

vk.com/club152685050 | vk.com/id446425943
Введение
Целью изучения дисциплины «Химия нефти и газа» является формирование у студентов комплекса знаний о составе и свойствах нефтяных систем различного происхождения, о влиянии состава нефтей и газов на эксплуатационные параметры оборудования, а также о методах их исследования и переработки.
ЛАБОРАТОРНАЯ РАБОТА 1
Определение фракционного состава нефти и нефтепродуктов путём прямой перегонки и ректификации.
Разделение нефти и нефтепродуктов методом перегонки
Первичными методами переработки нефти и нефтепродуктов являются перегонка (при атмосферном и пониженном давлениях) и ректификация. С помощью данных методов определяют фракционный состав нефти и нефтепродукта.
Перегонка (дистилляция) — это физический метод разделения, основанный на испарении жидкости и конденсации паров, обогащённых легколетучим компонентом. Термин «дистилляция» означает «разделение по каплям» или «стекание по каплям». История перегонки насчитывает около 3500 лет. В древности с помощью перегонки получали розовое и другие эфирные масла, дистиллированную (пресную) воду из морской, использовали перегонку для приготовления микстур и напитков. Современная нефтепереработка берёт свое начало с перегонного куба, целевым назначением которого было получение осветительного керосина.
Перегонку можно проводить периодически и непрерывно. При периодической перегонке содержимое перегонного аппарата частично или полностью отгоняется. В этом процессе происходит непрерывное изменение состава паровой и жидкой фаз, а также температуры отбора паров. При непрерывной перегонке продукт непрерывно вводят в перегонный аппарат. При этом обычно разделение паровой и жидкой фаз происходит однократно, поэтому такой процесс называется однократной перегонкой или однократным испарением. В этом процессе образующаяся паровая фаза остается в равновесии в смеси с жидкой до установления конечной (заданной) температуры. Фазы разделяются после установления этой температуры.
Прямой перегонкой можно разделить до определённой степени смесь компонентов, температуры кипения которых отличаются более, чем на 50°С.
Ректификация — физический метод разделения, основанный на многократном противоточном контакте жидкой и паровой фаз. При этом паровая фаза обогащается низкокипящим компонентом, а жидкая — высококипящим. Ректификация, как перегонка, может осуществляться периодически и непрерывно. Ректификацией можно разделять смесь компонентов, различающихся по температуре кипения всего на 0,5°С.
Прямая перегонка нефти
Обычно процесс простой перегонки проводят периодически; в принципе этот процесс можно организовать и непрерывным.
При периодической перегонке жидкость постепенно испаряется, и образующиеся при этом пары непрерывно удаляются из системы и конденсируются с получением дистиллята (иногда этот процесс называют простой дистилляцией). При этом содержание низкокипящей фракции (НК) в исходной жидкости уменьшается, что приводит к снижению содержания НК в дистилляте - в начале процесса содержание НК максимально, а в конце процесса - минимально.
Простую перегонку можно проводить при атмосферном давлении или под вакуумом (для снижения температуры перегонки). Для получения нужных фракций (или разного состава дистиллята) применяют фракционную, или дробную, перегонку.
Приборы, реактивы, материалы
Нефть, колба Вюрца, насадка Вюрца, прямой холодильник, алонж, приемники, термометр на 250°С, кипелки, электронагревательный прибор.
2
vk.com/club152685050 | vk.com/id446425943
Проведение работы
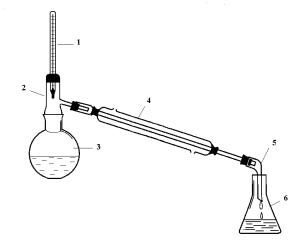
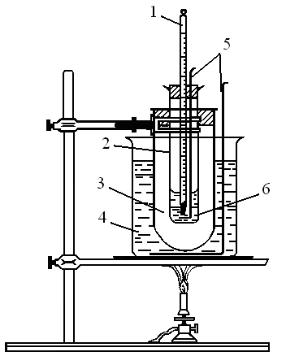
Собирают установку для перегонки (рис. 1). Во взвешенную колбу помещают 30 мл отфильтрованного нефтепродукта и снова взвешивают для определения массы нефтепродукта. В колбу помещают кипелки. При простой перегонке термометр следует помещать так, чтобы верхняя граница ртутного шарика находилась на уровне нижнего края отверстия отводной трубки. После того, как установка для перегонки полностью собрана, подставляют нагревательный прибор. Отбор фракций производится двумя способами: отбираются фракции с чёткими температурами кипения; если в течение перегонки не устанавливается чёткая температура кипения, то отбор фракций проводится через 5°С. Для каждой фракции определить выход и показатель преломления. По окончании перегонки, остывшую колбу взвешивают для определения массы кубового остатка.
Рис. 1. Установка для перегонки:
1 – термометр; 2 – насадка Вюрца; 3– круглодонная колба; 4 – холодильник Либиха; 5 – аллонж; 6 – приемник.
Задание: Определить фракционный состав нефти. Полученные результаты сравнить с данными, полученными при фракционировании нефти на ректификационной колонке.
Фракционирование нефтепродуктов на ректификационной колонке под атмосферным давлением
Перегонка с дефлегматором представляет собой простейшую ректификационной колонку.
Приборы и реактивы
Нефть или смесь органических веществ (углеводородов), колба Вюрца, дефлегматор, обратный холодильник, головка полной конденсации, приемники, термометр на 250°С, кипелки, электронагревательный прибор.
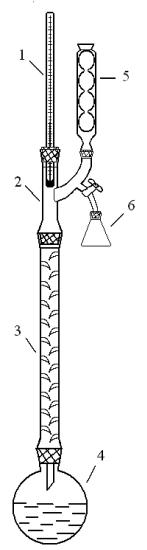
Перед перегонкой ректификационную колонку собирают так, как это изображено на рис.2.
В колбу 4 загружают взвешенные 30 мл хорошо высушенной исследуемой жидкости. Определение массы нефтепродукта проводят также, как и при прямой перегонке. Перед началом перегонки в колбу необходимо поместить кипелки. Колбу присоединяют к дефлегматору 3,
сообщённому с головкой полной конденсации 2, к которой присоединён приёмник 6. Шлифы прибора должны быть промазаны смазкой, нерастворимой в перегоняемой жидкости. Приёмниками служат пришлифованные колбы ёмкостью 15—25 мл. Термометр (с ценой деления 0,2— 0,5°С) устанавливают так, чтобы его шарик находился на 1—2 см ниже боковой трубки.
Кран головки полной конденсации держат закрытым, чтобы колонка работала с полным орошением, без отбора дистиллята. Необходимо дать колонке «захлебнуться» — заполнить центральную трубку и часть головки. Таким образом, из насадки удаляется воздух, и вся она смачивается флегмой и
3
vk.com/club152685050 | vk.com/id446425943
паром. При «захлебывании» желательно регулировать нагрев колбы так, чтобы до появления жидкости в головке прошло 15—20 мин. Через 5—10 мин после «захлебывания» уменьшают нагрев колбы. Поддерживая слабое кипение жидкости в колбе и не прекращая орошения, дают стечь избытку флегмы в колбу.
Рис. 2. Ректификационная колонна 1 – термометр, 2 – головка полной конденсации,
3 – дефлегматор,
4– круглодонная колба,
5– обратный холодильник,
6– приемник.
Затем устанавливают оптимальный режим перегонки. По достижении равновесия записывают температуру кипения жидкости, и слегка открывают кран для отбора дистиллята. При отборе дистиллята нужно не только поддерживать в колонке режим, близкий к только что установленному, но и следить за соотношением орошения, возвращаемого в колонку, и дистиллята, отбираемого в приемник, за один и тот же промежуток времени.
Для хорошего разделения важно, чтобы это соотношение (флегмовое число) было высоким, но чтобы количество флегмы не достигало величины, при которой колонка начинает «захлёбываться». Для большинства простейших колонок оптимальное флегмовое число должно лежать в интервале 20:1 - 45:1.
Перегонку ведут до заданной глубины отбора, после чего отставляют колбонагреватель, дают жидкости охладиться и стечь в колбу. Колбу отсоединяют от колонки и взвешивают. Суммируют массы отогнанных фракций и остатка и вычисляют потери при перегонке. Обычно их относят к массе первой фракции. В каждой из фракций в дальнейшем определяют показатель преломления и, если требуется, плотность.
При разделении углеводородов перегонкой следует выделять фракции в правильных температурных интервалах. Если фракционирование осуществляется для выделения заранее намеченных соединений с уже известными температурами кипения, то при первой перегонке фракции отбирают так, чтобы их низшие и высшие пределы отстояли одинаково от точек кипения соответствующих чистых веществ. Подвергая выделенные фракции повторному фракционированию, температурные пределы всё
4
vk.com/club152685050 | vk.com/id446425943
более сужают, пока не будут достигнуты постоянные точки кипения. Если же целью фракционирования является выделение возможно большего числа индивидуумов, то при первых перегонках отбирают фракции в пределах 10 или 5°С.
Отбор дистиллята
Отбор дистиллята можно проводить непрерывно, периодически и смешанным способом.
Непрерывный отбор дистиллята
При этом способе слегка открывают кран для отбора дистиллята у головки так, чтобы установить заранее заданное флегмовое число.
Отсчёт температуры производят через равные объёмы дистиллята. Величину флегмового числа уменьшают, увеличивая скорость отбора дистиллята, если в приемнике собирается индивидуальное вещество (или азеотропная смесь). Признаком этого является постоянство температуры паров в головке полной конденсации в течение некоторого времени. При переходе от фракции к фракции флегмовое число опять увеличивают, уменьшая скорость отбора дистиллята. Это процесс называется «отжимом» фракции.
Периодический отбор дистиллята
При этом способе периодически на 1—5 сек. полностью открывают кран для отбора дистиллята, спускают в приемник небольшое количество последнего (иногда 1—2 капли) и закрывают кран. Отбор дистиллята в дальнейшем производят таким же способом через равные промежутки времени, отбирая каждый раз одинаковое его количество. Температуру и объём дистиллята отсчитывают непосредственно перед очередным отбором.
Флегмовое число при этом определяют по отношению числа капель, падающих в колонку за известный промежуток времени, не меньший, чем требуется для того, чтобы 5 раз отобрать дистиллят, к числу капель, падающих в приемник за тот же промежуток. При периодическом отборе флегмовое число варьируют изменением отношения времени, в течение которого кран закрыт, ко времени, в течение которого кран открыт. Для уменьшения флегмового числа предпочтительно чаще открывать кран на короткий промежуток времени, нежели удлинять промежуток времени, в течение которого кран бывает открыт.
Отжим фракций при периодическом отборе дистиллята производят следующим образом. Открыв кран, отбирают небольшое количество дистиллята. Так как при этом удаляется часть легколетучего компонента, то равновесие в колонке нарушается и температура пара в головке начинает увеличиваться. Когда кран вновь закрывают, в колонке постепенно вновь устанавливается состояние равновесия, и пар в головке обогащается легколетучим компонентом; температура пара при этом понижается (до некоторого предела). Когда температура установится, вновь отбирают дистиллят.
Смешанный способ отбора дистиллята.
Перегонку отдельных фракций проводят по способу непрерывного отбора дистиллята, а при переходе от фракции к фракции прибегают к периодическому отбору дистиллята, «отжимая» фракцию.
Пример записи и расчёта
Фракционирование смеси (углеводородов или нефти). Масса колбы с продуктом, г.
Масса пустой колбы, г. Навеска, г.
5
vk.com/club152685050 | vk.com/id446425943
|
|
|
|
|
|
|
|
Таблица 1 |
|
|
|
Результаты опытов фракционирования исследуемого |
|
|
|
||||
|
|
|
|
нефтепродукта |
|
|
|
||
|
|
|
|
|
Масса, г |
|
|
|
|
|
|
|
|
|
|
|
|
n20 |
|
№ |
|
Ткип.° |
|
|
Приемника |
|
Вы |
|
|
С |
|
|
|
|
Фр |
ход, % |
фракции |
|
|
|
|
Пусто |
|
с |
ак-ции |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
го |
|
продуктом |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
и т.д. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Остат |
|
|
|
|
|
|
|
|
|
ок |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Потер |
|
|
|
|
|
|
|
|
|
и |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
При фракционировании смеси двух углеводородов наблюдается ряд промежуточных фракций. Они содержат смесь обоих компонентов. Процентное содержание компонентов в промежуточной фракции может быть определено на основании показателей преломления смеси и чистых углеводородов по формуле:
n смеси = n1х+ n2 (100 — х)
где х — содержание одного из компонентов, %.
