

96. Гемоглобины человека, структура. Транспорт кислорода и диоксида углерода. Гемоглобин плода и его физиологическое значение. Гемоглобинопатии.
Дыхательный пигмент, содержащийся в эритроцитах, с помощью которого осуществляется транспорт молекулярного кислорода из легких к тканям. Относится к сложным белкам - хромопротеидам. Молекула гемоглобина состоит из двух частей: простетической группы (гема), в состав которой входит атом железа, и белка типа альбуминов - глобина. На долю гема приходится 4 % молекулы гемоглобина, а глобина - 96 %. Гем относится к порфиринам, он содержит 4 пирроловые группы, соединенные в центре атомом железа. При потере железа гем превращается в гематопорфирин. Как в оксигемоглобине, так и в редуцированном гемоглобине железо находится в двухвалентной закисной форме. Трехвалентная (окисная) форма железа, не способная переносить молекулярный кислород, может образовываться при окислении в метгемоглобин. При разрушении эритроцитов (гемоглобина) в конечном счете в печени образуются желчные пигменты - билирубин и биливердин; в течение суток 3,6 г гемоглобина превращается в желчные пигменты. В капиллярах легких гемоглобин (гем) вступает с кислородом в непрочное соединение, образуя оксигемоглобин, а в капиллярах тканей происходит отщепление кислорода и восстановление гемоглобина. Последний вновь легко вступает в соединение с кислородом. Нарушение дыхательной функции крови может наблюдаться при уменьшении количества гемоглобина, эритроцитов в крови (анемии) и изменении качества гемоглобина: образовании карбоксигемоглобина (отравлении окисью углерода), метгемоглобина (отравлении гемолитическими ядами) сульфгемоглобина.
Типы нормального гемоглобина
С помощью физико-химических методов исследования (электрофорез, хроматография) удалось установить неоднородность человеческого гемоглобина, существование различных его типов как в нормальных, так и в патологических условиях. В настоящее время известны три формы нормального гемоглобина:
Примитивный гемоглобин (гемоглобин Р)
Это гемоглобин, который может быть обнаружен у трехсантиметрового зародыша, характеризуется высокой щелочной резистентностью и малой электрофоретической подвижностью. Находится в эритроцитах зародыша до 18 - недельного возраста (в основном между 7 - й и 12 - й неделями), затем сменяется фетальным гемоглобином.
Фетальный гемоглобин (гемоглобин F)
Представляет собой основную массу гемоглобина с 9 - 13 недельного возраста эмбриона. После третьего месяца - основной гемоглобин плода. Затем содержание его постепенно уменьшается за счет образования с 13 - й недели гемоглобина А. К моменту рождения гемоглобина F остается около 20 %, а 80 % приходится на гемоглобин А. К 4 - 5 месяцу жизни гемоглобина F остается всего 1 - 2 %. Существует метод цитологической дифференцировки гемоглобина F путем обработки мазка крови лимоннокислофосфатной буферной смесью с рН = 3,4. В этих условиях эритроциты, содержащие преимущественно гемоглобин А, подвергаются гемолизу и представляются на препарате в виде теней (стром). Эритроциты, в которых преобладает гемоглобин F, оказываются резистентными и контрастно окрашиваются.
Гемоглобин взрослых (гемоглобин А)
Представляет основную массу гемоглобина взрослых людей. С помощью электрофореза на крахмале установлено наличие нескольких фракций гемоглобина А:
гемоглобин A1 (основная фракция, на долю которой приходится 96 - 98 % всей массы гемоглобина).
гемоглобин A2 (2 - 5 %).
гемоглобин A3 (содержание менее 1 %).
Содержание гемоглобина F в крови взрослого человека составляет 1 - 2 %. Повышение этих величин возможно в условиях патологии. Преобладание того или иного типа гемоглобина совпадает во времени с периодами эмбрионального кроветворения: гемоглобин Р характерен для периода желточного кроветворения, гемоглобин F - для печеночного, гемоглобин А - для периода костномозгового кроветворения.
