

1.2 Обладнання газової хроматографії.
Особливості розподілу речовини, що аналізується
Теорія розглядає закони руху та розмиття аналізуємої речовини (АР) в колонці (ХК) після введення в випаровувач хроматографа під час аналізу (рис. 1.3).
Через хроматографічну колонку рухома фаза протікає з кінцевою швидкістю та переносить аналізуєму речовину від входу до виходу.
Швидкість переміщення аналізуємої речовини по колонці, залежить від двох швидкостей:
швидкості адсорбції та десорбції, або розчинення та випаровування;
від об'ємної швидкості рухомої фази в колонці.
 А В
С
А В
С
Рис. 1.5 Хроматограма розподілу речовини, що аналізується:
А , В , С ─ піки індивідуальних речовин.
Швидкість хроматографічного аналізу, якість розподілення більше залежить від першої швидкості. Будь-яке розподілення речовини між двома фазами, що не змішуються, характеризує ізотерма розподілу (рис. 1.4). В залежності від її виду розрізняють лінійну та нелінійну хроматографії. Взаємозв’язок між ними визначається коєфіцієнтом Генрі :
Кг= Са/Сг (1.1.)

Рис. 1.. Ізотерма розподілу концентрації речовини між двома фазами:
Са ─ нерухома фаза (адсорбент) ; Сг ─ рухома фаза (газ-носій); 1,3 ─ нелінійна хроматографія; 2 ─ лінійна хроматографія
В залежності від форми ізотерми розподілу піки розподілення мають вигляд, представлений на рис. 1.6
а) б) в)
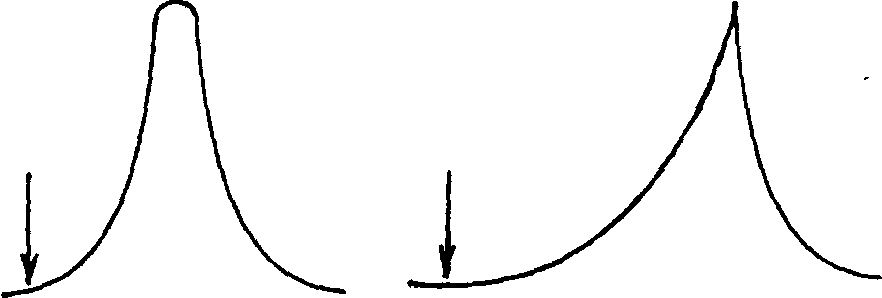

Рис. 1.6 Піки розподілення індивідуальних речовин:
а) ─ індивідуальні речовини утримуються нерухомою фазою; б) ─ рівномірне розподілення концентрації АР між рухомою та нерухомою фазами; в) ─ індивідуальні речовини виносяться газами-носіями.
Визначення швидкості руху аналізуємої речовини
Лінійна швидкість руху речовини, що аналізується Vлар по колонці знаходиться по формулі:
Vлар= Wгн/(Vрф+Vнф• Кг) , м/с (1.2)
де Wгн ─ швидкість газа-носія;
Vрф,Vнф ─ об'єм рухомої та нерухомої фази;
Кг─ коефіцієнт Генрі.
Через хроматографічну колонку газ протікає з кінцевою швидкістю і в колонці не встигає встановитися термодінамічна рівновага між концентрацією речовини, яка знаходиться в рухомій та нерухомій фазі. Якщо швидкість газу-носія постійна, температура колонки постійна, кількість фази в колонці та спосіб нанесення нерухомої фази і заповнення колонки не змінюється, то на окремій ділянці колонки можливе встановлення термодінамічної рівноваги. В цьому випадку швидкість аналізу буде залежати від об'єму та швидкості газу-носія, та об'єму в колонці, який займають рухома та нерухома фази. Рівняння пов'язує лінійну ізотерму розподілу кожної речовини в рухомій та нерухомій фазі.
Ефективність хроматографічного розподілу
Для характеристики ефективності роботи хроматографічної колонки використовують поняття "теоретична тарілка", яка визначається з хроматограми для одного піку:
n=5,54((tRj; lRj; VR)/μ)2 , (1.3)
де tRj, lRj, VR ─ параметри утримування аналізуємої речовини колонкою;
μ ─ ширина піка, яка береться на половині висоти піка.
Друга величина, що характерізує ефективність ─ висота, яка еквівалентна теоретичній тарілці:
H=L/n , (1.4)
де L─ довжина колонки;
n ─ кількість теоретичних тарілок.
Чим менше значення висоти, тим вище якість розділення.Можна сказати, що Н ─ це довжина ділянки колонки, на якій встановлюється термодінамічна рівновага між концентрацією речовини, яка знаходиться в двох фазах.
Параметри утримання колонкою аналізуємих речовин
До параметрів утримання (ПУ) відносяться (рис. 1.7):
- час утримання tR;
- відстань утримання lR;
- об'єм утримання VR;
- індекс утримання IR.