Задание 1. Провести фракционирование нефти. Определить содержание низкокипящих фракций нефти и показатель преломления каждой фракции.
Задание 2. Провести фракционирование смеси углеводородов. Определить процентный состав смеси углеводородов.
ЛАБОРАТОРНАЯ РАБОТА 2
Очистка нефтепродуктов от ароматических углеводородов адсорбцией на адсорбционной колонке
При изучении химического состава углеводородов важное место отводится хроматографическим методам исследования. Бензиновые фракции разделяют на группы углеводородов при помощи жидкостно-адсорбционной хроматографии, чаще называемой просто адсорбционной. Для исследования индивидуального химического состава фракций (особенно низкокипящих) часто применяется газожидкостная хроматография.
Адсорбционный анализ основан на способности адсорбентов избирательно извлекать из смесей соединения определённых типов. Для разделения углеводородов применяют различные адсорбенты: окись алюминия, активированный уголь, силикагель и др. Чаще всего используют силикагель. Ароматические углеводороды более прочно удерживаются на поверхности адсорбента, чем парафиновые и нафтеновые. Пропуская смесь углеводородов сверху вниз по колонке с адсорбентом, обычно с добавлением растворителя, выделяют из колонки вначале парафиновые и нафтеновые углеводороды, а затем ароматические. Для выделенных фракций измеряют объём и исследуют (определяют наличие ароматических углеводородов, показатель преломления, анилиновую точку и т.п.).
При адсорбционном разделении бензиновых фракций применяют два типа растворителей: вытесняющие и смещающие. Вытесняющие растворители — спирты (изопропиловый, этиловый, метиловый) — адсорбируются сильнее компонентов бензина и выделяются из колонки вслед за ароматическими углеводородами. В этом случае нельзя достигнуть полного разделения бензина на две фракции — парафино-нафтеновую и ароматическую, так как они движутся по колонке вплотную друг к другу. Поэтому приходится еще отбирать промежуточную фракцию, представляющую собой их смесь. Смещающие (разбавляющие) растворители — пентан, изопентан — близки по адсорбируемости к
6
vk.com/club152685050 | vk.com/id446425943
парафино-нафтеновой фракции. Такие растворители смешиваются в колонке с углеводородами, постепенно десорбируя их и заставляя двигаться вниз. Если вслед за смещающим растворителем (изопентан) ввести в колонку вытесняющий (метанол, этанол), то можно отделить парафинонафтеновую фракцию без отбора промежуточной. Измеряя показатель преломления фильтрата, можно
обнаружить компоненты смеси в такой последовательности: парафины + нафтены |
парафины + |
нафтены + изопентан изопентан + ароматические углеводороды метанол |
+ ароматические |
углеводороды метанол. Фракции парафино-нафтеновых и ароматических углеводородов выделяют из фильтрата отгонкой изопентана. Фракцию ароматических углеводородов отделяют от метанола промывкой водой, после чего обезвоживают СаСl2 и металлическим натрием.
Для бензинов, содержащих до 15 объёмных процентов ароматических углеводородов, удобно применять адсорбционное разделение с вытесняющим растворителем и отбором промежуточной фракции, при более высоком содержании в бензине ароматических углеводородов рекомендуется разделение при помощи смещающей жидкости + вытесняющий растворитель.
Приборы, реактивы, материалы
Исследуемый нефтепродукт или смесь, стеклянная колонка (рис. 3) высотой 200 мм, диаметром 8—10 мм с воронкой для подсадки пробы, рефрактометр типа ИРФ, изопропиловый спирт или пентан (изопентан), адсорбент – Al2O3, мерные цилиндры (градуированные пробирки) с ценой деления 0,1 мл.
Проведение анализа
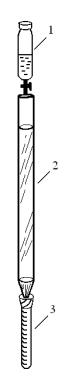
Колонку заполняют сухим адсорбентом, укрепляют в штативе, наверху закрепляют делительную воронку 1 для подсадки пробы и элюента, под нижний конец колонки 2 подводят градуированную пробирку 3.
Исследуемую фракцию в количестве 5 мл заливают в колонку и после того, как она полностью впитается в адсорбент, добавляют в качестве десорбирующей жидкости 15-20 мл изопропилового спирта. Сначала из колонки будет выходить насыщенная (алкано-циклоалкановая) часть исследуемой фракции, которая адсорбируется Al2O3 менее прочно. Первую порцию отбирают в количестве 1 мл, а все последующие — по 0.5 мл. Для каждой отобранной фракции определяют показатель преломления. Фракции, отличающиеся по показателю преломления не более чем на 0.0005, смешиваются.
Рис. 3. Адсорбционная колонка
1– делительная воронка,
2– колонка с адсорбентом,
3– градуированная пробирка.
Появление аренов замечают по формалитовой реакции: в пробирку помещают 1 мл 98 %-й бесцветной серной кислоты, добавляют 2—3 капли 10 %-го раствора формалина и столько же продукта.
7
vk.com/club152685050 | vk.com/id446425943
При отсутствии аренов смесь остается бесцветной или слегка желтеет. Ярко-красное окрашивание указывает на появление в отобранной фракции аренов.
ЛАБОРАТОРНАЯ РАБОТА 3 Определение плотности, кинематической вязкости и показателя преломления нефтепродуктов
I. Подготовка к анализу. Удаление механических примесей
Механические примеси содержатся в сырой нефти в виде песка, глинистых минералов и различных солей, которые находятся во взвешенном состоянии. При исследованиях нефтей, большое содержание механических примесей может в значительной степени повлиять на правильность определения таких показателей, как плотность, молекулярная масса, коксуемость, содержание серы, азота, смолисто-асфальтеновых веществ и микроэлементов. В процессе переработки нефти в нефтепродукты могут попасть продукты коррозии аппаратов и трубопроводов, мельчайшие частицы отбеливающей глины, минеральные соли. Загрязнение нефти и нефтепродуктов может происходить также при хранении и транспортировке.
Механические примеси в топливе могут привести к засорению топливопроводов, фильтров, увеличению износа топливной аппаратуры, нарушению питания двигателя. Эти же примеси в маслах и смазках могут вызвать закупорку маслопроводов, поломку смазочной аппаратуры, образование задиров на трущихся поверхностях.
Из механических примесей наиболее опасными являются песок и другие твёрдые частицы, истирающие металлические поверхности.
Методы определения общего содержания механических примесей основаны на способности всех органических компонентов нефти растворяться в органических растворителях. Нерастворившийся остаток, задерживаемый фильтром при фильтровании раствора нефти или нефтепродукта, и характеризует содержание в них механических примесей.
Перед началом проведения аналитических работ следует очистить нефть и нефтепродукты от механических примесей обычными методами фильтрования.
II. Плотность
Плотностью называется масса единицы объёма вещества (нефти, нефтепродукта). Единицей плотности в системе СИ является кг/м3.
В исследовательской практике определяется относительная плотность. Относительной плотностью называется отношение плотности нефти или нефтепродукта при 20°С к плотности дистиллированной воды (эталонного вещества) при 4°С, т.е. отношение массы нефти или нефтепродукта при 20°С к массе такого же объема дистиллированной воды при 4°С. Относительную плотность обозначают . Умножив значение относительной плотности на 1000, можно получить плотность в кг/м3. Плотность нефти и нефтепродукта зависит от температуры.
Значение плотности для характеристики нефтей и нефтепродуктов
Плотность принадлежит к числу наиболее распространённых показателей, применяемых при исследовании нефтей и нефтепродуктов. Особое значение этот показатель имеет при расчёте массы нефтепродуктов, занимающих данный объём, и, наоборот, объёма нефтепродуктов, имеющих определённую массу. Это очень важно как для конструктивно-расчётных исследований, так и для практической работы на местах производства, транспортировки и потребления нефтепродуктов.
Если принять во внимание, что основная масса нефтепродуктов падает на долю углеводородов, а последние в подавляющей части состоят из трёх основных классов — парафинов, нафтенов и ароматических углеводородов, заметно различающихся по величине плотности для соединений с одинаковым числом атомов в молекуле, то можно сделать вывод, что величина плотности будет до известной степени характеризовать не только состав и происхождение продукта, но также и его качество.
В самом деле, при прочих равных показателях качества нефтепродуктов можно считать, что
8
vk.com/club152685050 | vk.com/id446425943
более высокая плотность указывает на большее содержание ароматических компонентов, а более низкая
— на большее содержание углеводородов и с этим положением приходится считаться не только в процессе производства, но и в процессе потребления нефтепродуктов.
Экспериментально плотность нефти (нефтепродукта) определяют одним из трёх стандартных методов: ареометром (нефтеденсиметром), гидростатическими весами Вестфаля — Мора и пикнометром. Из них наиболее быстрым является ареометрический метод, а наиболее точным — пикнометрический. Преимуществом пикнометрического метода является использование сравнительно малых количеств анализируемой пробы.
Определение плотности пикнометром
Приборы, реактивы, материалы
Пикнометр, хромовая смесь, вода дистиллированная, этиловый спирт, пипетка, бумага фильтровальная.
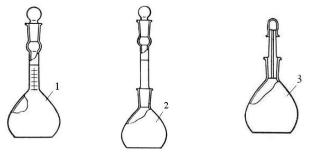
Стандартной температурой, при которой определяется плотность нефти и нефтепродукта, является 20°С. Для определения плотности применяют стеклянные пикнометры с меткой и капиллярной трубкой различной ёмкости (рис. 4). Каждый пикнометр характеризуется так называемым «водным числом», т.е. массой воды в объёме данного пикнометра при 20°С.
Рис. 4. Пикнометры. 1 – пикнометр Бирона, 2 – пикнометр с меткой, 3 – пикнометр с капилляром.
Перед определением водного числа пикнометр промывают последовательно хромовой смесью, дистиллированной водой, этиловым спиртом и сушат. Чистый и сухой пикнометр взвешивают с точностью до 0.0002 г. С помощью пипетки наполняют пикнометр дистиллированной свежекипяченой и охлаждённой до комнатной температуры водой (пикнометры с меткой заполняют по верхнему краю мениска). Пикнометр с установленным уровнем воды при 20±0,1°С тщательно вытирают снаружи и взвешивают с точностью до 0,0002 г.
Водное число m пикнометра вычисляют по формуле: m = m2 –m1
где m1, m 2 — масса пикнометра с водой и пустого, соответственно, г.
Плотность нефти (нефтепродукта) с вязкостью при 500С не более 75 мм2/с определяют следующим образом. Сухой и чистый пикнометр наполняют с помощью пипетки анализируемой нефтью (нефтепродуктом) при 18-20°С (уровень нефтепродуктов в пикнометре устанавливают по верхнему краю мениска), стараясь не замазать стенки пикнометра. Пикнометр с установленным уровнем тщательно вытирают и взвешивают с точностью до 0,0002 г.
«Видимую» плотность ρ' анализируемой нефти (нефтепродукта) вычисляют по формуле: ρ' = (m3 – m1)/ m2
где m3 — масса пикнометра с нефтью (нефтепродуктом), г; m1 - масса пустого пикнометра, г; m2 - водное число пикнометра, г.
«Видимую» плотность пересчитывают в плотность ρ420 по формуле:
ρ420 = (0. 99823 – 0.0012) ρ' + 0.0012 = 0.99703 ρ' + 0.0012,
где 0,99823 - значение плотности воды при 20°С; 0,0012 — значение плотности воздуха при 20°С и давлении 0,1 МПа (760 мм рт. ст.).
9
vk.com/club152685050 | vk.com/id446425943
Вычисленные по этой формуле поправки к «видимой» плотности приведены в табл. 1. Для получения плотности ρ420 анализируемой нефти (нефтепродукта) поправку вычитают из значения «видимой» плотности. Расхождение между параллельными определениями плотности не должно превышать 0,0004.
