

3. Соли, утворені слабкою кислотою і слабкою основою.
Наприклад, амоній ціанід гідролізуєтся по реакції
NH4 СN + HOH ↔ NH3.Н2О + HСN
або
NH4+ + СN- + HOH ↔ NH3.Н2О + HСN
Порівняння констант дисоціації кислоти [Ка (НСN) = 7,2.10-10] і основи [Кв (NН3.Н2О) = 1,79.10-5] дозволяє зробити висновок, що при гідролізі цієї солі буде лужна реакція (рН>7), оскільки Ка(НСN) < Кв (NН3.Н2О).
4. Соли, утворені сильною кислотою і сильними основою. Наприклад, NаС1, КNО3 гідролізу не піддаються. Це пов'язано з тим, що іони таких солей не утворюють з іонами води малодисоційовані сполуки. В цьому випадку рівновага дисоціації води у присутності солі майже не порушується, і розчини практично нейтральні (рН ≈7).
Механізм гідролізу солей полягає в поляризаційній взаємодії іонів соли з їх оболонкою гідрату. Чим сильніше це взаємодія, тим інтенсивніше протікає гідроліз.
Всі розглянуті випадки гідролізу стосувалися солей, утворених однокислотними основами і одноосновними кислотами. Солі багатоосновних кислот і багатокислотних основ гідролізуються ступінчасте, утворюючи при цьому кислі і основні солі.
Наприклад, соли Nа2СО3, К3РО4 гідролізуються ступінчасте, утворюючи кислі солі.
Соли А1С13, Сu (NО3) 2, СrСl3 гідролізуються ступінчасте з відтворенням на проміжних стадіях основних солей.
Іонні реакції
Багато хімічних реакцій протікають у водних розчинах. Якщо в цих реакціях беруть участь електроліти, то слід враховувати, що вони знаходяться у водному розчині в
дисоційованому стані, т.т. або тільки і вигляді іонів (сильні електроліти) і частково у вигляді молекул (слабкі електроліти). Таким чином, реакції між водними розчинами електролітів - це реакції, в яких беруть участь іони. Тому такі реакції називаються іонними реакціями.
Ці реакції можливі тільки в тому випадку, якщо між іонами відбувається хімічна взаємодія, тобто які-небудь іони одного електроліту і які-небудь іони іншого електроліту зв'язуються один з одним і утворюють:
а) нерозчинна речовина;
б) газоподібна речовина;
в) мало дисоціююча речовина (слабкий електроліт).
г) комплексні сполуки.
При складанні іонних рівнянь реакцій слід керуватися тим, що речовини мало дисоційовані, малорозчинні (випадні в осад) і газоподібні зображуються в молекулярній формі. Стрілка вниз ↓, що стоїть при формулі речовини, означає, що речовина йде з сфери реакції у вигляді осаду, а стрілка вгору↑означає, що речовина віддаляється з сфери реакції у вигляді газу. Сильні розчинні електроліти, як повністю дисоційовані, пишуться у вигляді іонів. Сума електричних зарядів в лівій частині рівняння повинна бути рівна сумі електричних зарядів в правій частині.
Напишемо рівняння реакції взаємодії розчинів хлориду барію і сульфату натрію в молекулярній, іонній і скороченій іонній формах.
Розіб'ємо рішення задачі на чотири етапи.
1. Записуємо рівняння реакції в молекулярній формі:
ВаС12 + Nа2SO4 = ВаSО4↓ + 2NаСl
2. Переписуємо це рівняння, зобразивши добре диссоціюючі речовини у вигляді
іонів, а реакції, що йдуть з сфери, - у вигляді молекул:
Ва2+ + 2С1- + 2Nа+ + SО42- = ВаSO4↓ + 2Nа+ + 2С1-
Це іонне рівняння реакції.
3. Виключаємо з обох частин рівності однакові іони, тобто іони, що не беруть
участь в реакції (підкреслені):
Ва2+ + 2С1- + 2Nа+ + SО42- = ВаSO4↓ + 2Nа+ + 2С1-
4. Виписуємо рівняння реакції в остаточному вигляді:
Ва2+ + SО42- = ВаSO4↓
Це скорочене іонне рівняння реакції.
Як видно з цього рівняння, суть реакції зводиться до взаємодії іонів Ва2+ і SО42- внаслідок чого утворюється осад ВаSO4↓ .
При цьому зовсім не має значення, до складу яких електролітів ці іони входили до їх взаємодії.
Іонними рівняннями можуть бути зображені будь-які реакції, що протікають в розчинах між електролітами. Якщо при таких реакціях не відбувається зміни зарядів іонів (не змінюється ступінь окислення), то вони називаються обмінними. Численні реакції обміну в розчинах електролітів діляться на необоротні і оборотні. Необоротними є реакції, якщо вони протікають по наступних схемах:
1. Реакція з відтворенням осадів, наприклад:
AgNO3 + HCl = AgCl ↓ + HNO3
Ag+ + NO3- + H+ + Cl- = AgCl ↓ + H+ + NO3-
Ag+ + Cl- = AgCl ↓
2. Реакція з утворенням газоподібних малорозчинних речовин, наприклад:
Nа2СO3 + Н2SO4 = Na2SO4 + СO2↑ + Н2О
2Nа+ + СО3- + 2Н+ + SО42- = 2Nа+ + SО42- + СO2↑ + Н2О
СО3- + 2Н+ = СO2↑ + Н2О
3. Реакції з утворенням малодисоційованих речовин (слабких електролітів), наприклад:
НСl + КОН = КС1 + Н2О
Н+ + С1- + К+ + ОН- = К+ + С1- + Н2О
Н+ + ОН- = Н2О
Таким чином, реакції обміну в розчинах електролітів практично необоротно протікають у бік утворення осадів (малорозчинних речовин), газів (легколетючих речовин), слабких електролітів (малодисоціюючих сполук).
Решта реакцій обміну є оборотними.
Роль гидролиза в биохимических процессах трудно переоценить. Прежде всего необходимо отметить ферментативный гидролиз, благодаря которому три основных компонента пищи — жиры, белки, углеводы — в желудочно-кишечном тракте расщепляются водой на более мелкие фрагменты. В общем виде гидролиз пищевых компонентов описывается уравнением
R1—О—R2 + Н2О → R1—ОН + R2—ОН
Где R1,R2 — фрагменты биоорганической молекулы, связанные через кислород.
Без этого процесса не было бы возможно усвоение пищевых продуктов, так как всасываться в кишечнике способны только относительно небольшие молекулы. Так, например, усвоение полисахаридов и дисахаридов становится возможным лишь после полного их гидролиза ферментами до моносахаридов. Точно так же белки и липиды гидролизуются до веществ, которые лишь потом могут усваиваться.
Гидролиз АТФ. Для роста и нормального функционирования всем животным необходима энергия. Человек получает энергию как за счет многостадийного процесса окисления пищи — белков, жиров и углеводов, так и за счет гидролиза некоторых сложных эфиров, амидов, пептидов и гликозидов. Однако главным источником энергии для многих биологических процессов — биосинтеза белка, ионного транспорта, сокращения мышц, электрической активности нервных клеток — является аденозинтрифосфат (АТФ).
АТФ принадлежит к бионеорганическим соединениям, так как состоит из органической части — аденозина и неорганической части — трех связанных в цепь фосфатных групп.
Энергия, необходимая для жизнедеятельности, высвобождается вследствие гидролиза АТФ. При рН ≥7,0 АТФ существует в виде аниона АТФ4-, так как все фосфатные группы при этом значении рН ионизированы.
Гидролиз АТФ записывают в виде кислотно-основного равновесия:
АТФ4- + Н20 ↔ АДФ3- + НРО42- + Н+; ΔG° = —30,5 кДж/моль
где АДФ3- — анион аденозиндифосфата.
Реакция сопровождается убылью энергии Гиббса ΔG0
Роль гідролізу в біохімічних процесах важко переоцінити. Перш за все необхідно відзначити ферментативний гідроліз, завдяки якому три основні компоненти пищи, - жири, білки, вуглеводи - в шлунково-кишковому тракті розщеплюються водою на дрібніші фрагменти. У загальному вигляді гідроліз харчових компонентів описується рівнянням
R1-О-R2 + Н2О → R1-ОН + R2-ОН
Де R1,R2 - фрагменти біоорганічної молекули, зв'язані через кисень.
Без цього процесу не було б можливе засвоєння харчових продуктів, оскільки всмоктуватися в кишечнику здатні тільки відносно невеликі молекули. Так, наприклад, засвоєння полісахаридів і дисахаридів стає можливим лише після повного їх гідролізу ферментами до моносахаридів. Так само білки і ліпіди гідролізуються до речовин, які лише потім можуть засвоюватися.
Гідроліз АТФ. Для зростання і нормального функціонування всією твариною необхідна енергія. Людина отримує енергію як за рахунок багатостадійного процесу окислення їжі - білків, жирів і вуглеводів, так і за рахунок гідролізу деяких складних ефірів, амідів, пептидів і глікозидів. Проте головним джерелом енергії для багатьох біологічних процесів - біосинтезу білка, іонного транспорту, скорочення м'язів, електричній активності нервових клітин - є аденозинтрифосфат (АТФ).
АТФ належить до біонеорганічних сполук, оскільки складається з органічної частини - аденозіна і неорганічної частини - трьох зв'язаних в ланцюг фосфатних груп.
Енергія, необхідна для життєдіяльності, вивільняється унаслідок гідролізу АТФ. При рн ≥7,0 АТФ існує у вигляді аніона АТФ4-, оскільки всі фосфатні групи при цьому значенні рН іонізовані.
Гідроліз АТФ записують у вигляді кислотний-основної рівноваги:
АТФ4- + Н20 ↔ АДФ3- + НРО42- + Н+; ΔG° = -30,5 кдж/моль
де АДФ3- - аніон аденозіндифосфату.
Реакція супроводжується спадом енергії Гіббса ΔG0
Самостійна робота: Окисно – відновні реакції
Окислювально-відновними реакціями називають хімічні процеси, що супроводжуються перенесенням електронів від одних молекул або іонів до інших.
Наприклад, при витісненні міді з розчину СuSО4 цинком
Zn (т) + СuSО4(р) = ZnSО4 (р) + Сu (т)
Електрони від цинку переходят до іонів міді:
Zn (т) + Сu2+ (р) = Zn2+ (р) + Сu (т) .
При окислювально-відновних реакціях протікають взаємозв'язаних два процеси: окислення і відновлення.
Окисленням називають процес втрати електронів. Відновленням - процес приєднання електронів.
Речовини, атоми або іони яких віддають електрони, називають відновниками Red.
Речовини, атоми або іони яких приєднують електрони (або зволікають до себе загальну пару електронів), називають окислювачами Ох.
У реакції цинку з СuSО4 іони Сu2+ приєднують електрони
Си2+ + 2е- = Си0 .