Аномальные гемоглобины
Наличие в эритроцитах людей аномальных или патологических гемоглобинов определяет состояния, обозначаемые как гемоглобинозы, или гемоглобинопатии. Это наследственные аномалии кроветворения, при которых молекулы патологических гемоглобинов имеют измененную структуру, поэтому подобные заболевания относятся к группе так называемых молекулярных болезней. Аномальные гемоглобины различаются своими физико - химическими свойствами (электрофоретической подвижностью, резистентностью к щелочам, растворимостью, изоэлектрической точкой), а также по молекулярной структуре глобиновой части (по одному из пептидов, в котором изменена последовательность аминокислот). Появление аномальных гемоглобинов объясняется мутационной теорией, передача потомкам аномального гена осуществляется по законам наследственности. У гетерозиготных особей (Аа) заболевание отсутствует или обнаруживаются субклинические признаки, у гомозиготных (от брака гетерозиготных особей) наблюдается развитие тяжелых анемий гемолитического типа. В настоящее время установлено более 200 аномальных гемоглобинов: B (S), С, D, Е, G, J, I, К, L, M, N, О, Р, Q и других, а также возможные их комбинации (SC, SD и др.). На XVI Международном конгрессе гематологов (Япония, 1976 г.) сделаны сообщения о новых аномальных гемоглобинах: Hb Beth (Nagel, США), Hb Austin (Moo-Peen, США), Hb Djelfa (Labie, Франция), Hb Hrosaki (Ohba, Япония), Hb Waco (Moo-Peen, США). Гемоглобинозы в гетерозиготной и гомозиготной форме имеют распространение в экваториальной Африке, странах, омываемых Средиземным морем, на Аравийском полуострове, в Южной Индии, на острове Шри-Ланка, в Юго-Восточной Азии, Южном Китае, южных районах США. Причину появления аномальных гемоглобинов объясняет малярийная гипотеза, согласно которой мутации в гене, контролирующем образование гемоглобина, возникли в странах с широким распространением тропической малярии. Было установлено, что наличие аномального гена в гетерозиготной форме повышает устойчивость людей к заболеванию, создает иммунитет к малярии, так как изменения молекулы гемоглобина препятствуют использованию его малярийным плазмодием.
Гемоглобин S
Отличается от гемоглобина А строением четвертого пептида, в котором на шестом месте вместо глутаминовой кислоты находится электрически нейтральный валин. Гемоглобин S менее растворим, нейтрален по заряду, электрофоретически менее подвижен. В капиллярах при отдаче кислорода гемоглобин S выпадает в осадок в форме веретенообразных кристаллоидов (тактоидов), которые растягивают оболочку и ведут к распаду эритроцитов. У гетерозиготов содержание гемоглобина S равняется 20 - 45 %, у гомозиготов - 60 - 90 %. Гетерозиготная форма аномалии протекает бессимптомно или сопровождается легкой гемолитической анемией. У гомозиготных особей уже с первых месяцев жизни развивается тяжелая форма серповидноклеточной анемии.
Гемоглобин F
Характерный для крови плода фетальный гемоглобин может быть обнаружен в повышенных количествах в эритроцитах крови недоношенных детей, при коклюше, серповидноклеточной анемии, талассемии, врожденной микроцитарной анемии, пернициозной анемии, острых и хронических лейкозах, миеломной болезни. Наибольшее содержание (до 97 %) наблюдается при большой талассемии.
Гемоглобин С
Отличается строением четвертого пептида молекулы гемоглобина, в котором на шестом месте вместо глутаминовой кислоты находится лизин. Центр распространения гена С - северная часть Ганы. Частота гетерозиготности по данным одних авторов, до 15 %, по данным других, - 16,5 - 28 %, среди негров США - 1,8 - 3% на Ямайке - 2,7 % (В. П. Эфроимсон). Наличие гена С в гомозиготном состоянии ведет к развитию выраженной спленомегалии, умеренной микроцитарной анемии с наличием эритроцитов мишеневидной формы. При наличии комбинации гемоглобинов С и S анемия оказывается более тяжелой.
Гемоглобин D
Обнаружен у 2 % берберов Марокко и у 0,4 % негров США. У гомозиготов наблюдается микроцитоз, слабый анизо- и пойкилоцитоз и мишеневидность эритроцитов. Описано несколько гемоглобинов D (в северо-западной Индии, среди сикхов в Индии, на острове Кипр, в Турции).