Таблица 1
Поправки к «видимой» плотности
«Видимая» |
Поп |
«Видимая» |
Попр |
|
плотность |
||||
равка |
плотность ρ' |
авка |
||
ρ' |
||||
|
|
|
||
|
|
|
|
|
0,6900…0, |
0,00 |
0,8500…0,85 |
0, |
|
|
|
|
|
|
0,7000…0, |
0,00 |
0,8600…0,86 |
0, |
|
|
|
|
|
|
0,7100…0, |
0,00 |
0,8700…0,87 |
0, |
|
|
|
|
|
|
0,7200…0, |
0,00 |
0,8800…0,88 |
0, |
|
|
|
|
|
|
0,7300…0, |
0,00 |
0,8900…0,89 |
0, |
|
|
|
|
|
|
0,7400…0, |
0,00 |
0,9000…0,90 |
0, |
|
|
|
|
|
|
0,7500…0, |
0,00 |
0,9100…0,91 |
0, |
|
|
|
|
|
|
0,7600…0, |
0,00 |
0,9200…0,92 |
0, |
|
|
|
|
|
|
0,7700…0, |
0,00 |
0,9300…0,93 |
0, |
|
|
|
|
|
|
0,7800…0, |
0,00 |
0,9400…0,94 |
0, |
|
|
|
|
|
|
0,7900…0, |
0,00 |
0,9500…0,95 |
0, |
|
|
|
|
|
|
0,8000…0, |
0,00 |
0,9600…0,96 |
0, |
|
|
|
|
|
|
0,8100…0, |
0,00 |
0,9700…0,97 |
0, |
|
|
|
|
|
|
0,8200…0, |
0,00 |
0,9800…0,98 |
0, |
|
|
|
|
|
|
0,8300…0, |
0,00 |
0,9900…1,00 |
0, |
|
|
|
|
|
|
0,8400…0, |
0,00 |
|
|
|
|
|
|
|
Следует иметь в виду, что результаты определения плотности искажаются при наличии в исходной пробе нефти (нефтепродукта) воды и механических примесей.
III. Показатель преломления
Показатель преломления (коэффициент рефракции) также является одной из основных характеристик нефтепродуктов. Он характеризует способность нефтепродукта преломлять падающий на него световой луч. При этом отношение синуса угла падения луча к синусу угла преломления луча для каждого нефтепродукта постоянно и называется показателем преломления. Определение показателя преломления основано на явлении предельного угла, при котором наступает полное внутреннее отражение. Показатель преломления определяют с помощью прибора, который называется
рефрактометр.
Показатель преломления зависит от температуры и длины световой волны. Чем больше длина волны светового луча, тем меньше показатель преломления.
Показатель преломления нефтепродукта обычно определяют для желтой линии натрия при 20°С и обозначают n20D.
По показателю преломления можно оценить чистоту индивидуального углеводорода, углеводородный состав нефтяной фракции. Из углеводородов наименьшее значение показателя преломления имеют н-алканы. С утяжелением фракционного состава нефтяной фракции повышается её показатель преломления.
10
vk.com/club152685050 | vk.com/id446425943
По изменению показателя преломления можно судить о фазовых превращениях твердых углеводородов. При этом анизотропная жидкая фаза характеризуется одним значением показателя преломления, а анизотропная твёрдая фаза — двумя значениями показателя преломления. Область появления твёрдой фазы в некотором интервале температур характеризуется двумя показателями преломления: жидкой и твёрдой фаз.
Для определения показателя преломления применяют два типа рефрактометров: Аббе и Пульфриха. К первому типу относятся отечественные рефрактометры РЛУ, ИРФ-22, ИРФ-454. Рефрактометром типа Пульфриха является прибор ИРФ-23. В лабораторной практике наиболее часто применяют рефрактометры типа Аббе. Для более точных определений показателя преломления и дисперсии необходимо использовать рефрактометр типа Пульфриха.
11
vk.com/club152685050 | vk.com/id446425943
Определение показателя преломления рефрактометром ИРФ-454
Приборы, реактивы, материалы
Рефрактометр типа ИРФ-454, стеклянная палочка или пипетка, петролейный эфир или этиловый спирт, салфетка или вата.
Рефрактометр ИРФ-454 позволяет определять показатель преломления нефтепродукта в интервале от 1,2 до 2,0 для линии D с точностью ±2.10-4. Принцип действия рефрактометра основан на явлении полного внутреннего отражения при прохождении светом границы раздела двух сред с разными показателями преломления.
Определение показателя преломления проводят при дневном или электрическом свете. Рефрактометр и источник света устанавливают так, чтобы свет падал на входное окно осветительной призмы или на зеркало, которым направляют свет во входное окно измерительной призмы. Перед началом работы следует откинуть осветительную призму и очистить поверхность измерительной призмы. Поверхность призмы очищают путём протирки чистой мягкой неворсистой салфеткой (тканью), смоченной петролейным эфиром или спиртом. Затем по дистиллированной воде или по контрольному образцу проверяют юстировку рефрактометра при 20°С.
После юстировки на чистую полированную поверхность измерительной призмы стеклянной палочкой или пипеткой осторожно, не касаясь призмы, наносят две-три капли исследуемого нефтепродукта и опускают осветительную призму. Измерения прозрачных нефтепродуктов проводят в проходящем свете, когда он проходит через открытое окно осветительной призмы, при этом окно измерительной призмы закрыто зеркалом. Окуляр устанавливают на отчётливую видимость перекрестия. Поворотом зеркала добиваются наилучшей освещенности шкалы. Вращением нижнего маховика границу светотени следует ввести в поле зрения окуляра. Верхний маховик необходимо вращать до исчезновения окраски граничной линии. Наблюдая в окуляр, нижним маховиком наводят границу светотени точно на перекрестие и по шкале показателей преломления снимают отсчёт. Цена деления шкалы 1*10-3. Целые, десятые, сотые и тысячные доли отсчитывают по шкале, а десятитысячные доли оценивают на глаз.
Для окрашенных и тёмных нефтепродуктов измерения проводят в отражённом свете.
IV. Вязкость
Вязкость, как и плотность, — важный физико-химический параметр, используемый при подсчёте запасов нефти, проектировании разработки нефтяных месторождений, выбора способа транспорта и схемы переработки нефти.
Различают динамическую, кинематическую и условную вязкость. Динамическая вязкость η — это отношение действующего касательного напряжения к градиенту скорости при заданной температуре. Единица измерения динамической вязкости паскаль-секунда — Па•с, на практике используют обычно мПа•с.
Необходимость определения кинематической и условной вязкости связана с тем, что для определения динамической вязкости требуется источник постоянного давления (постоянно приложенного напряжения) на жидкость. Это условие предопределяет дополнительные технические трудности, сложность воспроизведения и трудоёмкость анализа. Кинематическая вязкость ν — это отношение динамической вязкости жидкости к плотности при той же температуре:
ν = η/ρ
Размерность измерения кинематической вязкости - м2/с, на практике используют обычно см2/с. Сущность метода определения кинематической вязкости заключается в замене постоянного
давления (внешней силы) давлением столба жидкости, равным произведению высоты столба жидкости, плотности жидкости и ускорения силы тяжести. Эта замена привела к значительному упрощению и распространению метода определения кинематической вязкости в стеклянных капиллярных вискозиметрах.
Определение условной вязкости также основано на истечении жидкости (как правило, для этих целей используют трубку с диаметром отверстия 5 мм) под влиянием силы тяжести. Условная вязкость
— отношение времени истечения нефтепродукта при заданной температуре ко времени истечения дистиллированной воды при 20°С. Единица измерения — условные градусы (°ВУ). Метод определения условной вязкости применяется для нефтепродуктов, дающих непрерывную струю в течение всего
12
vk.com/club152685050 | vk.com/id446425943
испытания и для которых нельзя определить кинематическую вязкость по ГОСТ 33—82. Условную вязкость определяют для нефтяных топлив (мазутов).
Определение кинематической вязкости обязательно для таких товарных нефтепродуктов, как дизельные топлива и смазочные масла (ньютоновские жидкости).
Согласно унифицированной программе исследования, для нефтей определяют кинематическую (или динамическую) вязкость при температурах от 0 до 50°С (через 10°С). Для маловязких нефтей определение начинают с 20°С. Для керосиновых дистиллятов определяют кинематическую вязкость при 20 — 40°С. Для дизельных — при 20°С, для масляных — при 40, 50 и 100°С. Для остатков, выкипающих выше 350°С, определяют условную вязкость при 50, 80 и 1000С.
На вязкость нефти и нефтепродуктов существенное влияние оказывает температура. С её понижением вязкость увеличивается. Вязкостно-температурные свойства нефтепродуктов зависят от их фракционного и углеводородного состава. Наименьшей вязкостью и наиболее пологой кривой вязкости обладают алифатические углеводороды. Наибольшей вязкостью и наиболее крутой кривой вязкости — ароматические (особенно би- и полициклические) углеводороды.
Важным эксплуатационным показателем топлив и масел является прокачиваемость. Прокачиваемость моторных топлив и топлив для газотурбинных и котельных установок существенно зависит от их вязкости. Например, количество бензина вязкостью 0.65 мм2/с, поступающего в двигатель за одну минуту, составляет 100 г, а бензина вязкостью 1,0 мм2/с — 95 г. В технических требованиях на товарные топлива и смазочные масла предусмотрены соответствующие ограничения значения вязкости. Так, топлива для быстроходных дизелей должны иметь кинематическую вязкость при 20°С в пределах
1,5—6,0 мм2/с.
С понижением температуры высоковязкие нефти, природные битумы и остаточные нефтепродукты (мазут, гудрон) могут проявлять аномалию вязкости, так называемую структурную вязкость. При этом их течение перестает быть пропорциональным приложенному напряжению, т.е. они становятся неньютоновскими жидкостями. Причиной структурной вязкости является содержание в нефти и нефтепродукте смолисто-асфальтеновых веществ и парафинов.
Для оценки вязкостно-температурных свойств масел имеются соответствующие зависимости для расчёта температурного коэффициента вязкости (ТКВ) и индекса вязкости (ИВ).
Определение кинематической вязкости
Приборы, реактивы, материалы
Вискозиметр стеклянный Пинкевича (ВПЖ–2), термостат, резиновая трубка, резиновая груша, секундомер.
Сущность метода заключается в измерении времени истечения определённого объёма испытуемой жидкости под влиянием силы тяжести. Испытание проводят в капиллярных стеклянных вискозиметрах. Для проведения анализа подбирают вискозиметр с таким диаметром капилляра, чтобы время истечения жидкости составляло не менее 200 с (рис. 5).
Проведение анализа
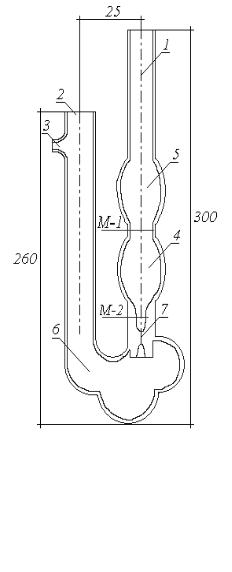
Вискозиметр ВПЖ-2 представляет собой U-образную трубку, в колено 1 которой впаян капилляр 7. При измерении вязкости жидкость из резервуара 4 течет по капилляру 7 в расширение 6.
Вискозиметр заполняют следующим образом: на отводную трубку 3 надевают резиновый шланг. Далее, закрыв пальцем колено 2 и повернув вискозиметр, опускают колено 1 в сосуд с исследуемой
жидкостью и отбирают её с помощью груши до отметки образовались пузырьки воздуха.
М |
2 |
|
, следя за тем, чтобы в жидкости не
Втот момент, когда уровень жидкости достигнет отметки М 2 , вискозиметр вынимают из сосуда
ибыстро переворачивают в нормальное положение, снимают с внешней стороны с конца колена 1 избыток жидкости и надевают на него резиновую трубку.
13
vk.com/club152685050 | vk.com/id446425943
Вискозиметр устанавливают в термостат так, чтобы расширение 5 было ниже уровня жидкости в термостате. Термостатируют не менее 5 мин. при заданной температуре. Грушей затягивают жидкость в колено 1, примерно до одной трети расширения 5. Сообщают колено 1 с атмосферой и определяют время опускания мениска жидкости от отметки М1 до отметки М 2 .
Вязкость нефти определяют при двух – трёх температурах. Если результаты трёх последовательных измерений не отличаются более, чем на 0,2%, кинематическую вязкость ν
(мм2/с), вычисляют как среднее арифметическое по формуле: ν = k·τ,
где k — постоянная вискозиметра (k=0,2711 см2/с2); τ — среднее время истечения нефти (нефтепродукта) в вискозиметре, с.
Динамическую вязкость η, (мПа·с) исследуемой нефти (нефтепродукта) вычисляют по формуле:
η = ν·ρ
где ν — кинематическая вязкость, см2/с; ρ — плотность при той же температуре, при которой
определялась вязкость, г/см3.