Атоми цинку вітдают електрони:
Zn°= Zn2+ + 2е-
Відповідно СuSО4 - окислювач, Zn - відновник. Ступінь окислення. Одним з основних понять неорганічної хімії є поняття про ступінь окислення (СО).
Ступенем окислення елементу в сполуці називають формальний заряд атома елементу, обчислений з припущення, що валентні електрони переходять до атомів з більшою відносною електронегативністю (ВЕН) і всі зв'язки в молекулі сполуки є іонними.
Ступінь окислення елементу Е указують вгорі над символом елементу із знаком
+7
«-» або «+» перед цифрою, наприклад Мn . Ступінь окислення іонів, що реально існують в розчині або кристалах, співпадає з їх зарядовим числом і позначається аналогічно із знаком «+» або «-» після цифри, наприклад Сl-, Са2+.
Застосовують також метод Штока позначення ступеня окислення римськими цифрами після символу елементу: Мn(VII), Fе (III).
Питання про знак ступеня окислення атомів в молекулі вирішується на підставі зіставлення електронегативностей зв'язаних між собою атомів, які утворюють молекулу. При цьому атом з меншою електронегативністю має позитивний ступінь окислення, а з більшою електронегативністю - негативну.
Слід зазначити, що не можна ототожнювати ступінь окислення з валентністю елементу. Валентність, визначувана як число хімічних зв'язків, якими даний атом сполучений з іншими атомами, не може дорівнювати нулю і не має знаку «+» або «-». Ступінь окислення може мати як позитивні, так і негативні значення, а також приймати нульове і навіть дробне значення. Так, в молекулі СО2 ступінь окислення С рівна +4, а в молекулі СН4 ступінь окислення С рівна -4. Валентність же вуглецю і в тому і в іншому з'єднанні рівна 4.
При окисленні елементу ступінь окислення збільшується, інакше кажучи, відновник при реакції підвищує ступінь окислення:
+ 2 +7
Мп = Мп + 5е-
Навпаки, при відновленні елементу ступінь окислення знижується, тобто при реакції окислювач зменшує ступінь окислення:
+5 -1
С1 + 6е- = С1
Таким чином, можна дати і таке формулювання окислювально-відновним реакціям:
окислювально-відновними реакціями називають реакції, що протікають із зміною ступеню окислення атомів елементів, що входять до складу реагуючих речовин.
Окислювачі і відновники. Для прогнозу продуктів напряму окислювально-відновних реакцій корисно пам'ятати, що типовими окислювачами є прості речовини, атоми яких мають велику ВЕН > 3,0 (елементи VIA і VIIА-груп). З них найбільш сильні окислювачі фтор (ВЕН = 4,0), кисень (ВЕН =3,0), хлор (ВЕН = 3,5). До важливих окислювачів відносяться РbО2, КМnО4, Се(SО4) 2, K2Cr2O7, К2СгО4, НС1О, НСlOз, КСlO4, NаВiOз, Н2SО4 (конц.), HNO3(конц.), Nа2О2, (NН4)2S2О8, КVОз, КС1О3, Н2О2, МnО2 і інші речовини, які містять атоми з вищою або високою СО.
До типових відновників відносяться прості речовини, яких мають малі ВЕН<1,5 (метали Iа- і IIА-груп і деякі інші метали). До важливих відновників відносяться : Н2S, NН3, НI, КI, SnС12, FеSО4, С, Н2,СО, Н2SО3, Cr2(SО4)3, СuС1, Na2S2О3 і інші речовини, які містять атоми з низькими СО.
Речовини, що містять атоми в максимальному і мінімальному ступенях окислення, можуть бути, відповідно, тільки окислювачами, наприклад К2Сг2О7, КМnО4, РbО2, НС1О4, або тільки відновниками, наприклад NНз, Н2S, НI.
Важливими процесами в тваринних організмах є реакції ферментативного окислення речовин - субстратів: вуглеводів, жирів, амінокислот. В результаті цих процесів організми отримують велику кількість енергії. Приблизно 90 % всій потребі дорослого чоловіка в енергії покривається за рахунок енергії, що виробляється в тканинах при окисленні вуглеводів і жирів. Решту частини енергії ~10% дає окислювальне розщеплювання амінокислот.
Біологічне окислення протікає по складних механізмах за участю великого числа ферментів. У мітохондріях окислення відбувається в результаті перенесення електронів від органічних субстратів - інтермедіатов на елементний кисень, який при цьому відновлюється до води:
½О2 + 2Н+ + 2е- → Н2О
Як переносники електронів в дихальний ланцюг мітохондрій входять різні білки, що містять різноманітні функціональні групи, які призначені для перенесення електронів. У міру просування по ланцюгу від одного інтермедіата до іншого електрони втрачають вільну енергію. На кожну пару електронів, переданих по дихальному ланцюгу кисню синтезуються три молекули АТФ. Вільна енергія, що вивільняється при перенесенні двох електронів на кисень, складає 220 кдж/моль.
На синтез однієї молекули АТФ в стандартних умовах витрачається 30,5 кдж. Звідси ясно, що досить значна частина вільної енергії, що виділяється при перенесенні однієї пари електронів, запасається в молекулах АТФ., З цих даних стає зрозумілою і роль багатостадійної передачі електронів початкового відновника до кисню. Велика енергія (220 кдж), що виділяється при перенесенні однієї пари електронів до кисню, розбивається на ряд порцій, відповідних окремим стадіям окислення. На трьох таких стадіях кількість енергії, що виділяється, приблизно відповідає енергії, необхідній для синтезу однієї молекули АТФ.
Окислювально-відновні реакції лежать в основі методів оксидиметрії, які застосовують в клінічному аналізі для визначення в крові іонів кальцію, сечової кислоти, ферментів каталази і пероксидази, цукру, а в санітарно-гігієнічному - для визначення окислюваності води, вміст активного хлора в хлорному вапні, залишкового хлора в господарський-питній воді.
Біонеоганічна хімія
Лекція № 5 . Буферні системи, класифікація та механізм дії
5.1 Буферні розчини, їх класифікація. Рівняння Гендерсона—Гассельбаха.
5.2 Механізм буферної дії.
5.3 Буферна ємність. Буферні системи організму.
Кислотно-основний стан крові.
Самостійна робота: Визначення рН середовища розчинів солей.
5.1 Буферні розчини, їх класифікація. Рівняння Гендерсона—Гассельбаха.
Однією з характерних властивостей внутрішнього середовища організмів є постійність концентрації водневих іонів (ізогидрія). Так, наприклад, рН крові людини дорівнює 7,36. Збереження цього показника забезпечується сумісною дією ряду фізико-хімічних і фізіологічних механізмів, з яких дуже важлива роль належить буферним системам. .
Буферними системами (буферами) називають розчини, що володіють властивістю достатньо стійко зберігати постійність концентрації водневих іонів як при додаванні кислот або лугів, - так і при розведенні.
З погляду протонної теорії буферна дія розчинів обумовлена наявністю кислотно-основної рівноваги загального типу:
В + Н+ ↔ ВН+ .
Основа зв'язана кислота
НА ↔ Н+ + А- .
кислота зв'язана основа
Зв'язані кислотний-основні пари В/ВН+ і А- / НА називають буферними системами.
Буферні розчини грають велику роль в життєдіяльності. До виняткових властивостей живих організмів належить їх здатність підтримувати постійність рн біологічних рідин, тканин і органів - кислотний-основний гомеостаз. Ця постійність обумовлена наявністю декількох буферних систем, що входять до складу цих тканин.
Класифікація кислотний-основних буферних систем. Буферні системи можуть бути чотирьох типів.
1. Слабка кислота і її аніон А-/НА. Наприклад, ацетатна буферна система СН3СОО-/СН3СООН в розчині СН3СОONa і СНЗСООН, область дії - інтервал рН 3,8-5,8. Водень - карбонатна система НСО3-/Н2СОЗ в розчині NaНСОЗ і Н2СОЗ, область її дії - рН 5,4-7,4.
2. Слабка основа і її катіон В/ВН+. Наприклад, аміачна буферна система NНЗ/NН4+ в розчині NН3 і NН4Сl область її дії - рН 8,2-10,2.
3.Аніони кислої і середньої солі або двох кислих солей. Наприклад, карбонатна буферна система СОЗ2- /НСО3- в розчині Nа2СО3 і NаНСО3 (область її дії рН 9,3-11,3). Фосатная буферна система НРО42-/Н2РО4- в розчині Na2НРО4
NаН2РО4, область її дії рН 6,2-8,2. Ці сольові буферні системи можна віднести і до 1-го типу, оскільки одна з солей цих буферних систем виконує функцію слабкої кислоти. Так, у фосфатній буферній системі аніон Н2РО4- є слабкою кислотою.
4.Іони і молекули амфолітів. До них відносять амінокислотні і білкові буферні системи. Якщо амінокислоти або білки знаходяться в ізоелектричному стані (сумарний заряд молекули рівний нулю), то розчини цих сполук не є буферними. Вони починають проявляти буферну дію, коли до них додають деяку кількість кислоти або лугу. Тоді частина білка (амінокислоти) переходить з ізоелектричного стану у форму «білок-кислота» або відповідно у форму «білок-основа». При цьому утворюється суміш двох форм білка: а) слабка «білок - кислота» + сіль цієї слабкої кислоти; б) слабка «білок-основа» + сіль цієї слабкої основи:

Склад буферних сумішей. Буферні системи (суміші або розчини) по складу бувають двох основних типів: а) із слабкої кислоти і солі, утвореною слабкою кислотою і сильною основою; б) із слабкої основи і солі утвореною слабкою основою і сильною кислотою.
Як приклад приводяться наступні буферні суміші:
СН3СООН + СН3СООNа — ацетатний буфер ;
Н2СO3+NаНСО3 — бікарбонатний буфер ;
NН4ОН + NH4С1 — аміачний буфер ;
Білок-кислота (Рt —СООН) + білок-сіль (Pt -— СООNа) — білковий буфер;
NaН2РО4 + Nа2НРО4 — фосфатний буфер.
Фосфатна буферна суміш складається з двох солей, одна з яких є однометалевою, а друга - двохметалевою сіллю фосфорної кислоти.
Потрібно вказати, що буферні розчини можуть носити і змішаний характер, включаючи аніони різних слабких кислот, наприклад фосфатно - цитратний NaН2РО4 + С6Н8О7
(лимонна кислота).
Визначення кислотний-основної рівноваги. На практиці порушення кислотний-основної рівноваги класифікують на підставі їх вимірювань в системі гідрокарбонат-вугільна кислота (основна буферна система позаклітинної рідини). Оскільки унутрі- і позаклітинна буферні системи функціонально зв'язані між собою, вимірювання тільки концентрації гідрокарбонату в плазмі дозволяє отримати інформацію про загальну буферну систему організму. Взаємозв'язок окремих елементів гідрокарбонатної системи зазвичай описують рівнянням Гендерсона-Гассельбаха:
[НСО3-]
рН = рК + lg -------------
[Н2СО3]
де рK (від’ємний десятинний логарифм константи дисоціації )вугільної кислоти складає 6,1. Величина [Н2СОЗ] розраховується як добуток a•ρСO2, де а-коеффіциент розчинності двоокису вуглецю в рідких середовищах організму, рівний 0,031 ммоль/л на 1 мм рт. ст. парціального тиску двоокису вуглецю. При нормальному ρСО2 (тобто 40 мм рт. ст.) розрахункова величина [Н2СО3] складатиме 40•0,031=1,2 ммоль/л.