Рис. 1.7 Параметри утримання
На приведеному рисунку (рис.1.7) 1 ─ пік несорбційної речовини, для якої коефіцієнт розподілу дорівнює 0. Він служить для визначення неефективного часу вимірювання.
Чим менша площа від вводу проби до піка, тим ефективніша хроматографічна система.
2,3 ─ піки індивідуальних речовин ─ первинні параметри утримання.
tR ─час від моменту вводу проби, до виникнення максимуму в хроматограмі;
lR ─ відстань на хроматограмі від моменту вводу проби до виникнення максимуму;
VR ─ кількість рухомої фази, необхідної для вимивання з колонки речовини, що аналізується, для виникнення максимуму на хроматограмі.
tR, lR, VR ─ характеризують речовину.
t0, l0, V0 ─ "мертвий" час, відстань, об'єм.
Виправленні параметри утримання розраховуються за наступними формулами:
tR'=tR-t0 ; (1.5)
lR'=lR-l0 ; (1.6)
VR'=VR-V0 ; (1.7)
tR'= lR'/ Wдс; (1.8)
VR'=tR' Wгн , (1.9)
де Wдс ─ швидкість діаграмної стрічки.
Оцінка якості хроматографічного розподілу
Кількість піків на хроматограмі визначає детальність параметрів. Повнота їх розподілу говорить про точність результатів. Про якість розподілення роблять висновки по кількості піків та по відстані їх максимумів на хроматограмі. При розробці методик для їх порівняння необхідно кількісно визначити з хроматограми ступень розділення двох речовин, який залежить від коефіцієнту розподілення та величин В,А,С в рівнянні Ван-Деємтра.
У випадку добре розподілених піків ступінь розділення Kр визначається як різниця відстані утримання двох піків розділена на суму ширин, які беруться на половині висоти (рис.9.29).
Kр=(l2-l1)/(M1+M2)= ∆l/(M1+M2) (1.10)
Kр=0 до ∞.
У випадку, якщо піки розподіленні не повністю (рис.9.30), якість розподілення визначають по формулі:
Kр=(h2-hmin)/h2 (1.11)
Kр=2 до ∞.


Рис.9.29. До розрахунку ступеня розділення добре розподілених піків
Рис.9.30. До розрахунку ступеня розділення не розподілених піків
Селективність хроматографічної колонки вказує на відстань між піками та їх кількість. Вона характеризується коефіцієнтом селективності Kс:
Kс=(l2-l1)/(l1+l2 ) (1.12)
Коефіцієнт селективності залежить:
- від температури колонки (якщо температура збільшується, Кс зменьшується);
- від співвідношення в колонці об’ємів рухомої фази та нерухомої;
- від пористості сорбенту;
- при використанні капілярних колонок від кількості фази в колонці.
Взаємозв’язок між Kс і Kр можна виразити формулою:
![]()