Допускаемые расхождения последовательных определений кинематической вязкости от среднего арифметического значения не должны превышать следующих значений:
Рис 5. Вискозиметр Пинкевича (ВПЖ-2)
1 и 2 – колени
3 – отводная трубка
4 – резервуар
5 и 6 – расширения
7 – капилляр
Температура измерения, °С |
от -60 до -30 |
от-30 до 15 |
от 15 до 150 |
|
|
|
|
Допускаемое расхождение, % |
±2,5 |
±1,5 |
±1,2 |
|
|
|
|
ЛАБОРАТОРНАЯ РАБОТА 4 Определение содержания воды в нефтях и нефтепродуктах
Диапазон содержания воды в нефтях весьма широк и может изменяться от десятых долей до 60 % и более.
Содержащаяся в нефтях вода может быть в трёх формах: растворённая, диспергированная и свободная. Содержание растворённой воды зависит в основном от химического состава нефти, нефтепродуктов и температуры. С повышением температуры растворимость воды увеличивается во всех углеводородах. Наибольшей растворяющей способностью по отношению к воде обладают ароматические углеводороды. Чем выше содержание в нефти ароматических углеводородов, тем выше в ней растворимость воды.
При снижении температуры, растворимость воды в нефти и нефтепродуктах уменьшается и вода может выделяться в виде дисперсных частиц, образуя водонефтяные эмульсии. В монодисперсных эмульсиях содержание воды может доходить до 74%. В реальных условиях водонефтяные эмульсии являются полидисперсными. В нефтях, поступающих со сборных пунктов на установки обезвоживания и обессоливания, размеры глобул воды находятся в пределах от 3—5 до 7—10 мкм. Эти размеры зависят
14
vk.com/club152685050 | vk.com/id446425943
от гидродинамических и других условий добычи нефти, а также от степени обводнённости пласта. Размеры глобул в течение года для одной и той же скважины могут меняться в пределах 5—12 мкм. Содержание воды в нефти может доходить до 97 %, однако большинство нефтей образуют с водой достаточно устойчивые эмульсии с содержанием воды не более 60 %. Остальная часть воды находится в свободном состоянии и легко отстаивается.
Важным показателем нефтяных эмульсий является их устойчивость, т.е. способность в течение длительного времени не разрушаться. Агрегативная устойчивость нефтяных эмульсий измеряется продолжительностью их существования и для различных нефтяных эмульсий колеблется от нескольких секунд до нескольких часов и даже месяцев. Устойчивость водонефтяных эмульсий зависит от ряда факторов, в том числе от наличия в них веществ, называемых эмульгаторами. Эти вещества, адсорбируясь на поверхности раздела фаз, снижают межфазное поверхностное натяжение и таким образом повышают её устойчивость. Известны десятки подобных веществ, содержащихся в нефтях. Большая их часть принадлежит к классу поверхностно-активных веществ. Такими компонентами нефти являются различные нефтяные кислоты, смолистые соединения.
Впроцессе образования и стабилизации водонефтяных эмульсий, наряду с поверхностноактивными веществами, важную роль играют тонкодисперсные нерастворимые твёрдые продукты, находящиеся в нефти в коллоидном состоянии.
К ним относятся асфальтены, микрокристаллы парафина, сульфид железа и другие механические примеси. Эти продукты образуют на поверхности капель механически прочные оболочки, препятствующие их коалесценции.
Стабилизация водонефтяных эмульсий определяется закономерностями адсорбции на поверхности капель различных эмульгирующих веществ. Вначале этот процесс идёт быстро, а затем, по мере заполнения свободной поверхности капель, постепенно затухает и скорость его стремится к нулю.
Вэтот период состав и структура бронирующих оболочек стабилизируется. Время, необходимое для такой стабилизации, называется временем старения эмульсии. Время старения эмульсии зависит от многих факторов и для большинства нефтей изменяется от двух-трёх до десятков часов. Во время старения повышается и устойчивость эмульсий к расслоению.
Стойкость эмульсий существенно зависит от фракционного состава нефтей. Чем больше содержание в нефти светлых фракций, тем менее устойчивы водонефтяные эмульсии, так как при этом увеличивается разность плотностей воды и нефти. Эмульсии высоковязких нефтей имеют более высокую стойкость, так как более высокая вязкость дисперсной среды препятствует столкновению частиц воды и их укрупнению, т.е. коалесценции.
Повышение концентрации солей в пластовой воде, которая образует с нефтью водонефтяную эмульсию, приводит к уменьшению стойкости эмульсии, так как в этом случае возрастает разность плотности воды и нефти.
Внефтепродуктах содержание воды значительно меньше, чем в нефтях. Большинство нефтепродуктов по отношению к воде обладает очень низкой растворяющей способностью. Кроме того, нефтяные дистиллятные топлива обладают и меньшей, чем нефть, эмульгирующей способностью, так как в процессе переработки удаляется значительная часть смолистых веществ, нафтеновых кислот и их солей, серосодержащих соединений, которые, как сказано выше, играют роль эмульгаторов.
Наличие воды в моторных топливах, смазочных маслах крайне нежелательно. Содержание воды в смазочных маслах усиливает их склонность к окислению и ускоряет коррозию металлических поверхностей, соприкасающихся с маслом. Присутствие воды в моторных топливах может привести при низких температурах к прекращению подачи топлива из-за забивки топливных фильтров кристаллами льда.
Методы определения воды в нефти и нефтепродуктах могут быть разбиты на две группы: качественные и количественные.
Качественные испытания позволяют определять не только эмульсионную, но и растворенную воду. К этим методам относятся пробы на прозрачность, Клиффорда, на потрескивание и на реактивную бумагу. Первые два из этих методов используют для определения воды в прозрачных нефтепродуктах. Наиболее часто применяемым методом качественного определения воды является проба на потрескивание.
Для количественного определения воды в нефти и нефтепродуктах можно использовать различные их свойства, функционально связанные с содержанием в них воды: плотность, вязкость,
15
vk.com/club152685050 | vk.com/id446425943
поверхностное натяжение, диэлектрическую проницаемость, электропроводимость, теплопроводность и т.д. Заранее рассчитать вид функции, как правило, невозможно из-за неаддитивного вклада воды в измеряемый параметр. Неаддитивность обусловлена химическим взаимодействием молекул воды и вещества. По этой причине математическую зависимость обычно находят, используя экспериментальные данные.
Другая группа методов основана на использовании химических и физико-химических свойств самой воды. К ним, например, относятся метод титрования реактивом Фишера, гидридкальциевый метод и др.
Существующие количественные методы определения воды в жидких продуктах, кроме того, делят на прямые и косвенные. К прямым методам относят метод Дина и Старка, титрование реактивом Фишера, гидридкальциевый метод и центрифугирование, к косвенным — ИК-спектрофотометрический, кондуктометрический, колориметрический и др.
Определение содержания воды по методу Дина и Старка
Это наиболее распространенный и достаточно точный метод определения количественного содержания воды в нефтях и нефтепродуктах. Он основан на азеотропной перегонке пробы нефти или нефтепродукта с растворителями.
Приборы, реактивы, материалы
Колбонагреватель или электроплитка, приемник-ловушка, обратный холодильник, колба круглодонная вместимостью 200 мл, мерный цилиндр, кипелки, петролейный эфир (Ткип=80-100°С), нефтепродукт.
Подготовка к анализу. По этому методу в качестве растворителя используют бензинрастворитель (петролейный эфир), выкипающий при 80…100°С и содержащий не более 3% ароматических углеводородов.
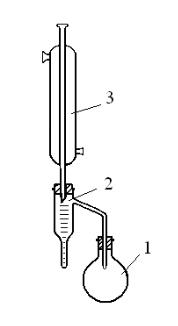
Пробу нефти тщательно перемешивают встряхиванием в склянке в течение 5 мин. Высоковязкие нефти и нефтепродукты предварительно нагревают до 40—50°С. Из перемешанной пробы нефти или нефтепродукта берут 30 мл мерным цилиндром в чистую сухую, предварительно взвешенную стеклянную колбу 1 (рис. 6). Затем в колбу 1 этим же цилиндром отмеряют 50 мл растворителя и содержимое перемешивают. Для равномерного кипения перед началом нагревания в колбу бросают несколько кипелок.
Рис. 6. Прибор Дина и Старка: 1 – круглодонная колба, 2 – насадка Дина –Старка,
3 – холодильник.
Колбу при помощи шлифа присоединяют к отводной трубке 2 (насадке Дина – Старка), а к верхней части приёмника-ловушки на шлифе присоединяют холодильник 3. Приёмник-ловушку (пробирку) присоединяют к отводной трубке 2. Пробирка и холодильник должны быть чистыми и
16
vk.com/club152685050 | vk.com/id446425943
сухими. Во избежание конденсации паров воды из воздуха верхний конец холодильника необходимо закрыть ватой.
Проведение анализа.
Содержимое колбы нагревают с помощью колбонагревателя или на электрической плитке. Перегонку ведут так, чтобы из трубки холодильника в приёмник-ловушку падали 2—4 капли в секунду. Нагрев прекращают после того, как объём воды в приемнике-ловушке перестанет увеличиваться и верхний слой растворителя станет совершенно прозрачным. Продолжительность перегонки должна быть не менее 30 и не более 60 мин. Если на стенках трубки холодильника имеются капельки воды, то их сталкивают в приёмник-ловушку стеклянной палочкой. После охлаждения испытуемого продукта до комнатной температуры прибор разбирают. Если количество воды в приёмнике-ловушке не более 0,3 мл и растворитель мутный, то приёмник помещают на 20—30 мин в горячую воду для осветления и снова охлаждают до комнатной температуры. После охлаждения определяют объём воды в приёмникеловушке с точностью до одного верхнего деления.
Объёмную долю воды X (%) рассчитывают по формуле
X (%) = V·100)/V1
где V — объём воды, собравшейся в приёмнике-ловушке мл; V1— объём нефти или нефтепродукта, взятый для испытания, мл.
Количество воды в приёмнике-ловушке 0,03 мл и меньше считается следами.
ЛАБОРАТОРНАЯ РАБОТА 5
Определение температуры вспышки и воспламенения нефтепродуктов
Большинство нефтей имеют температуру вспышки паров ниже 0°С. Например, температура вспышки усть-балыкской и самотлорской нефтей равна соответственно -30°С и ниже -35°С. Природный битум Мордово-Кармальского месторождения, добытый методом внутрипластового горения, имеет температуру вспышки 59°С. Фракции 120…230°С и 180…350°С мордово-кармальского природного битума имеют температуру вспышки соответственно 32 и 91°С.
По температуре вспышки нефтепродукты делятся на легковоспламеняющиеся и горючие. К легковоспламеняющимся относятся нефтепродукты, имеющие температуру вспышки паров не более 61°С в закрытом тигле (не более 66°С в открытом тигле). К горючему классу относятся нефтепродукты с температурой вспышки более 61°С в закрытом тигле (более 66°С в открытом тигле).
Легковоспламеняющимися нефтепродуктами являются моторные топлива. Так, автомобильный бензин имеет температуру вспышки в закрытом тигле -50°С, авиационный -30°С. Топлива для реактивных двигателей, в зависимости от сортности, должны иметь температуру вспышки не ниже 28…60°С, а топлива для быстроходных дизелей 35…61°С.
Температура воспламенения дизельных топлив находится в пределах 57…119°С. Температура воспламенения всегда выше температуры вспышки.
Температура самовоспламенения нефтепродукта с увеличением его молекулярной массы уменьшается: если бензины самовоспламеняются при температурах выше 500°С, то дизельные топлива при 300…330°С.
По температурам вспышки, воспламенения и самовоспламенения оценивают пожаро- и взрывоопасность нефти и нефтепродукта. Температуру вспышки нефти, лёгких нефтяных фракций и моторных топлив определяют в закрытом и открытом тиглях. Определение в открытом тигле применяют для масел и тёмных нефтепродуктов.
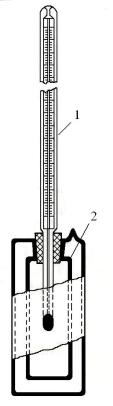
Определение температур вспышки и воспламенения в открытом тигле по методу Бренкена
Приборы, реактивы, материалы
17
vk.com/club152685050 | vk.com/id446425943
Нефтепродукт, железный тигель, песчаная баня, электронагревательный прибор, термометр до 360°С, лучины, защитная маска.
