдвиги в организме. При этой патологии ГХС частота встречаемости аллеля апо Е 4 выше, чем при других вариантах ГХС. Его присутствие сочетается с умеренным повышенным уровнем ХС ЛПНП и общего ХС в крови. Клинически проявляется преждевременным развитием атеросклероза, в том числе коронарного. Это наиболее частая форма у людей, перенесших инфаркт миокарда, а также страдающих другими проявлениями атеросклероза.
Семейная гиперальфа-липопротеидемия относится к патологии условно, так как частота обнаружения сердечно-сосудистых заболеваний и других проявлений нарушений обмена липидов и ЛП при высоком уровне ЛПВП меньше, чем у лиц с низким содержанием их. Гиперальфа-липопротеидемия рассматривается как синдром долголетия, так как у людей с этим вариантом ЛП - спектра увеличена продолжительность жизни. Одна из разновидностей гиперальфа - липопротеидемии связана с активацией синтеза апо А –1 без изменения синтеза апо – II. Симптомов клинической патологии не отмечается. Другая разновидность гиперальфа-липопротеидемии развивается при наследственном дефиците белка переносящего эфиры холестерина (ЭХС - ПБ).
Недостаточность ЭХС - ПБ проявляется также понижением содержания ХС во фракциях ЛПОНП, ЛППП и ЛПНП. Отмечаются выраженные изменения в структуре ЛП, а именно – образование более мелких, чем в норме, частиц ЛПНП и более крупных частиц ЛПВП. Нарастание ЛПВП в крови при дефиците ЭХС - ПБ обусловлено в первую очередь замедлением скорости катаболизма апо–I и апо–II, а скорость образования этих белков остается в пределах нормальных величин.
Все типы липопротеидемий имеют различную картину распределения полос липопротеидов при их разделении в полиакриламидном геле (рис.22), содержание липопротеидов можно определить с помощью денситометрии (рис.23)
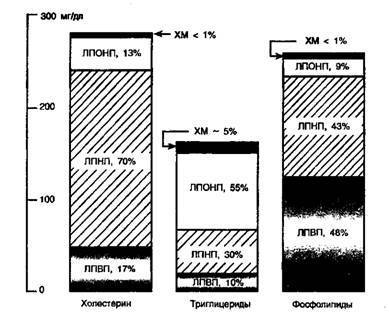
Рис. 22. Электрофоретическое разделение липопротеидов в полиакриламидном геле

Рис. 23. Количественное определение содержания липопротеидов