5.2 Механізм буферної дії.
Лужний резерв виміряється об'ємом хімічно зв'язаної СО2 (головним чином, у вигляді гідрокарбонатів) з 100 мл плазми крові, насиченої газом з парціальним тиском СО2 53,3 кПа (як в альвеолярному повітрі). Резервну лужність виражають в об'ємних частках хімічно зв'язаного в крові диоксида вуглецю, у нормі вона відповідає об'ємній частці 50-70% (або 25-30 моль/м3).
Буферні системи, що перешкоджають різкому зниженню рН крові при надходженні в неї кислот, обмежують також і зростання рН у випадку надходження в кров лугів. У цьому випадку іони гідроксилу, що утворюються при дисоціації лугів або при гідролізі відповідних солей, взаємодіють з вільними кислотами Н2СО3, ННb і ННbО2 і іонами дигідрофосфата по схемах:
Н2СО3 + NаОН ↔ NаНСО3 + Н2О
NаН2РО4 + NаОН ↔ Nа2НРО4 + Н2О
ННb + NаОН ↔ NaНb + Н2О
ННbО2 + NaОН ↔ NаНbО2 + Н2О зі зрушенням рівноваг (1), (3) - (8) справа наліво.(див. пункт 5,3)
З останніх двох співвідношень випливає, що при нормальному значенні рН крові тільки 13% із загального числа молекул гемоглобіну дисоційовано на іони Н+ і Нb-, у той час як оксигемоглобін у тих же самих умовах дисоційований на 72%.
У більшій мірі буферним системам крові приходиться протидіяти зміні рН убік зменшення його значення, оскільки в процесі засвоєння їжі в організмі генерується значна кількість диоксида вуглецю (550-775 г/добу), при взаємодії якого з вологою утворюється вугільна кислота в кількості, еквівалентній надходженню від 25 до 35 моль/добу іонів гідрогену. Зниження рН у крові з відповідним зменшенням її лужного резерву сприяє також процес, який відбувається в легенях при перетворенні гемоглобіну в оксигемогло-бін :
ННb + О2 ↔ ННbО2,
оскільки ННbО2 більш сильна кислота, ніж ННb. Протидіє цьому збільшенню кислотності зворотний процес перетворення оксигемоглобіну в гемоглобін, що відбувається в капілярах тканин організму і спрямований на збільшення лужного резерву.
Таким чином, буферні системи дозволяють живому організму, як відкритій стаціонарній системі, реалізувати принцип консервативності Ле Шателье - Брауна, протидіяти впливу зовнішніх факторів, спрямованих як на зниження, так і на збільшення рН рідких середовищ, тобто зберігати гомеостазис.
Усі буферні системи крові і тканинних рідин утворюють єдину взаємозалежну систему. З огляду на цей взаємозв'язок, можна, наприклад, пояснити, чому на процеси поглинання кисню гемоглобіном і звільнення його оксигемоглобіном істотний вплив має диоксид вуглецю.
Усередині еритроцитів існують рівноваги, які безпосередньо впливають одна на одну:
ННbО2 ↔ ННb + О2 (9)
ННbO2 ↔ Н+ + НbО2 (10)
НbО2 ↔ Нb- + О2 (11)
Недисоційовані молекули ННbО2 віддають кисень (9) легше, ніж утворені ними іони НbО2 (11). Тому, коли у внутрішньому рідкому середовищі еритроцита через поглинання СО2 і утворення Н2СО3, що відбувається в тканинах, зростає концентрація Н+-іонів, це приводить до зрушення рівноваги (10) уліво - убік збільшення концентрації недисоційованих молекул ННbО2, і виділення в газову фазу О2 (9) підсилюється. Наявні в цьому ж водному середовищі іони Нb- реагують з вугільною кислотою з утворенням малодисоційованих молекул ННb і гідрокарбонат-іонів
Нb- + Н2СО3 ↔ ННb + HCO3- ,
причому останні, дифундуючи через оболонку еритроцитів у плазму, виносяться зі струмом крові.
Коли венозна кров, збагачена гемоглобіном і містить в собі надлишок розчиненого диоксида вуглецю, повертається в легені, гемоглобін реагує з киснем і утворює оксигемоглобін. Причому Н+-іони, порівняно легко звільняються від оксигемоглобіну і зв'язуються з бікарбонат-іонами, утворюючи вільну вугільну кислоту. Значна частина цієї кислоти розпадається потім (термічна дисоціація) на воду і диоксид вуглецю:
ННbО2 + НСО3- ↔ НbО2 + Н2СО3
Н2СО3 ↔ СО2 + Н2О
Із легень СО2 видаляється в атмосферу за рахунок легеневої вентиляції.
5.3 Буферна ємність. Буферні системи організму.
Кислотно - основний стан крові.
Буферна суміш підтримує постійним рН тільки при умові, що кількість сильної кислоти або сильної основи, що додаються до розчину, не перевищує визначеної фізичної величини. Межа, в якій виявляється буферна дія, називається буферною ємністю.
Чисельне значення буферної ємності (В) визначається числом моль-еквівалентів сильної кислоти або основи, яку необхідно додати до 1 м3 буферної суміші, щоб змінити значення рН на одиницю, що можна виразити рівнянням:
С
В = -----------------
рН2 - рН1
д е
С - кількість лугу або кислоти
(кмоль-екв/м3),
що
додається до буферного
розчину, а рН2
– рН1
- водневий
показник до і після додавання кислоти
або
лугу.
е
С - кількість лугу або кислоти
(кмоль-екв/м3),
що
додається до буферного
розчину, а рН2
– рН1
- водневий
показник до і після додавання кислоти
або
лугу.
Величину, що характеризує здатність буферного розчину протидіяти зсуву реакції середовища при додаванні сильних кислот або сильних підстав, називають буферною ємністю розчину.
На мал. 3.4 показаний типовий графік залежності буферної ємності від рН на прикладі ацетатної кислотно-основної системи СНЗСОО-/СНЗСООН. З даних, представлених на мал. 3.4, видно, що максимальна буферна емність, т. т. найбільша здатність цієї системи протистояти зміні рН, відповідає значенню рН = рКа = 4,76. Це витікає з рівняння Гендерсона - Гассельбаха. При рН == рКа відношення с(сіль) /с(кисл.) = 1, тобто в розчині є однакова кількість соли і кислоти. При такому співвідношенні концентрацій рН розчину змінюється у меншій мірі, чим при інших, і, отже, буферна ємність максимальна при рівних концентраціях компонентів буферної системи і зменшується з відхиленням від цього співвідношення.
Мал. 3.4 демонструє і інший важливий момент. Робоча ділянка буферної системи, тобто здатність протидіяти зміні рН при додаванні кислот і лугів, має протяжність приблизно одну одиницю рН з кожного боку від точки рН = рКа. Поза цим інтервалом буферна ємність швидко падає до 0. Інтервал рН= рКа ± 1 називається зоною буферної дії.
Величина буферної ємності залежить від:
а) концентрації компонентів буферної суміші;
б) співвідношення між цими концентраціями.
Зі збільшенням концентрації компонентів буферної суміші збільшується резерв, за допомогою якого утримується сталість рН. Найбільша величина буферної ємності будь-якої буферної суміші досягається при рівності еквівалентних концентрацій обох компонентів:
С солі = С кислоти або Ссолі = С лугу
У такому буферному розчині концентрація іонів гідрогену дорівнює константі дисоціації [Н+] = К або рН = рК.
Стійкість буферного розчину, де [Н+] = К, визначається тим, що перехід визначеної кількості С лугу або кислоти з чисельника у знаменник або навпаки у цих умовах викликає мінімальну зміну у співвідношенні С кислота/Ссіль
Таким чином, буферні суміші мають наступні властивості:
Концентрація іонів гідрогену буферних сумішей мало залежить від розведення.
Додавання до буферних сумішей невеликих кількостей сильної кислоти або сильної основи дуже мало змінює концентрацію іонів гідрогену (у межах буферної ємності розчину).
Величина буферної ємності залежить від концентрації ком- понентів буферної суміші і від співвідношення між цими концентраціями. Зі збільшенням концентрації компонентів буферної суміші збільшується її буферна ємність.
Максимальна буферна дія виявляється у випадку, якщо кис- лота і сіль присутні в розчині в еквівалентних кількостях
Буферні суміші мають велике значення для живих організмів, підтримуючи сталість рН у крові і тканинах. Буферною сумішшю крові є карбонатна суміш, що складається з NaНСО3 і СO2; фосфатна суміш, що складається з дигідрофосфату і гідрофосфату (NaН2РО4 + Na2НРО4); білкові буферні суміші - гемоглобін, альбуміни, глобуліни тощо. Велике значення для організмів має також сталість рН розчинів зовнішнього середовища
Ґрунтові розчини також мають буферність, обумовлену вмістом у них гідрокарбонатів, фосфатів і ін.
Буферні суміші знаходять застосування в хімічних лабораторіях при визначенні рН розчинів колориметричним (індикаторним) методом.
Кислотно-лужна рівновага і головні буферні системи в організмі людини
Організм людини має у своєму розпорядженні тонкі механізми координації фізіологічних і біохімічних процесів, що відбуваються в ньому, і підтримки сталості внутрішнього середовища (оптимальних значень рН і рівнів вмісту різних речовин у рідинах організму, температури, кров'яного тиску і тощо). Ця координація названа, за пропозицією В.Кеннона (1929), гомеостазисом (з грецької "гомео" - подібний; "стазис" - сталість, стан). Вона здійснюється шляхом гуморальної регуляції (з лат. "гумор" - рідина), тобто через кров, тканинну рідину, лімфу і т.д. за допомогою біологічно активних речовин (ферментів, гормонів і ін.) при участі нервових регулюючих механізмів. Гуморальні і нервові компоненти тісно взаємозалежні між собою, утворюючи єдиний комплекс нейрогуморальної регуляції
Деякі сторони гомеостазису були вже розглянуті. Наприклад, прагнення організму до збереження сталості температури, ентропії, енергії Гіббса, вмісту в крові і міжтканинних рідинах різних катіонів, аніонів, розчинених газів, величини осмотичного тиску тощо. Зупинимося на ще одній з найважливіших сторін гомеостазису - на прагненні організму підтримувати для кожної з його рідин визначену оптимальну концентрацію іонів гідрогену Н+ (вірніше іонів гідроксонію Н3О+). Збереження сталості кислотності рідких середовищ має для життєдіяльності людського організму першорядне значення, тому що, по-перше, іони Н+ діють каталітичне на багато біохімічних перетворень; по-друге, ферменти і гормони виявляють біологічну активність тільки в строго визначеному інтервалі значень рН середовища; по-третє, навіть невеликі зміни концентрації іонів гідрогену в крові і міжтканинних рідинах відчутно впливають на величину осмотичного тиску в цих рідинах.