Подготовка к анализу. Для определения температур вспышки и воспламенения берут обезвоженный нефтепродукт. Тигель промывают бензином, затем при наличии углеродистых отложений очищают металлической щеткой, промывают дистиллированной водой и высушивают.
Прибор для определения температуры вспышки и воспламенения устанавливают в таком месте, где нет заметного движения воздуха и попадания дневного света на поверхность тигля, и защищают от движения воздуха щитом или экраном.
Для проведения анализа тигель охлаждают до 15—25°С и ставят в наружный тигель аппарата с прокаленным песком. При этом песок должен быть на высоте около 12 мм от верхнего края внутреннего тигля, а между дном этого тигля и наружным тиглем толщина песка должна быть 5…8 мм.
Анализируемый нефтепродукт наливают во внутренний тигель так, чтобы его уровень был ниже края тигля на 12 мм для нефтепродуктов со вспышкой до 210°С включительно и на 18 мм для нефтепродуктов со вспышкой выше 210°С.
Внимание! Не допускается заливание нефтепродукта выше уровня, отмеченного на внутренней стороне тигля и разбрызгивания нефтепродукта.
Термометр устанавливают во внутренний тигель с нефтепродуктом в строго вертикальном положении, так чтобы ртутный шарик находился в центре тигля приблизительно на одинаковом расстоянии от дна тигля и уровня нефтепродукта.
Проведение анализа.
При анализе наружный тигель аппарата нагревают на электроплитке. За 40°С до ожидаемой температуры вспышки скорость нагрева ограничивают до 4°С/мин. За 10°С до ожидаемой температуры вспышки проводят медленно по краю тигля на расстоянии 10—14 мм от поверхности анализируемого нефтепродукта и параллельно этой поверхности пламенем лучины. Длина пламени должна быть 3…4 мм, время продвижения пламени от одной стороны тигля до другой 2…3 с. Определение повторяют через 2°С подъёма температуры.
За температуру вспышки принимают температуру, которую показывает термометр при появлении первого синего пламени над частью или над всей поверхностью анализируемого нефтепродукта. При этом не следует смешивать истинную вспышку с отблеском от пламени лучины. В случае появления неясной вспышки она должна быть подтверждена последующей вспышкой через 2°С.
После установления температуры вспышки нефтепродукта, если требуется определить температуру его воспламенения, продолжают нагревание наружного тигля так, чтобы анализируемый нефтепродукт нагревался со скоростью 4°С/мин. Через каждые 2°С подъёма температуры к нефтепродукту подносят пламя лучины. За температуру воспламенения принимают температуру, показываемую термометром в тот момент, в который анализируемый нефтепродукт при поднесении к нему пламени загорается и продолжает гореть не менее 5 с.
Расхождения между двумя последовательными определениями температуры вспышки не должны превышать при температуре вспышки до 150°С - 4°С, выше 150°С - 6°С. Расхождения между двумя последовательными определениями температуры воспламенения не должны превышать 6°С. Внимание!
Данную работу следует проводить только в защитных масках.
18
vk.com/club152685050 | vk.com/id446425943
ЛАБОРАТОРНАЯ РАБОТА 6 Определение температуры плавления твердых парафинов
Выделяемые при анализе нефтяные парафины представляют собой сложную смесь твёрдых УВ, среди которых основную массу составляют н-алканы общей формулы СnН2n+2. Однако в зависимости от типа нефти, чистоты и способа выделения в парафинах могут присутствовать в виде примеси разветвлённые алканы и полициклические нафтены.
Начиная с гексадекана C16H34 и выше, парафиновые УВ при комнатной температуре находятся в твёрдом состоянии. Температура плавления нефтяных парафинов тем выше, чем больше молекулярная масса н-алканов и чем меньше в них разветвлённых алканов и циклопарафинов как изгрязняющих примесей.
Парафин нефти в условиях залежи находится в растворённом состоянии, тогда как в дегазированной нефти на поверхности он может быть в растворённом и во взвешенном состоянии, а частично может выпадать в осадок. При высоком содержании твёрдые парафины застывают при обычных условиях, вследствие чего нефть теряет подвижность и приобретает мазеобразную консистенцию.
Данные по составу нефтей показывают, что твёрдые парафины содержатся во всех нефтях, однако содержание их колеблется в широких пределах. Условно принято, что высокопарафинистые нефти содержат твёрдых парафинов от 10 % и выше, парафинистые — от 2,5 до 10, малопарафинистые
— от 0,5 до 2,5 и беспарафиновые — до 0,5 %.
Между содержанием твёрдых парафинов в нефти и количеством других её компонентов есть определённая взаимосвязь. Нефти с высоким содержанием парафиновых УВ в дистиллятных фракциях (свыше 60 %) содержат значительное количество твёрдых парафинов и могут быть отнесены к группе высокопарафинистых нефтей. Нефти одного и того же района содержат твёрдых парафинов тем больше, чем меньше в них смолистых веществ и больше моноциклических ароматических УВ. С увеличением глубины залегания (геологического возраста) наблюдается уменьшение смолистости нефтей, что в свою очередь сопровождается увеличением общего количества парафиновых УВ, и в том числе твёрдых парафинов. Установлено, что в общем случае геологически древние нефти содержат больше твёрдых парафинов, чем молодые.
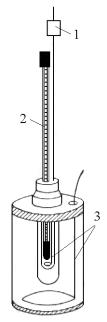
Метод Жукова. Температуру плавления парафинов определяют обычно по методу Жукова в приборе, изображенном на рис. 7. Прибор представляет собой сосуд Дьюара из прозрачного стекла, снабжённый термометром на 100°С с ценой деления 0,2°С. Расплавленный и нагретый на 15…20°С выше предполагаемой температуры плавления парафин наливают на ¾ высоты и подогретый сосуд Жукова. В отверстие прибора вставляют термометр, укреплённый на плотно пригнанной пробке так, чтобы ртутный шарик находился на оси прибора и в центре расплавленного парафина. Прибор с расплавленным парафином оставляют в покое до тех пор, пока температура не будет превышать предполагаемую температуру плавления продукта на 3…4 °С. Во избежание переохлаждения содержимое прибора встряхивают; как
только парафин начнет мутнеть и пениться, встряхивание прекращают, прибор ставят на стол, включают секундомер и начинают записывать через каждую минуту
Рис.7. Прибор Жукова |
показания термометра с точностью до 0,1°С до тех пор, пока весь парафин не |
|
1 |
– Термометр |
затвердеет. Так как разреженный воздух между двойными стенками сосуда |
2 |
– сосуд Дьюара |
Дьюара является плохим проводником тепла, то расплавленный парафин |
|
|
охлаждается очень медленно и равномерно. За температуру плавления |
|
|
принимают ту, которая не менее трёх отсчетов сохраняется постоянной. |
Расхождение между повторными определениями для одного и того же продукта не должно превышать
0,2°С.
19
vk.com/club152685050 | vk.com/id446425943
Галицийский метод. Существует ускоренный (галицийский) метод определения температуры плавления парафина. Галицийский метод является менее точным, чем метод Жукова, однако более быстрым и удобным в случае небольших количеств парафина.
Температуру плавления по этому методу определяют следующим образом. Берут термометр с большим круглым шариком для ртути и опускают в расплавленный и несколько перегретый парафин. Вынув термометр, на ртутном шарике фиксируют каплю парафина. Затем термометр вводят в
горизонтально расположенную коническую колбу и, сохраняя горизонтальное положение термометра, вращают его вокруг оси. Пока капля жидкая, она все время висит на шарике и не участвует в его движении. Застынув, капля начнет вращаться с шариком термометра. Наблюдаемая при этом температура и будет температурой плавления или застывания парафина.
ЛАБОРАТОРНАЯ РАБОТА 7 Определение молекулярной массы нефтяных фракций
Молекулярная масса — одна из важнейших физико-химических характеристик вещества. В случае смесей химических соединений, каковыми являются фракции нефти, она не теряет своего значения как аддитивно складывающаяся молекулярная масса отдельных компонентов. Знание молекулярной массы необходимо при определении структурно-группового состава нефтяных фракций, молекулярной рефракции и многих других параметров.
Криоскопический метод (в бензоле)
Существует большая группа методов, связанных с осмотическим давлением и предусматривающих определение молекулярной массы в растворе. Так как для разбавленных растворов справедливо правило Рауля - Вант-Гоффа, согласно которому осмотическое давление прямо пропорционально молярной концентрации, то для определения молекулярной массы принципиально пригодны все величины, находящиеся в простой зависимости от осмотического давления. Обычно пользуются такими величинами, которые поддаются простому и легкому измерению: понижение точки замерзания растворов, повышение точки кипения растворов и депрессия точки плавления смесей (твердых растворов). В нефтяной практике наиболее широкое распространение получил криоскопический метод, основанный на измерении понижения температуры замерзания растворителя при добавлении к нему исследуемого вещества.
Криоскопический метод определения молекулярной массы, однако, не свободен от погрешностей. Во-первых, в основу этого метода положен закон Рауля, применимый лишь к разбавленным растворам; поэтому с повышением концентрации исследуемого вещества в растворителе наблюдается отклонение от действия этого закона. Кроме того, в применяемых растворителях многие вещества, например ароматические УВ, соединения кислого характера, проявляют склонность к ассоциации, которая тем сильнее, чем выше концентрация раствора. Поэтому истинную молекулярную массу можно определить только в сильно
разбавленных растворах, т.е. при бесконечно большом разведении.
Проведение анализа
Сначала определяют температуру замерзания чистого бензола. Для этого в пробирку наливают 20…25 мл криоскопического бензола и вставляют мешалку и термометр так, чтобы шарик последнего находился в середине жидкости. Затем пробирку вместе с воздушной муфтой помещают в охладительную баню, в которую предварительно наливают холодную воду с несколькими кусочками льда (температура смеси поддерживается на уровне 2…3°С).
Постоянно помешивая бензол, наблюдают по термометру за понижением температуры. Точку замерзания можно установить в момент образования твёрдой фазы по постоянству температуры. Но многие растворители, в том числе и бензол, переохлаждаются до начала образования твёрдой фазы в них. В результате постепенно и равномерно опускающийся столбик ртути на термометре Бекмана
20

vk.com/club152685050 | vk.com/id446425943
внезапно поднимается на несколько десятых долей градуса вверх, а затем эта максимальная температура остается в течение длительного времени постоянной. Наивысшая температура, которую показывает термометр после начала выделения кристаллов, и есть точка замерзания жидкости. Отсчитывают деление термометра с погрешностью до 0,01°С. Вынув пробирку из муфты, подогревают её рукой на 1…2°С выше температуры замерзания. Затем снова вставляют пробирку в муфту и повторяют определение. Расхождение между параллельными определениями температуры замерзания бензола не должно превышать 0,02°С.
Затем приступают к определению температуры замерзания растворов исследуемых фракций в бензоле. Готовят растворы различных концентраций — от 0,2 до 1 %. Для этого в конические колбы с притёртыми пробками берут три навески (с погрешностью до 0,0002 г) обезвоженного нефтепродукта: 0,16…0,25; 0,25…0,35 и 0,35…0,50 г. В каждую колбочку наливают 20…25 мл криоскопического бензола и снова их взвешивают, устанавливая таким образом массу взятого бензола. Навески хорошо перемешивают с бензолом до получения однородного раствора. Для каждого раствора определяют температуру замерзания точно так же, как и температуру замерзания чистого растворителя.
Относительная молекулярная масса рассчитывается по формуле:
М=
5,12 1000
а (B t)
,
где а — навеска исследуемой фракции, г; В — масса бензола, г; 5,12 — криоскопическая постоянная бензола; t — разность между температурами замерзания чистого бензола и раствора нефтепродукта в бензоле.
Результатом определения является среднее из двух полученных значений молекулярной массы. Для светлых нефтепродуктов между двумя параллельными определениями допускается расхождение ±1,5, для масел и других высокомолекулярных продуктов - до ±3 %.
ЛАБОРАТОРНАЯ РАБОТА 8 Определение анилиновой точки
Свойства нефтепродуктов в значительной степени определяется их углеводородным составом. Его знание необходимо для правильного применения нефтепродуктов, в частности, знание углеводородного состава бензинов необходимо для их рационального применения в качестве моторного топлива и смазочных масел.