Гемоглобин Е
Обнаружен у жителей Юго-Восточной Азии: в Кампучии, Таиланде, Бирме, Бенгалии, у веддов Шри-Ланки, в северо-восточной Малайе, у населения Калимантана и Сулавеси. Частота распространения гена С в разных местностях колеблется от 1 - 3 до 13 (Таиланд) - 20 (Бирма) - 28 - 37 % (Кампучия). У гомозиготов ЕЕ наблюдается микроцитоз, компенсируемый развитием эритроцитоза (до 7 - 8 x 1012 /л). Отмечены комбинации генов ES и ЕТ, дающие сублетальный эффект. Клинические проявления при других гемоглобинозах выражены слабо, а распространение более ограниченное (гены G, I, J, К, L, M, N, О, Р, Q).
Гемоглобинопатии
Серповидноклеточная анемия - тяжёлое наследственное заболевание, обусловленное точечной мутацией гена, кодирующего структуру β-цепи гемоглобина (см. раздел 4). В результате в эритроцитах больных присутствует HbS, β-цепи которого в шестом положении вместо гидрофильной глутаминовой кислоты содержат гидрофобную аминокислоту валин. Появление гидрофобной аминокислоты недалеко от начала молекулы способствует возникновению нового центра связывания, поэтому при низком парциальном давлении кислорода тетрамеры дезокси-HbS ассоциируют, образуя длинные микротрубчатые образования, которые полимеризуются внутри эритроцитов. Полимеризация приводит к нарушению структуры эритроцитов, они приобретают серповидную форму и легко разрушаются. При этом заболевании отмечают анемию, прогрессирующую слабость, отставание в развитии и желтуху.
Носители гена серповидноклеточной анемии чаще всего встречаются среди африканского населения, так как они приобретают некоторое преимущество при заболевании малярией, часто встречающейся в странах с тропическим климатом. Причина сохранения гена серповидноклеточной анемии в популяции связана с тем, что в эритроцитах гетерозигот хуже развивается малярийный плазмодий, часть жизненного цикла которого проходит в эритроцитах человека. В связи с этим гетерозиготные носители дефектного
гена выживали при эпидемиях малярии, однако четверть их потомства погибала от серповиднок-леточной анемии.
Талассемии - наследственные заболевания, обусловленные отсутствием или снижением скорости синтеза α- или β-цепей гемоглобина. В результате несбалансированного образования глобиновых цепей образуются тетрамеры гемоглобина, состоящие из одинаковых протомеров. Это приводит к нарушению основной функции гемоглобина - транспорту кислорода к тканям. Нарушение эритропоэза и ускоренный гемолиз эритроцитов и клеток-предшественников при талассемиях приводит к анемии.
При β-талассемии не синтезируются β-цепи гемоглобина. Это вызывает образование нестабильных тетрамеров, содержащих только α-цепи. При этом заболевании в костном мозге из-за преципитации нестабильных α-цепей усиливается разрушение эритробластов, а ускорение разрушения эритроцитов в циркулирующей крови приводит к внутрисосудистому гемолизу. Как известно, для образования фетального гемоглобина р-цепи не требуются (см. раздел 4), поэтому клинически β-талассемия не проявляется до рождения, после чего происходит переключение синтеза HbF на НBА.
В случае α-талассемии недостаток образования α-глобиновых цепей приводит к нарушению образования HbF у плода. Избыточные γ-цепи образуют тетрамеры, называемые гемоглобином Барта. Этот гемоглобин при физиологических условиях имеет повышенное сродство к кислороду и не проявляет кооперативных взаимодействий между протомерами. В результате гемоглобин Барта не обеспечивает развивающийся плод необходимым количеством кислорода, что приводит к тяжёлой гипоксии. При α-талассемии отмечают высокий процент внутриутробной гибели плода. Выжившие новорождённые при переключении с γ- на β-ген синтезируют β-тетрамеры или НBН, который, подобно гемоглобину Барта, имеет слишком высокое сродство к кислороду, менее стабилен, чем НBА и быстро разрушается. Это ведёт к развитию у больных тканевой гипоксии и к смерти вскоре после рождения.
Наследственный сфероцитоз. Причиной этой патологии чаще всего является дефект белков цитоскелета эритроцитов - спектрина или ан-кирина, которые обеспечивают поддержание двояковогнутой формы клетки и эластичности мембраны. Эритроциты приобретают шарообразную форму, что приводит к уменьшению площади их поверхности и снижению скорости газообмена. Потеря эластичности клеточной мембраны приводит к повышению хрупкости и травматичности клеток и, как следствие, к ускорению их разрушения в сосудистом русле и селезёнке. Заболевание сопровождается анемией и желтухой. Удаление селезёнки (спленэктомия) при наследственном сфероцитозе улучшает состояние больных, так как предотвращает разрушение сфероцитов в селезёнке.