Нерідко відхилення рН крові від нормального для неї значення 7,36 усього лише на декілька сотих призводить до неприємних наслідкіа При відхиленнях порядку 0,3 одиниці в ту або іншу сторону може наступити важкий коматозний стан, а відхилення порядку 0,4 одиниці можуть спричинити навіть смертельний результат. Утім, у деяких випадках, при ослабленому організмі, для цього виявляються достатніми і відхилення порядку 0,1 одиниці рН.
Підтримка постійного рівня рН у крові і тканинних рідинах досягається за допомогою регуляторних механізмів, що включають кілька буферних систем. Самими головними з них є наступні.
1. Гідрокарбонатна (бікарбонатна) буферна система, що характеризується рівновагою між молекулами слабкої вугільної кислоти з утвореними при її дисоціації гідрокарбонат-іонами:
Н2СО3 ↔ НСОз - + Н+ (1)
НСОз - + Н2О ↔ Н2СО3 + Н+ (2)
В організмі вугільна кислота виникає в результаті гідратації диоксиду вуглецю - продукту окиснення вуглеводів, білків і жирів. Причому процес цей прискорюється під дією ферменту карбоангідрази:
СО2 + Н2О ↔ Н2СО3
Рівноважна молярна концентрація в розчині вільного диоксиду вуглецю при 298,15 К приблизно у 400 разів вище, ніж концентрація вугільної кислоти [Н2СО3]/[СО2] = 0,00258.
2. Фосфатна буферна система, сталість рН в якій підтримується завдяки наявності рівноваги між гідрофосфат- і дигідрофосфат-іонами:
НРО42- + Н+ ↔ Н2РО4- (3)
НРО42- + Н2О ↔ Н2РО4- + ОН- (4)
3. Буферна система оксигемоглобін - гемоглобін, на частку якої приходиться близько 75% буферної ємності крові, що характеризується рівновагою між іонами гемоглобіну Нb- і самим гемоглобіном ННb, що є дуже слабкою кислотою :
Нb- + Н+ ↔ ННb (5)
Нb- + Н2O ↔ ННb + ОН- (6)
а також між іонами оксигемоглобіну НbО2- і самим оксигемоглобіном ННbО2, який є трохи сильнішою кислотою, ніж сам гемоглобін :
НbO2- + Н+ ↔ ННbO2 (7)
НbO2- + Н2O ↔ ННbO2 + ОН- (8)
Гемоглобін ННb, приєднуючи кисень, утворює оксигемоглобін ННbО2
ННb + О2 ↔ ННbО2
і таким чином рівноваги (5) і (6) взаємозалежні із рівновагами (7) і (8).
Іони НСО- , НРO42- , Нb- і НbО2- , будучи аніонами дуже слабких кислот, є (за Бренстедом) основами, тобто досить ефективними акцепторами іонів Н+. Тому, якщо в кров надходять сильні кислоти, то значна частина виникаючих при їхній дисоціації іонів негайно ж зв'язується з іонами НСО- , НРO42- , Нb- і НbО2- з утворенням дуже слабо дисоційованих молекул вугільної кислоти, гемоглобіну і оксигемоглобіну і дигідрофосфат іонів, наприклад:
NаНСОз + НС1 ↔ Н2СО3 + NaCl
Na2НРО4 + НС1 ↔ NaН2РО4 + NaС1
КНb + НСІ ↔ ННb + KCl
NaНbО2 + НС1 ↔ ННbО2 + NаСІ,
а також з іонами гідроксилу, що виникають у силу гідролізу по рівняннях (4) і (6) з утворенням слабодисоційованих молекул води, і з відповідним зрушенням цих гідролітичних рівноваг вправо.
Таким чином, завдяки буферній дії вище зазначених систем відбувається лише невелике зниження рН крові, незважаючи на надходження в неї значних кількостей сильних кислот. Викликане цим надходженням кислот зсув рівноваг (1), (2) і (3) - (8) праворуч приводить до зниження вмісту в крові іонів НСО- , НРО- , НЬ- і, у меншому ступені НbО2 ; у зв'язку з чим знижується буферна ємність розглянутих систем (лужний резерв крові).
Самостійна робота: Визначення рН середовища розчинів солей.
Зсув кислотно-лужної рівноваги крові убік підвищення концентрації іонів гідрогену (зниження рН) і зменшення резервної лужності називається ацидозом, а зсув його убік зниження концентрації іонів гідрогену (підвищення рН) і збільшення резервної лужності - алкалозом. Ацидоз або алкалоз можуть виникати в результаті безпосереднього надходження в організм через харчовий тракт або органи дихання надлишкових кількостей
продуктів з підвищеною кислотністю або лужністю (їжа, питво, медикаменти, забруднення повітря), в результаті аномальної генерації або евакуації з організму такого роду речовин при різних патологічних станах організму, зв'язаних з порушеннями нормального обміну речовин, функцій дихання і кровообігу.
У сучасній клінічній практиці кислотно-лужну рівновагу (КЛР) організму звичайно визначають шляхом дослідження крові по мікрометоду Аструпа і виражають в одиницях ВЕ (з латинської "bі-ехсеss" - "надлишок основ"). При нормальному кислотно-лужному стані організму ВЕ = О (в апараті Аструп цьому значенню ВЕ відповідає рН = 7,40).
При значеннях ВЕ від 0 до ±3 кислотно-лужний стан організму вважається нормальним, при ВЕ = ± (3-5) - стрес-нормальним, при ВЕ = ± (6-9) -тривожним, при ВЕ = ±(10-14) - загрозливим, а при абсолютному значенні ВЕ, що перевищує 14 - критичним.
Для корекції кислотно-лужної рівноваги при ВЕ< 0 (ацидоз) найчастіше використовують 4%-ний розчин гідрокарбонату натрію, який вводять внутрішньовенне. Необхідний об'єм цього розчину в мл розраховують за емпіричною формулою v = 0,5тВЕ, де т - маса тіла, кг.
Якщо стан ацидозу виник у результаті короткочасної зупинки серця, то об'єм 4%-ного розчину NаНСО3 (v, мл), необхідний для компенсації зрушення КЛР у кислу область, розраховують за формул v = т τ, де τ -тривалість зупинки серця, хв.
Корекція КЛР при алкалозі більш складна і вимагає обліку багатьох вихідних даних. У якості однієї з тимчасових мір доцільне уведення від 5 до 15мл 5%-ного розчину аскорбінової кислоти.
Метод кислотно-основного титрування в одному зі своїх варіантів (алкаліметрія) дозволяє визначати кількості кислот і кислотоутворюючих речовин (солей, складених з катіона слабкої основи й аніона сильної кислоти і т.п.) за допомогою робочих розчинів лугів відомої концентрації В іншому варіанті (ацидиметрія) цей метод дозволяє визначати кількості основ і речовин основного характеру (оксидів, гідридів і нітридів металів, органічних амінів, солей, складених з катіонів сильних основ і аніонів слабких кислот і т.п.) за допомогою робочих розчинів кислот.
Метод кислотно-основного титрування використовують у практиці клінічних, судово-експертних і санітарно-гігієничних досліджень, а також при оцінці якості лікарських препаратів.
Колориметричний метод визначення активної концентрації іонів гідрогену заснований на застосуванні речовин, названих індикаторами, забарвлення яких залежить від рН розчину.
Індикаторами є деякі органічні речовини, що містять хроматофорні групи. При зміні рН середовища в молекулах індикаторів відбуваються таутомерні (внутрішньомолекулярні) перегрупування, у результаті яких з'являються або руйнуються забарвлені групи.
Формально індикатор можна розглядати так само, як слабку кислоту НІnd або слабку основу ІndOН, у яких молекулярна й іонна форми мають різне забарвлення. Наприклад, якщо індикатор є слабкою кислотою, то в кислому розчині він практично весь буде знаходитися в молекулярній формі Нind, а в лужному середовищі - у вигляді іонів Іnd-. Відповідно буде розрізнятися і його забарвлення.
Так, для реакції дисоціації індикатора

Якщо прийняти, що забарвлення індикатора змінюється, коли половина індикатора розпадається на іони, тобто коли [Іnd-] = [НInd], тоді [Н+] = Ка, i рН = рКа.
Водневий показник, рівний рКа індикатора, прийнято вважати за межу переходу забарвлення індикатора
При однаковій інтенсивності забарвлення обох форм індикатор приймає на межі переходу проміжне забарвлення. Наприклад, індикатор метиловий оранжевий у недисоційованій формі забарвлений в оранжевий колір, у лужному розчині має жовте забарвлення й у кислому розчині - рожеве. В обидва боки від цієї межі забарвлення поступово зростає до червоного -в один бік і до жовтого - в інший. Повна зміна забарвлення індикаторів від НInd до Ind- відбувається приблизно в межах двох одиниць рН. Область значень рН, у якій відбувається помітна оком зміна кольору індикатора, називається інтервалом або областю переходу забарвлення індикатора. У фенолфталеїну цей інтервал лежить у межах рН 8,0-9,8, у метилового оранжевого - при рН 3,1-4,4, у лакмусу в у межах рН 5-8. Звідси випливає, що причиною різного положення області переходу у різних індикаторів є різниця у величині їх констант дисоціації.
Індикатори, що мають великі константи дисоціації (тобто більш схильні до розпаду на іони), змінюють своє забарвлення при менших значеннях рН, і навпаки. Наприклад, константа дисоціації метилового оранжевого Ка = =4,6.10-4 набагато більше, ніж у фенолфталеїна Ка = 8.10-10 . Область переходу і константа дисоціації найважливіших індикаторів представлені в таблиці 4.6.
С еред
наведених у таблиці індикаторів є такі,
що змінюють забарвлення
тільки
еред
наведених у таблиці індикаторів є такі,
що змінюють забарвлення
тільки
в кислому середовищі (метиловий червоний), інші - у лужному (фенолфталеїн) і тільки деякі з них (лакмус і феноловий червоний) мають одне забарвлення в кислому середовищі, інше - у лужному. Тому про індикатори не можна говорити як про речовини, зміна кольору яких показує кисле або лужне середовище, тому що це речовини, що показують різне значення рН розчину. Для визначення рН розчину потрібно досліджувати його декількома індикаторами. Останні підбирають таким чином, щоб зміна забарвлення відбувалася при поступово зростаючому рН. Так, наприклад, якщо метиловий оранжевий у досліджуваному розчині здобуває жовтий колір, то це означає, що рН цього розчину не нижче 4,4. Якщо при випробуванні цього ж розчину за допомогою метилового червоного забарвлення його залишається червоним, то це означає, що рН не вище 6,2. Звідси можна зробити висновок, що рН досліджуваного розчину лежить між 4 і 6,2. Підбираючи індикатори, що мають різні константи дисоціації, можна звузити межі і більш точно знайти значення рН розчину.