Различные классы углеводородов ведут себя в автомобильных и авиационных двигателях карбюраторного типа неодинаково. Например, парафины нормального строения вызывают при сгорании нежелательное явление – детонацию, в то время, как ароматические углеводороды и изопарафины отличаются высокой детонационной способностью. Нафтены занимают в отношении детонационной способности промежуточное положение. Сейчас можно считать установленным, что все основные характеристики качеств масел - вязкость, индекс вязкости, стабильность против окисления, термическая стабильность, зависят от содержания и состава ароматических углеводородов.
Черножуков и Крейн показали, что нафтеновые углеводороды весьма подвержены окислительному воздействию кислорода, причем окисляемость их возрастает с повышением среднего молекулярного веса фракций. Ароматические углеводороды в определенных концентрациях тормозят окисление нафтенов.
Вязкость масел и пологость кривой вязкости также до известной степени объясняются наличием и концентрацией ароматических углеводородов различного строения.
При определении группового состава нефти и нефтепродуктов следует учитывать, что эти соединения состоят из чрезвычайно большого числа индивидуальных углеводородов и других соединений. Вследствие сложности и длительности определения индивидуального углеводородного состава нефтепродуктов определяют суммарное количество углеводородов определённых классов, содержащихся в нефтепродуктах.
Анилиновый метод
21
vk.com/club152685050 | vk.com/id446425943
Среди неинструментальных методов определения группового химического состава бензиновых фракций наиболее широкое распространение получил анилиновый метод, основанный на неодинаковой растворимости углеводородов различных классов в анилине. При смешении нефтяной фракции с анилином при комнатной температуре обычно образуются два слоя, т.е. не происходит полного растворения нефтепродукта в анилине. Если эту смесь нагревать, постоянно перемешивая, то при достижении определённой температуры произойдет полное взаимное растворение анилина и нефтепродукта, слои исчезнут, и жидкость станет однородной. Температуру, соответствующую полному взаимному растворению анилина и нефтепродукта, называют анилиновой точкой или критической температурой растворения (КТР) данного нефтепродукта в анилине. Наиболее низкими анилиновыми точками среди углеводородов характеризуются арены, наиболее высокими — алканы; циклоалканы занимают промежуточное положение. Алкены и циклоалкены имеют несколько более низкое значение анилиновых точек по сравнению с циклоалканами близкой молекулярной массы. В пределах одного гомологического ряда анилиновые точки, как правило, возрастают с увеличением массы и температуры кипения углеводорода. Такая же закономерность наблюдается и для фракций, выделенных из одной и той же нефти.
Существуют два метода определения анилиновых точек: метод равных объемов и метод максимальных анилиновых точек. В первом случае берут равные объёмы анилина и исследуемой фракции и определяют температуру их полного смешения. Полученную температуру называют анилиновой точкой. Во втором случае находят температуру, называемую максимальной анилиновой точкой или истинной критической температурой растворения в анилине. Её получают после нескольких определений температуры растворения продукта в возрастающих количествах анилина. При увеличении количества анилина температура полного растворения сначала повышается и при некотором соотношении фракции и анилина достигает максимума, после чего при дальнейшем увеличении количества анилина начинает падать. Максимальную температуру полного растворения принимают за максимальную анилиновую точку (истинную КТР в анилине). Обычно разница между анилиновыми точками фракций и их максимальными анилиновыми точками невелика, причем она увеличивается с ростом температур кипения фракций и увеличением содержания в них аренов.
Приборы, реактивы, материалы
Пробирка с муфтой объемом 15 мл, стакан из термостойкого стекла вместимостью 750— 1000 мл, термометр с длинной ножкой, анилин свежеперегнанный марки ч., пипетки на 2 мл.
Метод равных объёмов. В чистую и сухую пробирку 2 (рис. 9) помещают по 2 мл анилина и анализируемой бензиновой фракции, плотно закрывают пробкой со вставленным в нее термометром 1 и мешалкой 5 и укрепляют в муфте 3, погружённой в водяную баню 4. Продукт и анилин берут с помощью пипеток вместимостью 2 мл. Термометр помещают так, чтобы середина ртутного шарика находилась на уровне линии раздела слоёв анилина и продукта. Температуру водяной бани медленно повышают, при этом непрерывно перемешивают мешалкой продукт с анилином. Отмечают температуру полного смешения жидкостей (при этом раствор становится прозрачным), прекращают нагревание и дают воде медленно остывать. Когда в пробирке 2 появляется муть, что свидетельствует о начале разделения фаз, снова начинают перемешивать раствор мешалкой. Вначале при перемешивании муть исчезает, но затем наступает момент неисчезающего помутнения. За анилиновую точку принимают наивысшую температуру, при которой муть при перемешивании не исчезает.
22
vk.com/club152685050 | vk.com/id446425943
Рис. 9. Прибор для определения анилиновой точки:
1 — термометр, 2 — пробирка, 3 — муфта, 4 — водяная баня, 5 — мешалки, 6—смесь анилина с исследуемой фракцией
Температуры полного смешения и помутнения не должны расходиться более, чем на 0,1°С. Определение анилиновой точки повторяют с новым образцом исследуемой фракции. Расхождение анилиновых точек в параллельных опытах не должно превышать 0,2°С.
Метод максимальной анилиновой точки (истинной КТР в анилине). В пробирку 2
помещают 2 мл исследуемой фракции, 1,6 мл анилина и определяют температуру полного растворения, как описано выше. После этого к смеси добавляют еще 0,2 мл анилина и снова определяют температуру растворения. Обычно она бывает выше, чем при первом определении. Анилин прибавляют по 0,2 мл до тех пор, пока после некоторого максимума температуры растворения не наметится её снижение. Максимальную анилиновую точку фиксируют; она соответствует истинной критической температуре растворения исследуемого продукта в анилине. Таким образом определяют максимальную анилиновую точку исходной и деароматизированной фракции (до и после удаления ароматических углеводородов). Содержание ароматических углеводородов (вес. %) определяют по формуле:
x=K(t-t0),
где К — коэффициент, показывающий количество ароматических УВ, которое соответствует понижению анилиновой точки смеси насыщенных УВ на 1°С; t — максимальная анилиновая точка фракции после удаления ароматических УВ, °С; t0 — максимальная анилиновая точка исходной фракции, °С.
Значения анилиновых коэффициентов К для различных фракций и различных концентраций ароматических УВ в них приведены в табл. 2 и 3.
Содержание нафтеновых УВ, вес. %, в деароматизированной фракции
X1 K1 t1 t / d420
где К1 — анилиновый коэффициент для |
нафтеновых |
УВ; t1 — средняя максимальная |
анилиновая точка чистых парафиновых УВ, |
°С; t — |
максимальная анилиновая точка |
деароматизированной фракции, °С; d420 — плотность деароматизированной фракции.
Таблица 2 Анилиновые коэффициенты К для количественного определения ароматических УВ в бензиновых
фракциях, выкипающих до 150°С
23
vk.com/club152685050 | vk.com/id446425943
Пределы выкипания |
Конц. ароматических УВ, вес. % |
|
||
фракции, °С |
До 20 |
20 -40 |
40-60 |
60 - 80 |
60…95 (бензольная) |
1,15 |
1,14 |
1,08 |
0,95 |
95…122 (толуольная) |
1,20 |
1,18 |
1,08 |
0,96 |
122…150 (ксилольная) |
1,26 |
1,22 |
1,14 |
1,00 |
Таблица 3. Анилиновые коэффициенты К для различных концентраций ароматических УВ во фракциях, выкипающих выше 150°С
|
Депрессия |
|
Пределы выкипания фракций, °С |
|
||
|
максимальной анилиновой |
150-200 |
200-250 |
|
250-300 |
800-380 |
|
точки |
|
||||
|
|
|
|
|
|
|
2 |
1,58 |
1,76 |
|
1,94 |
2,10 |
|
4 |
1,56 |
1,74 |
|
1,91 |
2,06 |
|
6 |
1,54 |
1,72 |
|
1,88 |
2,03 |
|
8 |
1,52 |
1,70 |
|
1 ,85 |
1,99 |
|
10 |
1,50 |
1,68 |
|
1,82 |
1,95 |
|
12 |
1,49 |
1,66 |
|
1,79 |
1,92 |
|
14 |
1,48 |
1,64 |
|
1,76 |
1,88 |
|
16 |
1,47 |
1,62 |
|
1,74 |
1,85 |
|
18 |
1,46 |
1,60 |
|
1,71 |
1,82 |
|
20 |
1,45 |
1,57 |
|
1,68 |
1,79 |
|
22 |
1,44 |
1,55 |
|
1,66 |
1,77 |
|
24 |
1,43 |
1,54 |
|
1,64 |
1,74 |
|
26 |
1,42 |
1,52 |
|
1,62 |
1,71 |
|
28 |
1,41 |
1,51 |
|
1,60 |
1,68 |
|
|
30 |
1,40 |
1,49 |
|
1,58 |
1,66 |
|
32 |
1,39 |
1,48 |
|
1,56 |
.1 ,64 |
|
34 |
1,38 |
1,46 |
|
1,54 |
1,62 |
|
36 |
1,37 |
1,45 |
|
1,52 |
1,59 |
|
38 |
1,36 |
1,43 |
|
1,50 |
1,57 |
|
40 |
1,35 |
1,42 |
|
1,48 |
1,54 |
|
42 |
1,34 |
1,40 |
|
1,46 |
1,51 |
|
44 |
1,33 |
1,39 |
|
1,44 |
1,49 |
|
46 |
1,32 |
1,37 |
|
1,42 |
1,46 |
|
48 |
1,31 |
1,35 |
|
1,39 |
1,43 |
|
50 |
1,30 |
1,34 |
|
1,37 |
1,40 |
|
70 |
1,15 |
1,14 |
|
— |
— |
Значения коэффициента К1 и максимальной анилиновой точки чистых парафиновых УВ t1 приведены в табл. 4. Так как для фракций, кипящих выше 150°С, используется анилиновый коэффициент нафтеновых УВ, учитывающий плотность, расчет для этих фракций проводят по формуле
|
|
|
X |
1 |
K |
1 |
t |
t |
|
|
|
|
|
|
|
|
1 |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
Таблица 4 |
|
Анилиновые коэффициенты нафтеновых УВ К1 и средняя |
|
|
||||||||
|
максимальная анилиновая точка чистых парафиновых УВ t1 |
|
|||||||||
Пределы |
|
|
|
|
|
|
|
Пределы |
|
|
|
выкипания фрак- |
К1 |
t1 |
|
|
|
|
|
|
К1 |
t1 |
|
выкипания фракций, °С |
|
||||||||||
ций, °С |
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
40-60 |
1,51 |
72 |
|
|
|
|
|
150—200 |
|
5,00 |
78 |
60—95 |
2,13 |
71 |
|
|
|
|
|
200—250 |
|
5,00 |
85,8 |
95—122 |
2,76 |
71 |
|
|
|
|
|
250—300 |
|
5,00 |
93,0 |
122—150 |
3,26 |
73 |
|
|
|
|
|
300—350 |
|
5,00 |
99,5 |
Пересчёт содержания нафтеновых УВ (вес. %) на исходную фракцию производят по формуле
X 2 X1 100 X /100 ,
где X - содержание ароматических УВ во фракции, вес. %; X1 - содержание нафтеновых УВ в деароматизированной фракции, вес. %.
24
vk.com/club152685050 | vk.com/id446425943
Содержание парафиновых УВ (вес. %) определяют по разности
100-(X+X2).
ЛАБОРАТОРНАЯ РАБОТА 9 Определение кислотности нефтепродуктов
Кислотность нефти обусловлена содержанием в ней алифатических и циклических карбоновых кислот, фенолов и других кислородных соединений кислотного характера. Все кислые вещества, выделяемые из нефти реакциями щелочей, объединяют термином «нефтяные кислоты».
Соединение кислорода в элементном составе нефти находится в прямой зависимости от соединения нефтяных кислот.
Вероятное происхождение нефтяных кислот – биохимическое окисление нефтей. Представление об искусственном происхождении нефтяных кислот оказалось не состоятельным. Однако, кислородосодержащие соединения могут образоваться в некоторых процессах добычи и переработки нефти (например, в результате окисления кислородом воздуха нефти при её добыче методом внутрипластового горения и остатков нефти при производстве битумов). Содержание нефтяных кислот в нефтях колеблется от следов до 1% (по массе) и более. В больших количествах нефтяные кислоты содержатся в нефтях нафтенового типа, в наименьших – в высокопарафинового типа.