Мегалобластная (макроцитарная) анемия развивается при дефиците фолиевой кислоты или витамина В12.
Фолиевая кислота в виде кофермента (Н4-фолата) участвует в синтезе нуклеотидов. Недостаток фолиевой кислоты приводит к снижению скорости синтеза ДНК в быстроделящихся клетках, и в первую очередь в предшественниках эритроцитов. Клетки дольше пребывают в интерфазе, синтезируя гемоглобин, и становятся крупнее. Кроме того, из-за недостатка нуклеотидов они реже делятся, и количество эритроцитов снижается, а крупные мегалобласты быстрее разрушаются. Всё это в конечном итоге приводит к развитию анемии.
Аналогичная симптоматика развивается при недостатке в организме витамина В12. Этот витамин участвует в переносе метальной группы с N5-метил-Н4-фолата на гомоцистеин с образованием метионина и Н4-фолата (см. раздел 10). Недостаточность витамина В12 приводит к накоплению N5-метил-Н4-фолата в клетках. Дефицит Н4-фолата приводит к нарушению деления клеток и развитию анемии.
97. Биосинтез гема. Схема процесса, химизм первых двух реакций, место протекания. Регуляция активности ферментов АЛК-синтазы и АЛК-дегидратазы. Источники железа для синтеза гема, всасывание железа, транспорт в крови, депонирование.
Гем синтезируется во всех тканях, но с наибольшей скоростью в костном мозге и печени (рис. 13-2). В костном мозге гем необходим для синтеза гемоглобина в ретикулоцитах, в гепатоцитах - для образования цитохрома Р450.

Рис. 13-2. Синтез гема. Цифрами на схеме указаны ферменты: 1 - аминолевулинатсинтаза; 2 - аминолевулинатдегидратаза; 3 - порфобилиногендезаминаза; 4 - уропорфириноген III косинтаза; 5 - уропорфириногендекарбоксилаза; 6 - копропорфи-риноген III оксидаза; 7 - протопорфириногеноксидаза; 8 - феррохелатаза. Буквами обозначены заместители в пиррольных кольцах: М - метил, В - винил, П - остатки пропионовой кислоты, А - ацетил, ПФ - пиридоксальфосфат. Донором железа служит депонирующий железо в клетках белок ферритин.
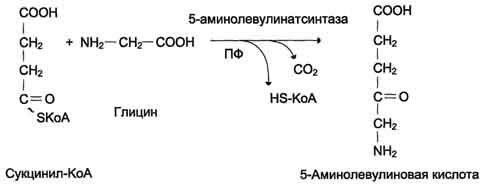
Рис. 13-3. Реакция образования 5-аминолевулиновой кислоты.

Реакция образования порфобилиногена.
В. Регуляция биосинтеза гема

Регуляция синтеза гема и гемоглобина. Гем по принципу отрицательной обратной связи ингибирует аминолевулинатсинтазу и аминолевулинатдегидратазу и является индуктором трансляции α- и β-цепей гемоглобина.

Регуляция синтеза аминолевулинатсинтазы. А - при высокой концентрации железа в ретикулоцитах оно присоединяется к железосвязывающему белку и снижает сродство этого белка к железочувствительному элементу (IRE) матричной РНК, кодирующей аминолевулинатсинтазу. Белковые факторы инициации трансляции связываются с мРНК и инициируют трансляцию аминолевулинатсинтазы. Б - при низком содержании железа в ретикулоцитах железосвязывающий белок обладает высоким сродством к IRE и взаимодействует с ним. Белковые факторы инициации трансляции не могут присоединиться к мРНК, и трансляция прекращается.
Аминолевулинатдегидратаза также аллостерически ингибируется гемом, но так как активность этого фермента почти в 80 раз превышает активность аминолевулинатсинтазы, то это не имеет большого физиологического значения.
Дефицит пиридоксальфосфата и лекарственные препараты, которые являются его структурными аналогами, снижают активность аминолевулинатсинтазы.