Колориметричне визначення рН полягає в наступному виготовляють ряд стандартних буферних розчинів зі зростаючими значеннями рН і додають до кожного з них декілька краплин індикатора Таким чином, в області переходу забарвлення індикатора одержують кольорову шкалу. Потім таку ж кількість індикатора додають до досліджуваного розчину і порівнюють у компараторі його забарвлення із забарвленням стандартних розчинів. Збіг забарвлення розчинів указує на збіг рН досліджуваного розчину зі значенням рН стандартного розчину.
Колориметричний метод визначення рН має ряд недолікіа По-перше, він недостатньо точний: рН виміряється з точністю до 0,2. По-друге, індикатор сам є слабкою кислотою або лугом, і додавання його до досліджуваного розчину викликає зсув рівноваги.
Виявляється, що в присутності багатьох нейтральних солей і білків область переходу забарвлення індикатора часто міняється, що приводить до істотного перекручування результатів (сольова похибка і білкова похибка). Білкова похибка обумовлена тим, що білок, що є присутнім у досліджуваному розчині, адсорбує індикатор. Перекручування результатів відбувається і внаслідок зміни температури. З підвищенням температури змінюється константа дисоціації індикатора Крім того, при роботі цим методом виникають утруднення, якщо досліджувані розчини мають забарвлення або мутні. Тому для визначення рН розчинів більш широко застосовуються електрометричні методи, які мають велику перевагу як по точності визначення, так і по швидкості, а головне, їм не завадить забарвлення або наявність муті у розчинах.
Задача 1. Розрахуйте рН буферного розчину. Три розчини містять оцтову кислоту
(Ка=1,80.10-5 моль.дм-3) концентрації 0,10 моль.дм-3 та ацетат натрію концентрацією 0,10(а), 0,20(б) та 0,50(в) моль.дм-3. Розрахуйте значення рН цих розчинів.
[сіль]
рН = рКа + lg ------------ рівняння Гендерсона - Гассельбаха
[кислота]
а. рН = 4,75+ lg(0,10/0,10) = 4,75 + lg 1,0 = 4,75
б. рН = 4,75+ lg(0,20/0,10) = 4,75 + lg 2,0 = 5,05
в. рН = 4,75+ lg(0,50/0,10) = 4,75 + lg 5,0 = 5,45
Біонеоганічна хімія
Лекція № 6 . Комплексні сполуки
6.1 Реакції комплексоутворення. Координаційна теорія А. Вернера.
6.2 Поняття про комплексоутворювач (центральний йон). Поняття про ліганди.
6.3 Координаційна ємність (дентатність) лігандів. Внутрішня та зовнішня сфери
комплексів. Природа хімічного зв'язку в комплексних сполуках.
6.4 Застосування комплексних сполук у медицині.
Самостійна робота: Ізомерія комплексних сполук.
Будова та номенклатура.
6.1. Реакції комплексоутворення. Координаційна теорія А.Вернера.
У визначенні поняття "комплексна сполука" ("координаційна сполука") у хіміків немає єдності. Засновник координаційної теорії комплексних сполук А. Вернер поділяв всі хімічні речовини на сполуки першого порядку (прості, або атомні типу СоС13, NН3, Н2О) і сполуки вищого порядку (або молекулярні), які є адуктами з'єднання сполук першого порядку (наприклад, СоС13 . NНз)- Вернер називав комплексними найбільш стійкі сполуки вищого порядку, які у водних розчинах або взагалі не розпадаються на свої складові частини, або розпадаються в дуже незначних кількостях. Пізніше різниця між термінами "комплексна сполука" і "сполука вищого порядку" стерлася і вони стали ідентичними.
Труднощі, що виникають при спробі розмежування комплексних сполук і сполук першого порядку в значній мірі обумовлені тим, що в залежності від умов одну і ту ж саму речовину можна розглядати як просту і як комплексну сполуку. Так, багато речовин (зокрема, галогеніди лужних і лужноземельних металів), які асоційовані у твердому стані, у газоподібному стані існують у вигляді окремих молекул, а в розчині дисоціюють на складаючі їх іони. Оскільки у лабораторній практиці звичайно мають справу з комплексними сполуками в твердому або розчиненому станах, важливо узагальнити їх здатність до існування саме в цих умовах.
Комплексною сполукою називається речовина, у вузлах кристалічної ґратки якої знаходяться комплексні іони, здатні до існування у розчині.
Комплексним іоном називається складний іон, який містить атом металу (іноді неметалу) у певному валентному стані, зв'язаний з одним або декількома здатними до самостійного існування молекулами або іонами.
Комплексні сполуки можуть містити комплексний аніон, комплексний катіон, або взагалі не дисоціювати на іони (сполуки типу неелек-тролітів). В іноземній літературі (особливо в англійській або американській) комплексний іон, незалежно від його заряду, називають "комплекс". Цей термін одержав достатньо широке поширення й у вітчизняній літературі.
Наприкінці XIX ст. був накопичений великий експериментальний матеріал, який показував, що багато молекул з реалізованими хімічними зв'язками здатні вступати в подальшу взаємодію з утворенням більш складних молекул другого порядку. А.Вернер (1893) був, очевидно, першим з хіміків, хто зрозумів, що для пояснення будови і властивостей цих незвичайних сполук необхідно відмовитися від уявлень про сталість валентності. Ним була створена так звана координаційна теорія, в основу якої покладене наступне:
Крім головних валентностей, у атомів існують також побічні валентності, які виявляють себе при деяких реакціях.
Насичення головних валентностей лежить в основі утворення сполук першого порядку, наприклад найпростіших бінарних сполук, типу НС1, Н2О, NН3, СuС12 і т.д.
Насичення побічних валентностей лежить в основі утворення сполук вищого порядку, що є продуктами сполучення сполук першого порядку, наприклад NH4СІ, Fе(СN)2 . 4КСN, А1С13 . 6Н2О і т.д.
Теорія Вернера носила чисто формальний характер і не торкалася питання про природу сил, що обумовлюють координаційний зв'язок, і про механізм утворення комплексних сполук. Ці питання були уперше висунуті основоположником вітчизняної школи дослідників комплексних сполук Л.А.Чугаєвим і потім розроблялися в працях В.Косселя, В.Магнуса, Н.В.Сиджвіка, І.І.Черняєва, В.В.Лебединського, А.А.Грінберга, К.Б.ЯІШ-мирського, Б.В.Некрасова й ін.
Відповідно до сучасних поглядів у структурі молекул комплексних сполук (названих також координаційними сполуками) варто розрізняти:
внутрішню координаційну сферу - центральний атом (комплексоутворювач), навколо якого знаходяться тісно зв'язані з ним ліганди (аденди) - молекули або іони (Н2О, NН3, СN-, F-, I-, Сl- і інші);
зовнішню координаційну сферу - сукупність всіх іонів, безпосередньо не зв'язаних з центральним атомом і внутрішньою координаційною сферою, що знаходяться за межами координаційної сфери.
Центральний іон – комплексоутворювач

K+[Fe(CN)6]4- [Co3+(NH3)6]3+Cl3-






Зовнішня внутрішня внутрішня зовнішня Коорденаційна коорденаційна коорденаційна коорденаційна
Сфера сфера сфера сфера
Комплексне угруповання, що несе надлишковий позитивний або негативний заряд (указується праворуч від квадратної дужки), називається комплексним іоном. Наприклад: [Fе(СN)6]4- - комплексний аніон, а [Со(NН3)6]3+ - комплексний катіон.
Відомі комплексні сполуки, що складаються тільки з центрального атома-комплексоутворювача і навколишніх його лігандів і не мають зовнішньої координаційної сфери. Такі, наприклад, карбоніли Ніколу [Ni(СО)4] і Феруму [Fе(СО)5], дихлордіамінплатина [Рt(NН3)2С12] та ін. Подібні їм сполуки не є електролітами.
А.А.Грінбергом, а потім К.Б.Яцимирським було встановлено, що найбільшу здатність до комплексоутворювання мають атоми елементів трьох побічних груп VIII групи, а також атоми лантаноїдів і актиноїдів. Комплексоутворювачами можуть бути також
неметали в позитивному ступені окиснення (В3+, Sі4+, Р5+, S6+, І7+ та ін.), рідше - у негативному ступені окиснення (I-, S2- та ін.).
В якості лігандів у комплексних сполуках найчастіше фігурують гідроксильна група ОН- і оксогруппа О2-, аніони кислот, полярні молекули і молекули, що легко поляризуються, найрізноманітніших неорганічних і органічних сполук (Н2О, NH3, СО, NO, С3Н5N та ін.). Заряд комплексного іона (або нейтрального комплексу) є алгебраїчна сума заряду центрального атома (його ступеня окиснення) і зарядів лігандів.
Іони зовнішньої сфери більш рухливі і легше вступають в обмінні реакції, ніж іони внутрішньої сфери. Слід, однак, мати на увазі, що хоча іони-ліганди, що утворюють внутрішню координаційну сферу, зв'язані з центральним атомом-комплексоутворювачем набагато більш міцними зв'язками, ніж іони зовнішньої сфери, усе-таки міцність цих зв'язків обмежена.
Може відбутися так, що підібраний реагент буде утворювати з іонами-лігандами настільки міцну сполуку (важко розчинний осад, слабкий електроліт, ще більш міцний новий комплекс), що для його утворення буде достатньо навіть тої невеликої рівноважної концентрації іонів-лігандів, що створюються в розчині даним комплексним іоном при його частковому розпаді. У цьому випадку буде відбуватися руйнування комплексного іона.
Координаційна теорія А. Вернера та сучасні уявлення про будову комплексних сполук.
Властивості комплексних сполук визначаються їхньою геометричною конфігурацією і характером зв'язку в молекулі (міцністю, ступенем іоно-генності або ковалентності зв'язку). Координаційна теорія, висунута А.Вернером у 1891, лише загалом указувала на розходження в характері зв'язку окремих атомів у молекулах комплексних сполук. Відповідно до цієї теорії, молекула комплексної сполуки має центричну будову (мається на увазі розміщення окремих замісників біля іона металу - комішексоутворювача), у більшості випадків октаедричну, тетраедричну або плоску. Атом комплексоутворювача називається центральнім атомом або центральним іоном. Щоб підкреслити різницю між центральним іоном комплексної сполуки й іоном того ж металу у вільному стані, центральний іон позначають символом елемента, поруч з яким у дужках римськими цифрами ставиться його валентність. Молекули й іони, безпосередньо зв'язані з центральним іоном, називають координованими групами (СІ - хлорогрупа, NНз — аміногрупа, H2О — аквогрупа), або внутрішньосферними замісниками (інакше адендами, або лігандами). Центральний іон у сукупності з координованими групами утворює внутрішню сферу комплексної сполуки. При написанні формул комплексної сполуки центральний іон разом із внутрішньосферними заступниками беруть у квадратні дужки, наприклад
[ Со(NН3)6]С13.