Нафтеновые кислоты представляют собой монокарбоновые кислоты ряда циклопентана и циклогексана. Группа COOH почти во всех нафтеновых кислотах отделена от циклической части молекулы одной или несколькими метиленовыми группами.
В нефти обнаружено более 20 алифатических карбоновых кислот: муравьиная, уксусная, пропановая, масляная, капроновая, энантовая, пальмитиновая, стеариновая и другие кислоты. Найдены фенолы, алкилфенолы (крезолы, ксиленолы), нафтолы.
Нефтяные кислоты обладают значительной поверхностной активностью, влияют на такие процесс нефтедобычи и нефтепереработки, как нефтевытеснение, эмульгирование, отложение парафинов и солей, коррозия. Большой коррозийной активностью обладают низкомолекулярные алифатические кислоты.
Кислотность выражается количеством миллиграммов KOH, необходимого для нейтрализации 100 мл продукта. Метод заключается в потенциаметрическом титровании продукта, растворённого в неводном растворителе раствором KOH. Титрование ведут до скачка потенциала или при отсутствии последнего до значений ЭДС, установленных по буферным растворам.
Потенциометрическое титрование осуществляют в стеклянном стакане, в который помещен электрод, магнитная мешалка и конец бюретки для титрования.
Потенциал между стеклянным электродом и средой устанавливается не сразу, поэтому при титровании при добавлении каждой порции раствора KOH необходимо выждать некоторое время, пока pH не достигнет максимального значения (≈5 минут), после чего добавляют следующую порцию раствора KOH. На основе полученных экспериментальных данных вычерчивают кривую зависимости ЭДС от объёма добавленной щёлочи – кривую титрования и находят точку эквивалентности по изгибу этой кривой.
Приборы, реактивы, материалы
рН-метр; стеклянный электрод; хлорсеребряный электрод; мерный цилиндр объемом 50 мл; магнитная мешалка; стакан для титрования объемом 200 мл; стакан для щёлочи объёмом 50 мл; бюретка для титрования объёмом 25 мл; гидроксид калия, 0,05 М спиртовый раствор; разбавитель – смесь хлороформа и этилового или пропилового спирта; проба бензина.
Проведение анализа
1.В стакан для титрования отбирают мерным цилиндром пробу бензина 30 мл.
2.Стакан с пробой устанавливают на магнитной мешалке.
3.Подключают электроды к рН-метру: стеклянный электрод – к гнезду «ИЗМ.», хлорсеребряный –
кгнезду «ВСПОМ.»
4.Электроды погружают в стакан с пробой.
5.Доливают растворитель так, чтобы электроды были прогружены в раствор как минимум на 1,5-
2 см.
25

vk.com/club152685050 | vk.com/id446425943
6.Включают рН-метр в режим измерения ЭДС и записывают показания прибора в таблицу.
7.Титруют раствором КOH с шагом 0,5 мл до резкого изменения потенциала, после которого измерить значения еще 5 точек, прибавляя каждый раз по 0,5 мл щелочи. После добавления каждой
порции КOH фиксируют значение ЭДС.
8.Результаты эксперимента заносят в таблицу 1.
Объём |
Е, мВ |
|
титранта VT, мл |
||
|
||
0 |
Е0 |
|
1 |
Е1 |
|
2 |
Е2 |
|
… |
… |
|
|
|
Таблица 1
ΔE |
|
ΔV |
|
ΔE |
|
ΔV |
1 |
|
ΔE ΔV 2
По данным таблицы строят кривую потенциометрического титрования в координатах E – VNaOH.
Рассчитывают значения |
ΔE |
, где ΔV – шаг титрования (0,5 мл); |
||
ΔV |
||||
|
|
|
||
Е =Еn+1−En, например, |
Е1=Е1−Е0, Е2=Е2−Е1 и т. д. |
|
||
Строят дифференциальную кривую титрования в координатах |
ΔE |
|||
ΔV |
||||
|
|
|
||
По положению острого максимума дифференциальной кривой эквивалентности.
Рассчитывают кислотность по формуле:
СK CT VT ,
Va
−VNaOH.
титрования находят точку
где VT – объём КOH в точке эквивалентности; СТ – концентрация КOH (0,05 экв/л); Va – объём пробы, взятой для анализа (30 мл).
ЛАБОРАТОРНАЯ РАБОТА 10
Определение содержания солей в нефти индикаторным титрованием
Основным источником солей в нефти является пластовая вода. При обезвоживании нефти идет и её обессоливание.
Общее содержание солей в пластовой воде изменяется для разных месторождений в широких пределах. В наибольшем количестве содержатся ионы Cl-, SO42-, HCO3-; CO32-, Ca2+, Mg2+,Na+, K+. Ионы остальных солей встречаются в малых количествах.
Нефти делятся по содержанию соли:
1)до 300 мг/л;
2)до1800 мг/л;
3)3000 мг/л.
Для определения солей в нефти и нефтепродуктах применяются физические и химические методы.
Сущность химических методов заключается в извлечении хлоридов из нефти водой с последующим индикаторным или потенциометрическим титрованием водной вытяжки нитратом ртути.
Физические методы основаны на измерении электропроводимости.
Приборы, реактивы, материалы
Экстрактор с делительной воронкой;
26

vk.com/club152685050 | vk.com/id446425943
Мерный цилиндр объёмом 50 мл; Мерный цилиндр для воды объёмом 100 мл; Колба для титрования объёмом 200 мл;
Стакан для нитрата ртути объёмом 50 мл; Бюретка для титрования объёмом 25 мл; Пипетка объёмом 5 мл; Пипетка объёмом 1 мл; Воронка; Фильтр;
Нитрат ртути (II), 0,05 М раствор; Азотная кислота, 0,2 М раствор;
Индикатор (смесь дифенилкарбозида и бромфенолового синего) Вода для приготовления водной вытяжки; Толуол; Проба нефти.
Проведение анализа
I. Приготовление водяной вытяжки
1.В делительную воронку наливают:
пробу нефти 25 мл (мерным цилиндром, который затем ополаскивают 20 мл толуола, толуол выливают в делительную воронку).
100 мл воды для приготовления водной вытяжки.
2.Настроить блок управления мешалки:
нажать кнопку «сеть»;
нажать кнопку «установка» для входа в режим настройки блока управления;
кнопками «выбор» и ▲▼ установить число оборотов (800-1200) и продолжительность перемешивания (10 минут);
нажать кнопку «установка» для выхода из режима настройки.
3.Погрузить мешалку в делительную воронку, нажать кнопку «пуск».
4.Экстрагировать в течение 10 мин. Остановить мешалку (кнопкой «стоп» или она остановится автоматически).
5.Дать фазам расслоиться в течение 5-10 мин.
6.Водную фазу (внизу) через бумажный фильтр слить в коническую колбу. Органическую фазу вылить в ёмкость для органических отходов!!!.
7.Провести анализ содержания солей хлоридов.
8.Вычислить концентрацию хлорида в пробе нефти.
9.
II. Анализ содержания солей хлоридов меркурометрическим титрованием
1.В две конические колбы отобрать по пробе водной вытяжки объёмом 5 мл.
2.К каждой пробе прилить дистиллированной воды до уровня раствора в колбе 2-2,5 см.
3.Добавить индикатор 1-2 мл − раствор приобретет синий цвет.
4.Добавить азотную кислоту до изменения цвета индикатора на жёлтый.
5.Титровать нитратом ртути до изменения цвета индикатора на фиолетовый.
6.Записать объём нитрата ртути.
7.Повторить пп. 3-6 с другой колбой.
Содержание хлоридов в пробе нефти вычисляется по формуле:
[Cl ] CT VT, ср VH2O MCl ,
Va Vнефти
где Vнефти - объём нефти для приготовления водной вытяжки, мл; VH 2 O - объём воды для приготовления водной вытяжки, мл;
27

vk.com/club152685050 | vk.com/id446425943
Va - объём пробы водной вытяжки, взятый на проведение анализа, мл; СТ - концентрация раствора нитрата ртути (титранта), экв/л;
– средний объём нитрата ртути, израсходованный на титрование проб, мл.
ЛАБОРАТОРНАЯ РАБОТА 11
Определение непредельных и ароматических углеводородов в светлых нефтепродуктах
Нефтепродукты, содержащие непредельные углеводороды, недостаточно химически стабильны. Этим и объясняется нормирование предельно допустимых сравнительно невысоких иодных чисел (2-10 г йода на 100 г продукта) для многих авиационных бензинов и дизельных топлив. Требования к химической стабильности топлив для воздушно-реактивных двигателей ещё выше, чем для карбюраторных топлив. Поэтому йодное число топлив Т-1, Т-2, ТС-1 и Т-5 не должно превышать 2 – 3,5, а топлива РТ – 0,5 г йода на 100 г продукта.
Присутствие ароматических углеводородов в бензинах повышает их антидетонационные свойства, однако ухудшает многие другие эксплуатационные характеристики: повышается температура застывания и помутнения, увеличивается гигроскопичность, повышается склонность к нагарообразованию. Повышенное содержание ароматических углеводородов в реактивных топливах снижает их теплотворную способность (на единицу массы), ухудшает воспламеняемость и также способствует нагарообразованию. Поэтому содержание ароматических углеводородов в бензинах и в реактивном топливе нормируется. В авиационных бензинах содержание ароматических углеводородов допускается не свыше 35 %, в реактивных топливах – не более 22 %, а в топливе РТ – 18,5 %.
Определение содержания непредельных углеводородов
О содержании непредельных углеводородов в нефтяном топливе судят по йодному числу ИЧ, т.е. по количеству йода (в г), присоединившегося к 100 г анализируемого топлива. Массовую долю непредельных углеводородов Н (в %) рассчитывают по формуле:
Н |
ИЧ М |
|
254 |
||
|
где М – молекулярная масса анализируемого топлива; 254 – молекулярная масса йода. Молекулярную массу нефтяных топлив определяют либо экспериментально криоскопическим
методом, либо по формуле Войнова по средней температуре кипения. Для авиационных бензинов молекулярная масса может быть принята равной 100, а для реактивных топлив Т-1, ТС-1 и Т-2 – 175.
Для определения йодного числа спиртовой раствор анализируемого нефтепродукта обрабатывают спиртовым раствором йода в присутствии большого избытка воды. Йод реагирует с водой с образованием йодноватистой кислоты:
I |
2 |
HOH HIO HI |
|
|
Гидролиз усиливается в присутствии ненасыщенных соединений. Образовавшаяся йодноватистая кислота вступает во взаимодействие с ненасыщенными соединениями быстрее, чем йод, присоединяясь по месту двойных связей:
RCH CHRCHR 2HIO C |
H |
OH |
||
|
2 |
5 |
|
|
RCHI CHIR CH |
CHO 2H |
O |
||
3 |
|
|
2 |
|
Избыток йода оттитровывается тиосульфатом натрия: |
|
|
|
|
I2 2Na2 S2O3 2NaI Na2 S4O6
Определение необходимо выполнять очень быстро с соблюдением всех рекомендуемых условий. Однако точность метода недостаточна, так как возможно протекание побочных реакций замещения, которое приводит к завышенным результатам. С другой стороны, из-за плохой растворимости некоторых топлив в спирте, могут получаться заниженные результаты за счёт неполноты реакции присоединения йода.
28

vk.com/club152685050 | vk.com/id446425943
Реактивы
Йод, спиртовый раствор с концентрацией 20 г/л.
Этиловый спирт ректификованный или ацетон (при анализе дизельного топлива). Йодид калия, 20 %-ный раствор в воде.
Тиосульфат натрия, 0,1 н. титрованный раствор. Крахмал, 0,5 %-ный раствор.
Методика определения
Навеску испытуемого топлива в количестве 0,2 – 0,4 г с точностью до 0,0004 г отбирают либо в тонкостенную ампулу, либо в бюкс диметром 10 мм, либо с помощью капельницы в колбу объёмом 500 мл, в которую налито 15 мо спирта. Последний способ применяется для топлив Т-1 и ТС-1.
Если известна плотность испытуемого топлива, то пробу на анализ можно брать и по объёму с помощью микробюретки. Навеску в этом случае вычисляют, умножая объём на плотность. Основные операции анализа проводят в конической колбе с притертой пробкой.