Якщо заряд центрального іона не дорівнює по величині сумі зарядів усіх іонів, що входять у внутрішню сферу, то комплексна сполука містить іони, що утворюють його зовнішню сферу і компенсують його заряд. Заряд комплексу може нейтралізуватися або простими, або комплексними іонами:
[Рt(NН3)4]С12; К2[РtС14]; [Рt(NН3)4][РtС14].
Комплексні сполуки, що містять зовнішньосферні іони, дисоціюють у розчині наступним чином (на комплексний іон та іони зовнішньої сфери):
[Рt(NН3)4]С12 ↔ [Рt(NН3)4] 2+ + 2С1-
К2[РtС14] ↔ 2К+ + [РtС14]2-
[Рt(NН3)4][РtС14] ↔ [Рt(NН3)4] 2+ + [РtС14]2-
У комплексній сполуці центральний іон виявляє так звану головну і побічну валентності. Головною валентністю Вернер назвав сили, аналогічні тим, що виявляються при утворенні простих сполук першого порядку. За рахунок сил побічної валентності відбувається сполучення молекул першого порядку у сполуки вищого порядку. Часто при написанні структурних формул комплексних сполук головні валентності позначають суцільними лініями, а побічні - пунктиром. Так, у сполуці



 NH3
Cl-
NH3
Cl-

 Pt
Pt


NH3 Cl-
дві хлорогрупи приєднані до Рt (II) за рахунок головної валентності, а дві молекули NHз - за рахунок побічної. Ґрунтуючись на таких фактах, як відсутність ізомерії у К2РtС14 та інших сполуках, Вернер не проводив принципового розходження між замісникaми, приєднаними за рахунок головної і другорядної валентності.
Надалі це припущення було підтверджено рентгеноструктурним аналізом, вивченням ізотопного обміну і т.п.
Прояв побічної валентності часто приводить до зміцнення зв'язків, утворених за рахунок головної валентності. Частіше зустрічаються випадки стабілізації вищого валентного стану центрального атома, хоча іноді має місце зміцнення і його нижчого валентного стану. Наприклад, у сполуці [Со(NНз)6]Сl стан Со(ІІІ) більш стійкий, ніж тривалентний стан Кобальту у СоС13, СоВг3. Двовалентний стан для Аргентуму нехарактерний, але комплексна сполука Аg(II) з α,α'-дипіридином (dр) цілком стійка. Солі Феруму (II) досить легко піддаються окисненню, але [Fеdp3]С13 піддається окисненню дещо важко. З іншого боку, характер насичення головної валентності істотно впливає на міцність зв'язку за рахунок побічної валентності. Наприклад, у ряді випадків термічна стійкість комплексних сполук (температура видалення груп із внутрішніх сфер) істотно залежить від характеру аніона зовнішньої сфери. Так, [Nі(NНз)6]С1з відщеплює аміак при 437 К, тоді як [Nі(NН3)6]І2 стійкий до 494 К.
Кількість атомів або груп атомів, зв'язаних з центральним атомом, називається його координаційним числом (скорочено к. ч.). Координаційне число є характеристикою елемента у визначеному валентному стані. Двовалентна Рt - виявляє к. ч. 4, чотиривалентна - к. ч. 6. Координаційне число може бути рівним валентності (наприклад, у Карбону у багаточисельних органічних сполуках), більшим від валентності (найбільш частий випадок), і тільки дуже рідко буває менше валентності. Останнє найчастіше виявляється у сполуках, які містять координовані багатовалентні іони О2-, наприклад у сполуках К2СО3, КСlO4 і т.п. (к. ч. Карбону в іоні СО32- дорівнює трьом, а його валентність - чотирьом; к. ч. Хлору в іоні С1O4- дорівнює 4, а його валентність - 7. Деякі іони металів мають стале координаційне число, а деякі - змінне. До перших відносяться: РІt(II) (к. ч. 4), Рt (IV) (к. ч. 6), Со (III) (к. ч. 6), Rh (III) (к. ч. 6), Іг (III) (к. ч. 6), Іг (IV) (к. ч. 6), Рd (II) (к. ч. 4), Аu (III) (к. ч. 4). До другого типу відносяться: Ni (II) (к. ч. 4; 6), Сu (II) (к. ч. 6; 4; 3) і більшість інших елементів.
Координаційне число, яке проявляється даним іоном, залежить від природи внутрішньосферних і зовнішньосферних груп і від температури, а у випадку розчинів комплексних сполук, крім того, і від концентрації аденду і центрального іона, а також від природи розчинника. Зниження температури сприяє прояву більш високого координаційного числа. Так, при нагріванні [(Со(NН3)6]С12 у рівновазі знаходяться такі сполуки:
T = 423 K T = 473 K
[(Со(NН3)6]С13 ↔ [(Со(NН3)2Cl2] + 4NH3 ↔ CoCl2 + 6NH3
К.ч. Со(ІІ) = 6 К.ч. Со(ІІ) = 4
Найчастіше зустрічаються комплекси, що містять центральний іон з к. ч. 4 і 6, рідше з к. ч. З і 2. Припускають існування комплексних сполук з к. ч. 5 і 7. Нарешті, до сполук з координаційним числом, більшим за 6, відносяться Ме4[Мо(СN)8] і Ме4[W(СN)8], де Ме - однозарядний катіон.
6.2 Поняття про комплексоутворювач (центральний йон). Поняття
про ліганди.
Комплексні сполуки(координаційні сполуки) - складні хімічні речовини, у складі яких є комплексні іони, утворені центральним атомом і пов'язаними з ним лігандами.
Згідно теорії А. Вернера, в центрі комплексного з'єднання знаходиться центральний іон-комплексоутворювач. Іонамі-комплексоутворювачами є катіони металів. Найбільшу схильність до комплексоутворення проявляють іони d-елементів.
Навколо центрального іона-комплексоутворювача знаходяться протилежно заряджені іони або нейтральні молекули, які називають лігандами, або адeндами.
Іон-комплексоутворювач і ліганди складають внутрішню сферу комплексної сполуки, яку позначають квадратними дужками. Число лігандів (адендів), які координуються навколо центрального іона-комплексоутворювача, називається координаційним числом.
Заряд комплексного іона рівний алгебраїчній суммі зарядів іона-комплексоутворювача і лігандів.
Наприклад :
+2 0 2+ 1-
[ (Сu(NН3)4 ] С12
центр. лиганд
 ион
ион
внутрішня зовнішня
сфера сфера
Комплексні сполуки не завжди побудовані з іонів; заряди атомів і молекул, що входять до складу комплексу зазвичай невеликі. Тому правильніше користуватися терміном «центральний атом», а не «центральний іон».
Центральний атом
• Найчастіше центральними атомами є іони металів, d-елементів: Сu, Аg, Pt, Сг, Fе, Zn і т.д. Але до складу деяких комплексних сполук можуть входити і іони лужних і лужно-земельних металів (Nа, Са, Мg).
• Заряд центрального іона є основним чинником, що впливає на координаційне число)
+ 1 → 2
заряд + 2 → 4 , 6 Координаційні числа
центрального + 3 → 6 , 4 (видєлені найбільш
іона + 4 → 8 , 6 характерні)
Координаційне число не є незмінною величиною. Навіть для одних і тих же комплексоутворювачів і лігандів координаційне число залежить від агрегатного стану речовини, від концентрації, температури.
Ліганди (lig) внутрішньої сфери можуть заміщатися на інші молекули або іони, при цьому змінюється заряд комплексного іона, наприклад :
NO2- NO2- NO2-
[(Со3+(NН30)6]3+С13 → [(Со3+(NН30)5NO2]2 +С12 → [(Со3+(NН30)4(NO2)2]1 +С1 →
NO2- NO2-
[(Со3+(NН30)3(NO2)3]o → K [(Со3+(NН30)2(NO2)4]- → K 2 [(Со3+NН30(NO2)5]2- →
NO2-
→ K 3 [(Со3+(NO2)6]3-
В даний час прийнято класифікувати комплекси відповідно до типу утворюючих їх лігандів.
1. Комплекси, що містять молекулярні монодентатні ліганди. Найважливішими представниками цього класу комплексних сполук є:
а) гідрати, у яких лігандами служать тільки молекули води
(аквакомплекси), наприклад [Са(Н2О)6]2+СІ2 , [Са(Н2О)4]2+(NO3-)2 і ін.; кристалічні речовини, що включають такого роду комплекси, називають кристалогідратами;
б) аміакати, у яких лігандами є тільки молекули аміаку, наприклад, [Аg(NНз)2]+Сl- , [Сu(NН3)4]2+SO42- ,[Сг(NН3)6]3+Сl3- та ін.;
в) карбоніли металів - ліганди молекули монооксида карбону, при цьому комплекс не має електричного заряду, наприклад [Fе(СО)5], [Rе2(СО)10], [W(СО)6] та ін.
Комплекси, що містять іонні ліганди або ацидокомплекси, у яких лігандами є кислотні залишки, причому увесь комплекс несе негативний заряд і може розглядатися як аніон якої-небудь кислоти; відповідно назві ліганда ці комплекси називають гідроксокомплексами - [А1(ОН)6]3-, [Zn(ОН)4]2-, [Sn(ОН)6]2-,
фторокомплексами - [А1F6]3- , [ВеF4]-, [ВF4]-.
ціанокомплексами - [Аg(СN)2] [Fе(СN)6]4-, [Fе(СN)6]3-, [Нg(СN)4]2- і т.д.
Циклічні комплексні сполуки, утворені полідентатними лігандами, що складають групу так званих хелатних (клішнеподібних), наприклад типу три (етилендіамін) кобальт (III) хлориду з трьома п'ятичленними циклами:
NH2 (CH2)2 H2N



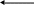

 N
Co
N
N
Co
N
H2 H2
(CH2)2 (CH2)2
H2 H2
N N
6.3 Координаційна ємність (дентатність) лігандів. Внутрішня та
зовнішня сфери комплексів. Природа хімічного зв'язку в
комплексних сполуках.
Одновалентні кислотні залишки, NН3, RNН2, С6Н5N і багато інших груп, що входять до складу комплексної сполуки, зв'язані з центральним іоном за допомогою одного зв'язку і займають одне координаційне місце. Але можливий випадок, коли один замісник зв'язаний з центральним іоном за допомогою двох або більше атомів, що входять до його складу. Такі комплексні сполуки називаються циклічними. Кількість координаційних місць, які може займати той або інший замісник біля центрального атома, називається координаційною ємністю, а самі аденди - одно-, дво-, три- і т.д. полікоординаційними замісниками. В якості полікоординаційних замісників можуть виступати нейтральні молекули, які містять два або декілька атомів із вільною електронною парою. Наприклад, NН2 - СН2 - СН2 - NН2, кислотні залишки багатоосновних кислот SО42- , С1O4- і т.п. Можливий випадок, коли один і той же замісник містить атом з донорною функцією і по способу свого утворення є кислотним залишком. Такими є кислотні залишки амінокислот, наприклад NН2СН2СОСl- .
Число іонів або молекул, безпосередньо зв'язаних з центральним атомом-комплексоутворювачем, тобто число лігандів, що координуються навколо центрального атома, називають координаційним числом останнього. Наприклад, у комплексних сполуках [Аg(NН3)2]+Сl-, К4+[Fе(СN)6]4-, [Си(NН3)4]2+Сl2-, [Fе(СО)5] і [Те(СО)7] координаційне число дорівнює, відповідно, 2, 6,4, 5 і 7.
У хімії комплексних сполук координаційне число грає приблизно ту ж роль, що і валентність у хімії сполук першого порядку. В залежності від хімічної природи і геометричних розмірів центрального атома і оточуючих його лігандів координаційне число може варіювати в межах від 1 до 12, причому найчастіше зустрічаються сполуки з координаційними числами 4 і 6.
Деякі ліганди приєднуються до центрального атома не одним, а двома і більшим числом координаційних зв'язків, займаючи у внутрішній координаційній сфері відповідно два і більше число місць. У таких випадках даний ліганд має координаційну ємність, або дентатність, більше одиниці, тобто є полідентатним. Так, якщо ліганди типу Н2О, NН3, іонів галогенів і іона СN- завжди монодентатні, то, наприклад, сульфат- і карбонат-іони і молекула етилендіаміну H2N-CH2-CH2-NH2 завжди бідентатні, а серед складних органічних сполук зустрічаються і такі, котрі можуть виступати в ролі три-, тетра-, пента-, гекса- і навіть октадентатних лігандів.
Полідентатні органічні ліганди, замикаючись двома або декількома координаційними зв'язками на центральний атом, можуть утворювати циклічні комплекси.
Зазвичай хімічний зв'язок між центральними атомами і лігандами в комплексних сполуках обумовлений донорно-акцепторною взаємодією, причому ліганди є донорами електронних пар, а центральні атоми -акцепторами цих пар. Дійсно, іони і молекули, які найчастіше виступають у ролі лігандів, мають вільні неподілені пари електронів. Так, наприклад, молекула води, молекула аміаку, молекула монооксиду Карбону, іон гідроксила, ціанід-іон та іон Флуору можуть бути зображені так:
H H
. . . . . . . .
: O : H : N : H : C ::: O: : O : H- : C ::: N- : : F- :
. . . . . . . .
H
В той же час центральні атоми-комплексоутворювачі, у ролі яких найчастіше виступають атоми перехідних елементів, мають вільні орбіталі для розміщення цих електронів у d - i f-підрівнях.
У зв'язку з цим комплексними сполуками іноді називають молекулярні сполуки визначеного складу, утворення яких з більш простих сполук не зв'язано з виникненням нових електронних пар.
Слід, однак, обмовитися, що зведення хімічного зв'язку в комплексах тільки до донорно-акцепторної взаємодії не завжди правомірно. У зв’язку з цим більш коректно наступне визначення:
комплексними називаються сполуки, у вузлах кристалічних ґраток яких знаходяться складні частинки (комплексні іони), що склаоаються з центрального атома і навколишніх його молекул або іонів причому цілісність цих комплексних іонів зберігається при переході сполуки в розплавлений або розчинений стан.
6.4 Застосування комплексних сполук у медицині.
Унікальне значення внутрішньокомплексних сполук для життєдіяльності людського організму виявляється вже з того, що складні молекули гемоглобінів (Мг = 67 000) здійснюють перенесення молекулярного кисню від легень до тканин. Гемоглобін містить у своєму складі так звані простетичні (небілкові) групи, або геми, побудовані по типу

Для життя рослин унікальне значення мають хлорофіли-пігменти, за допомогою яких вищими рослинами і зеленими водоростями здійснюється фотосинтез. Хлорофіли являють собою внутрішньокомплексні сполуки, у яких комплексоутворювачами є іони Магнію, зв'язані з органічною частиною молекули чотирма зв'язками:

По типу внутрішньокомплексних сполук побудовані поліатомні молекули багатьох ферментів, роль комплексоутворювачів у яких відіграють частіше атоми таких елементів, як Ферум, Цинк, Кобальт, Молібден, Купрум і Манган, які К.Б.Яцимирський (разом з Натрієм, Калієм, Кальцієм і Магнієм) образно назвав "металами життя ".
Так, простетична група гемоглобінів є також структурною складовою молекул пероксидази - ферменту, який каталізує окисно-відновні реакції за участю пероксида водню, а також каталази, яка каталізує розкладання пероксиду Гідрогену, що утворюється в процесах метаболізму.
За рахунок внутрішньокомплексних зв'язків атоми Цинку включаються в структуру карбоксипептидази, яка каталізує гідролітичне відщеплення кінцевих кислотних залишків у білках і пептидах. Атоми Молібдену і Феруму входять до складу ксантиоксидази, яка каталізує окисно-відновні реакції, зв'язані з обміном пуринів і утворенням сечової кислоти.
У медичній практиці комплексонометрію застосовують для визначення у різних рідинах і тканинах людського організму мікроелементів і вмісту різних катіонів металів у фармацевтичних препаратах. Запропоноване також використання комплексонів для маскування (зв'язування і знешкодження) іонів металів, що є присутніми у лікарських препаратах у вигляді забруднень. Є відомості про успішне застосування комплексонів для розчинення каменів, що утворюються в нирках, печінці і жовчному міхурі. Зрозуміло, таке застосування комплексонів вимагає дуже великої обережності, щоб разом з розчиненням каменів не відбувалося руйнування мінеральної основи кісткової й іншої тканин людського організму.
Внутрішньокомплексні сполуки набули великого значення для аналітичної хімії. На їхньому застосуванні заснований комплексонометричний метод кількісного визначення іонів металів, введений в аналітичну практику Г.Шварценбахом (1945). Як реагенти в комплексонометрії (хелатометрії) для кількісного зв'язування металів звичайно використовують похідні амінополікарбонових кислот, що одержали назву комплексонів, з яких найбільше значення набув комплексон IIІ (або трилон Б, ЕДТА, версен).
Комплексонометрія одержала велике поширення для кількісного визначення катіонів багатьох металів (Са2+, Мg2+, Sг2+, Zn2+, Рb2+, Сu2+, А13+ і ін.), а побічно і для визначення деяких аніонів (SО42- , РО42- , F- та ін.).
Поняття про металолігандний гомеостаз.
У живому організмі здійснюється цілий ряд зустрічних фізико-хімічних процесів (випаровування і конденсація, розчинення і кристалізація, електролітична дисоціація й утворення молекул з іонів і т.д.) -багато сотень тисяч біохімічних реакцій, що протікають у залежності від численних умов зовнішнього і внутрішнього середовища, з різними швидкостями. Проте завдяки тонкій нейрогуморальній регуляції досягається разюча сталість внутрішнього середовища (гомеостазис) - з погляду термодинаміки квазірівноважний, або стаціонарний стан. Особливе значення в гомеостазі мають d- елементи.
Тут було наведено досить прикладів, щоб оцінити, яке велике значення хімії для теоретичного озброєння медицини і її подальшого прогресу. Чим більш строгими і кількісно визначеними будуть наші знання щодо закономірностей і умов перебігу окремих хімічних і фізико-хімічних процесів, що здійснюються в організмі, тим повніше будуть наші уявлення про багатогранний процес обміну речовин і енергії, тим ясніше стануть фізико-хімічні механізми, що лежать в основі нейрогуморальної регуляції організму.
В організмі людини в якості лігандів (адендів) у біологічні комплекси входять органічні сполуки, що називаються біолігандами. Це білки, амінокислоти та їхні похідні, нуклеїнові кислоти, нуклеотіди, нуклеопро-теїди, азотисті основи, пептиди, жирні кислоти, вуглеводи, вітаміни, ферменти, гормони, вода, жовчні кислоти, кето- і оксикислоти та інші сполуки. Комплексоутворюючу здатність біологічних лігандів можна пояснити наявністю в їхніх молекулах декількох функціональних груп різного типу (звідси велика різноманітність цих сполук). Наприклад, нуклеїнові кислоти мають у своєму складі карбоксильні - СООН і аміногрупи - NН2, причому можуть бути первинні (-NН2), вторинні (-NН-), третинні (-N=) атоми Нітрогену, що здатні координувати іони металів. Характерно, що біологічні комплекси є відносно нестійкими при певних умовах (присутність розчинника, дія ферментів і т.д.) хімічні зв'язки розриваються, в результаті чого утворюються комплексні іони, активність яких у ряді випадків значно вища, ніж комплексних сполук.
Біологічна роль хімічних елементів в організмі людини дуже багатогранна. Макроелементи, в основному, відіграють роль пластичного матеріалу в побудові тканин, підтримують осмотичний тиск, рН середовища, іонну рівновагу, кислотно-лужну рівновагу, стан колоїдів і т.д. Мікроелементи разом з ферментами, гормонами, вітамінами та іншими біологічно активними речовинами беруть участь у процесах розмноження, росту, обміну білків, жирів, вуглеводів тощо. Біологічні функції мікроелементів у живому організмі зв'язані, головним чином, з процесами комплексоутворення між біологічними лігандами й іонами відповідних металів.
Участь мікроелементів у фізіологічних процесах може здійснюватися двома шляхами:
атом входить у структуру ферменту в якості комплексутворювача (метал - активатор);
елемент є сполучною ланкою між системами фермент - субстрат.
В даний час відомо близько 200 ферментів, до складу яких у якості комплексоутворювачів і активаторів входять мікроелементи. У той же час деякі мікроелементи можуть блокувати активні центри молекул ферментів. Вплив мікроелементів на біологічні процеси може здійснюватися й через нуклеїнові кислоти. Мікроелементи входять або до складу ферментів, що активують синтез і розпад нуклеїнових кислот, або безпосередньо в комплексні сполуки з нуклеїновими кислотами. Крім того, мікроелементи сприяють стабілізації структури нуклеїнових кислот, впливають на їхню просторову конфігурацію і т.д. Таку ж участь приймають мікроелементи в обміні вітамінів, гормонів та інших біологічно активних сполук.
Варто розрізняти специфічний і неспецифічний вплив мікроелементів на обмінні процеси. Приклади специфічного впливу: взаємозв'язок між обміном Цинку в тканині підшлункової залози і продукцією інсуліну, вплив Мангану на залози внутрішньої секреції, вплив Йоду на функцію щитовидної залози і т.д. Неспецифічний вплив, наприклад, на функцію щитовидної залози спостерігається в таких мікроелементів, як Купрум, Кобальт, Манган, Флуор та ін.
Властивість іонів металів утворювати міцні зв'язки з сірковмісними лігандами використовується і при підборі ліків, вживаних для лікування при отруєннях. Такі ліки мають загальну назву - антидоти.
У багатьох випадках фармацевтичні властивості медичних препаратів знаходяться в безпосередньому зв'язку з їх окисно-відновними властивостями. Так, багато з антисептичних, протимікробних і дезінфікуючих засобів, наприклад йод, перманганат Калію, пероксид Гідрогену, солі Купруму, Аргентуму і Меркурію, є в той же час і сильними окисниками.
Застосування тіосульфату Натрію (3,3 М Nа2S2О3) як універсального антидоту (протиотрути) засновано на його здатності брати участь в окисно-відновних реакціях у ролі як окиснювача, так і відновника.
У випадку отруєнь сполуками Арсену (III), Меркурію (II) і Плюмбуму (II) прийом усередину розчину Nа2S2О3 приводить до утворення важкорозчинних і тому практично неотруйних сульфатів, наприклад:
Рb (СН3СОО)2 + Na2S2О3 + Н2О = РbS+ Na2SO4 + 2СН3СООН.
Антіоксиданти - природні або синтетичні речовини що запобігають окисленню органічних сполук. Їх застосовують в харчовій промисловості для запобігання псуванню харчових продуктів.
Надзвичайно важлива роль в захисті організму на молекулярному і клітинному рівнях від ушкоджувальної дії сильного окислювача - радикала НО2- належить ферменту супероксидісмутазі. Цей Ф є метоллопротєїдом і може містити як кофактор іони Мn2+, Fе2+, Сu2+, Zn2+ . Реакція , що каталізується супероксидісмутазою має наступний вигляд :
НО2- + НО2- → Н2О2 + О2
Подібні сполуки використовують у фармацевтичній промисловості для збереження лікарських засобів.
Самостійна робота: Ізомерія комплексних сполук.
Будова та номенклатура.
Велике поширення в хімії комплексних сполук має ізомерія - існування декількох сполук з однаковими складом і молекулярною масою, але молекули яких відрізняються за будовою, фізичними і хімічними властивостями. Для комплексних сполук особливо характерні наступні види ізомерії: сольватна (у водних середовищах - гідратна), іонізаційна, координаційна, геометрична і оптична (дзеркальна).
Сольватна (гідратна) ізомерія полягає в різному розподілі молекул розчинника (води) між внутрішніми і зовнішньою координаційними сферами комплексу. Так, наприклад, гексагідрат хлориду Хрому (III) СгС13-ЗН2О існує у вигляді трьох ізомерів:
[Сг(Н2О)6]С13 [СгС1(Н2О)5]С12-Н2О [СгС12(Н2О)4]С1-2Н2О
трихлорид гексааква моногідрат дихлорида дигідрат монохлорида
хрому(ІII) пентааквахлор тетрааквадихлоро
хрому(ІII) хрому(ІII)
Іонізаційна ізомерія обумовлена різним розподілом неоднакових за своєю природою іонів між внутрішніми і зовнішньою координаційними сферами комплексної сполуки, у зв'язку з чим змінюється характер дисоціації цієї сполуки, наприклад:
(РtBr2(NHз)4]С12 [РtС12(NН3)4]Вг2
дихлорид тетраамінди- дибромід тетраамін-
бромоплатини (IV) дихлороплатини (IV)
Координаційна ізомерія обумовлена тим, що в сполуці шо складається з комплексного катіона і комплексного аніона, ліганди можуть бути розподілені різним чином між цими двома комплексними іонами, наприклад:
[Со(NH3)6] [Сг(СN)6] [Сг(NH3)6] [Со(СN)6]
гексаціанохромат (III) гексаціанокобальтат (III)
гексаамінокобальта (III) гексаамінхрома (III)
Суть геометричної ізомерії (цис-, транс-ізомерія) полягає у різному просторовому розташуванні адендів біля центрального іона і виявляється тільки в комплексних сполуках, які мають октаедричну або площинну будову, наприклад комплекс кобальту СоA4Ва2 існує у вигляді таких ізомерних форм:
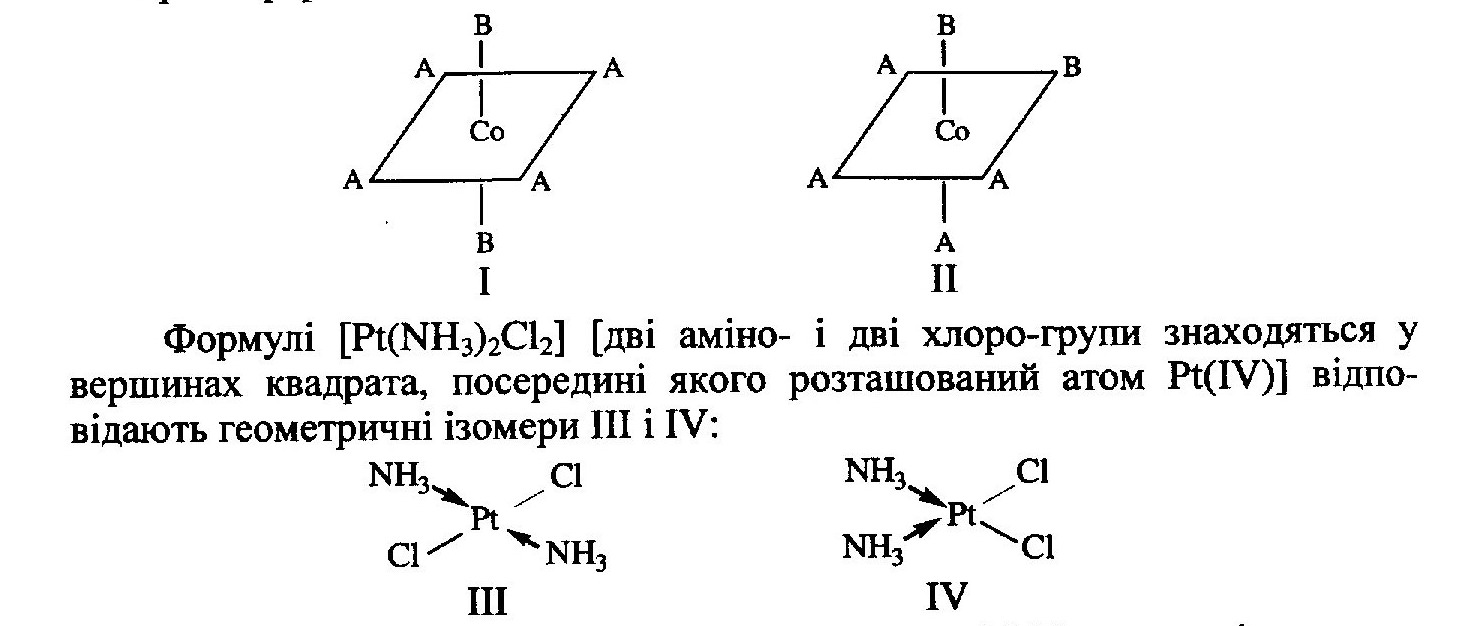
Сполуки II і IV називаються цис-ізомерами, а І і III — транс-ізомерами. Чим складніший хімічний склад молекули, тим більше можливостей для прояву ізомерії. Так, для комплексних сполук, що містять у внутрішній сфері 6 різних адендів, можуть існувати 15 геометрично ізомерних форм. У багатоядерних сполуках можливості прояву геометричної ізомерії особливо широкі. У більшості випадків геометричні ізомери розрізняються між собою хімічними властивостями, розчинністю, магнітним сприйняттям, дипольними моментами.
У випадку менш стійких сполук, наприклад похідних Со(ІІІ), Сг (II) та ін., в процесі хімічної взаємодії утворюється суміш двох або декількох ізомерів, які можна розділити перекристалізацією.
Оптична (дзеркальна) ізомерія спостерігається у комплексних сполуках, молекули яких не мають ні центру симетрії, ні площинну симетрію. Причиною появи оптичної активності може служити асиметрія центрального іона, наприклад іона Рt (IV) у сполуці
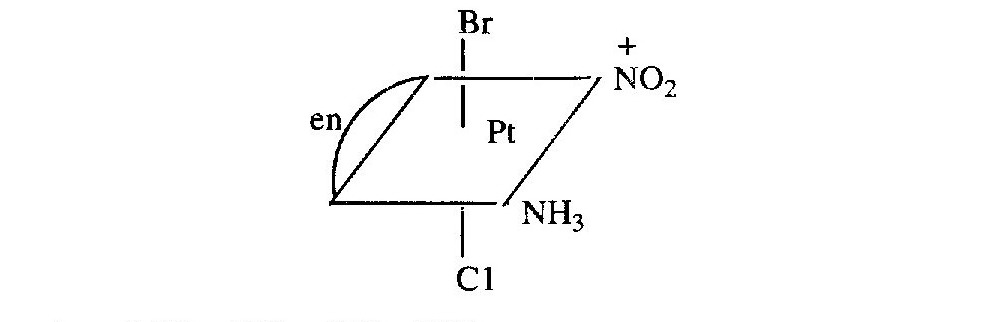
де еn відповідає NН2-СН2-СН2-NН2.
При синтезі оптично активних комплексних сполук у більшості випадків одержують їхні рацемати (суміш оптичних ізомерів), які можна розділити на окремі оптичні ізомери наступними способами: 1) кристалізацією; 2) додаванням до розчину рацемата затравки речовини тієї ж конфігурації, що і виділювана сполука; 3) одержанням діастереоізомерних форм, які можна далі розділити перекристалізацією; для поділу оптичних ізомерів, що містять оптично активний комплексний аніон, застосовують оптично активні основи (стрихнін, бруцин, цинхонін), а для поділу комплексних сполук катіонного типу - оптично активні кислоти (винну кислоту, камфорсульфонову кислоту і т.п.); 4) на основі різної адсорбуємості оптичних ізомерів на поверхні оптично активного кварцу; 5) користуючись різною реакційною здатністю ізомерів та ін.
Особливе місце серед інших комплексних сполук займають так звані внутрішньокомплексні сполуки. За типом цих сполук побудовані чисельні металопротеїди, білкова частина яких міцно зв'язана з іонами металів, їх значення для життєдіяльності людини, тварин і рослин настільки велике, що вони стали предметом особливої науки, виділеної з біохімії - біонеорганічної хімії.
Внутрішньокомплексні сполуки є одним із різновидів циклічних або так званих хелатних (від "хеле" - клішня), комплексних сполук, які відрізняються тим, що один з кінцевих атомів полідентатного ліганду зв'язаний з центральним атомом-комплексоутворювачем за рахунок неспареного електрона звичайним ковалентним або іонним зв'язком, а інший -за рахунок донорно-акцепторної взаємодії. Таким чином, іон металу за допомогою цих двох зв'язків виявляється як би затиснутим клішнею, а іноді і декількома такими клішнями і при переході такої комплексної сполуки в розчин він не виявляє своїх звичайних властивостей.
Номенклатура комплексних сполук
Назви солей, що містять комплексний катіон,, і солей, що містять комплексний аніон, відрізняються по своїй структурі.