В колбу наливают 15 мл спирта в качестве растворителя и вносят туда бюкс или ампулу с навеской. Крышку бюкса слегка приоткрывают и осторожными движениями обмывают бюкс спиртом, имеющимся в колбе. Ампулу разбивают стеклянной палочкой с направленной утолщённой головкой, следя при этом, чтобы и капилляр ампулы был измельчён. После этого палочку и стенки колбы обмывают 10 мл спирта.
К спиртовому раствору в колбу приливают из бюретки 25 мл спиртового раствора йода и плотно закрывают колбу пробкой, предварительно смоченной раствором йодида калия, во избежание больших потерь йода. Колбу осторожно встряхивают. Затем добавляют 150 мл дистиллированной воды, снова закрывают колбу пробкой, взбалтывают её содержимое в течение 5 мин и выдерживают в покое еще 5 мин. После этого промывают стенки колбы и пробку дистиллированной водой и оттитровывают избыток йода тиосульфатом натрия. Вначале титрование ведут до соломенно-жёлтого цвета, а затем, после добавления 1- 2 мл крахмала, - до исчезновения синевато-фиолетового окрашивания.
Точно таким же образом, но без навески испытуемого топлива проводят контрольный опыт. Йодное число вычисляют по формуле:
ИЧ |
(V V )T 100 |
|
1 |
||
|
||
|
G |
где V и V1 – объём 0,1 н. раствора тиосульфата натрия, израсходованного на титрование соответственно в контрольном и в целевом опыте, мл; Т – титр по йоду 0,1 н. раствора тиосульфата натрия, г/мл; G – навеска нефтепродукта, г.
Определение содержания ароматических углеводородов
Определение ароматических углеводородов, как правило, проводятся методами, основанными на существенной разнице в значениях физических констант (плотности, показателей преломления, анилиновых точек) ароматических углеводородов по сравнению с углеводородами других классов. Чаще всего применяется метод анилиновых точек, при котором мольную долю ароматических углеводородов А (в %) рассчитывают по формуле:
А К (Т |
2 |
Т |
) |
|
1 |
|
где К – расчётный коэффициент, показывающий, какое количество ароматических углеводородов данного продукта соответствует понижению анилиновой точки смеси предельных углеводородов на 1ºС; Т1 – анилиновая точка исходного продукта; Т2 – анилиновая точка деароматизированного продукта.
Значение коэффициента К зависит от строения ароматических углеводородов и их содержания в анализируемом продукте. Поэтому при анализе бензинов их предварительно разгоняют на бензольную (60…95ºС), ксилольную (122…150ºС) и остаточную фракции и каждую фракцию анализируют отдельно. Коэффициент К для этих фракций изменяется от 1,18 до 1,40:
Фракция, ºС |
60…95 |
95…122 |
122…150 |
150…175 |
|
К при содержании ароматических углеводородов |
|
||
менее 20%. |
1,20 |
1,22 |
1,30 |
1,40 |
20…40 % |
1,18 |
1,20 |
1,22 |
1,30 |
29
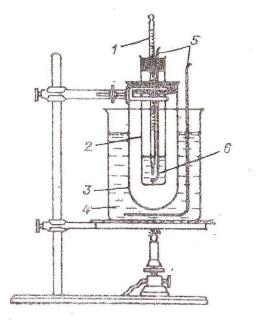
vk.com/club152685050 | vk.com/id446425943
Как видно из расчётной формулы, для определения содержания ароматических углеводородов необходимо найти анилиновую точку, т.е. критическую температуру растворения исследуемого нефтепродукта или фракции до и после удаления ароматических углеводородов. Удаление ароматических углеводородов проводится путём их адсорбции на силикагеле в стеклянной колонке. Для определения анилиновых точек применяют прибор, представленный на рис. 30
Реактивы
Анилин свежеперегнанный.
Серная кислота.
Этиловый спирт ректификованный.
Формалин технический, 40 %-ный водный раствор формальдегида.
Силикагель марки АСК, высушенный при 150ºС в течение
8 ч.
Подготовка к анализу. В нижнюю суженную часть стеклянной адсорбционной колонки высотой 700 мм и диаметром 100 мм помещают кусочек стеклянной ваты. Затем в колонку насыпают небольшими порциями около 20 г силикагеля, уплотняя его лёгким постукиванием по колонке. Подготавливают прибор для определения анилиновых точек и чистые сухие градуированные пробирки для приема деаромати зированных фракций.
Рис. 30. Прибор для определения анилиновых точек:
1 – термометр; 2 – внутренняя пробирка диаметром 25 мм и высотой 150 мм; 3 – внешняя пробирка диаметром 40 мм; 4 – жидкостная баня; 5 – мешалки; 6 – смесь анилина с испытуемым продуктом.
Проведение анализа
Определение анилиновой точки исходного продукта. Во внутреннюю пробирку наливают 3 мл анилина и 3 мл продукта. Вставляют мешалку и термометр так, чтобы ртутный шарик находился на границе раздела анилина и испытуемого продукта и не касался бы дна и стенок пробирки. Прибор погружают в стакан с водой и укрепляют в штативе. Воду нагревают при помешивании до полного растворения нефтепродукта в анилине (до получения однородного раствора).
Отставив затем нагревательный прибор, наблюдают при постоянном перемешивании смеси внутренней мешалкой за охлаждающейся смесью и за показаниями термометра. За анилиновую точку принимают температуру, при которой начинается расслоение раствора, выражающееся в его помутнении. Температуру с точностью до 0,1 ºС отмечают в тот момент, когда образовавшаяся равномерная муть скроет ртутный шарик термометра.
Не разбирая прибора, с той же порцией анилина и анализируемого продукта определение повторяют несколько раз до получения результатов, отличающихся не более чем на 0,2 ºС.
Удаление из продукта ароматических углеводородов.
В подготовленную адсорбционную колонку наливают около 15 мл исследуемого продукта. После того как он полностью впитается в силикагель, добавляют в колонку 25 – 35 мл спирта для вытеснения из колонки деароматизированной части исходного продукта. С низа колонки в градуированные пробирки отбирают вытекающие из колонки порции: первую объёмом 2 мл, а последующие - по 0,5 мл. Во всех отобранных порциях проводят качественную формалитовую реакцию на присутствие ароматических углеводородов. Для этого 1–2 капли продукта вводят в пробирку и туда же добавляют 1 мл серной кислоты и 2 – 3 мл формалина. Образование тёмного кольца (формалиты) на границе раздела «серная кислота – продукт» указывает на наличие ароматических углеводородов. Все порции, не
30

vk.com/club152685050 | vk.com/id446425943
содержащие ароматических углеводородов, соединяют вместе и из полученного деароматизированного продукта отбирают 3 мл для определения второй анилиновой точки.
Если анализируются отдельные фракции, то мольную долю ароматических углеводородов А (в %) в исходном продукте находят по формуле:
А |
А В А В |
... А B |
|||||
1 |
1 |
2 |
2 |
n |
n |
||
|
|||||||
|
|
|
|
100 |
|
||
где А1, А2 и т.д. – содержание ароматических углеводородов в отдельных фракциях; В1, В2 и т.д. – содержание фракций в исходном продукте.
Если анализу подвергается продукт, содержащий наряду с ароматическими и непредельные углеводороды, то их суммарное содержание находят весовым сернокислотным методом по ГОСТ 6994 – 74. Одновременно определяют содержание непредельных углеводородов по йодному числу. Содержание ароматических углеводородов рассчитывают по разности.
ЛАБОРАТОРНАЯ РАБОТА № 12 Комплексонометрическое титрование бензинов
Для определения содержания тетраэтилсвинца в бензинах применяется метод комплексонометрического титрования (ГОСТ 13210-72). Тетраэтилсвинец разлагается концентрированной соляной кислотой при нагревании. Получающийся хлорид свинца хорошо растворим в соляной кислоте, а соляная кислота легко отделяется от бензинового слоя в делительной воронке. В дальнейшем раствор хлорида свинца оттитровывают трилоном Б, по количеству которого рассчитывают содержание тетраэтилсвинца в граммах на 1 кг бензина. Протекающие при этом реакции выражаются следующими уравнениями:
Реактивы:
Соляная кислота, концентрированная и 0,1 н. раствор. Азотная кислота, концентрированная.
Пероксид водорода, 30%-ный раствор. Буферный раствор.
Смешанный индикатор. Трилон Б, 0,01 М раствор.
Проведение анализа
Разложение тетраэтилсвинца в колбе с испытуемым раствором, а также выпаривание избытка соляной кислоты из фарфоровой чашки проводят в вытяжном шкафу. Перед анализом определяют плотность испытуемого бензина. В сухую коническую колбу вместимостью 500 мл с пришлифованным обратным водяным холодильником наливают 50 мл испытуемого бензина и 50 мл концентрированной соляной кислоты. Когда в 1 кг испытуемого бензина содержится меньше 0,5 г или уменьшают в 2 раза объем пробы бензина.
Колбу укрепляют в штативе, помещают в водяную баню, нагревают и кипятят в течение 30 мин так, чтобы конденсат бензина стекал в колбу каплями. Затем колбу охлаждают и её содержимое
31
vk.com/club152685050 | vk.com/id446425943
переливают в делительную воронку. После расслоения нижний слой (раствор хлорида свинца в соляной кислоте) сливают в фарфоровую чашку, а верхний бензиновый слой переносят в коническую колбу, где происходило разложение тетраэтилсвинца. В колбу с бензиновым раствором приливают 50 мл дистиллированной воды и кипятят её содержимое 5-8 мин; после охлаждения водный слой отделяют в делительной воронке и переливают в фарфоровую чашку, присоединяя его к основному раствору хлорида свинца.
Так повторяют до трёх раз, выливая водные растворы в ту же фарфоровую чашку. Фарфоровую чашку с солянокислым раствором хлорида свинца устанавливают на кипящую водяную баню и выпаривают досуха. В случае покраснения раствора в процессе выпаривания к нему приливают 0,5 мл концентрированной азотной кислоты и 0,5 мл 30%-ного раствора пероксида водорода и продолжают выпаривать. При окислении должен исчезнуть красно-коричневый цвет. Для удаления выделяющихся оксидов азота сухой остаток выдерживают 5…10 мин. Затем к нему приливают 20 мл бидистиллированной воды, тщательно перемешивают и полученный раствор выливают в коническую колбу. Чашку промывают несколько раз 10…15 мл воды до полного перевода всего остатка в коническую колбу, расходуя на это не более 60 мл бидистиллированной воды. К полученному раствору хлорида свинца приливают 5 мл 0,1 н. раствора соляной кислоты и 2 мл буферного раствора и стеклянной лопаточкой насыпают около 0,1 г смешанного индикатора. Окрашенный в сиреневый цвет раствор титруют 0,01 М раствором трилона Б до устойчивого жёлтого цвета.
Параллельно проводят контрольный опыт, для чего в коническую колбу наливают 50 мл дистиллированной воды, 5 мл 0,1 н. раствора соляной кислоты, 2 мл буферного раствора и около 0,1 г смешанного индикатора и титруют раствором трилона Б до жёлтого цвета.
Содержание тетраэтилсвинца x (в г на 1 кг бензина) рассчитывают по формуле:
x
(V V ) 0,00207 |
||
1 |
2 |
|
|
V |
|
|
3 |
|
1.561
,
где V1 и
V2
- объём 0,01 М раствора трилона Б, израсходованного на титрование соответственно в
целевом и в контрольном опыте, мл; 0,00207 – количество свинца, эквивалентное 1 мл точно 0,01 М
раствора трилона Б, г; 1,561 – коэффициент для пересчёта свинца в тетраэтилсвинец;
испытуемого бензина, мл; |
- плотность испытуемого бензина, г/мл. |
V3
- объём
ЛАБОРАТОРНАЯ РАБОТА № 13 Анализ нефтяных масел
Общие сведения о маслах
Большинство нефтяных масел выпускается для смазки и поэтому они называются смазочными.
Кним относятся следующие группы и подгруппы масел, классифицированные по областям применения:
1)моторные масла (авиационные, автомобильные, автотракторные, дизельные, для реактивных двигателей);
2)индустриальные, масла, предназначенные для смазки станков, различных механизмов, холодильных машин, прокатных станов, насосов, прессов, приборов;
3)турбинные масла;
4) компрессорные масла;
5)масла для паровых машин (цилиндровые и судовое);
6)трансмиссионные и осевые масла.
Кроме того, вырабатываются масла различного специального назначения, которые принято относить к несмазочным нефтяным маслам: трансформаторное, конденсаторное, парфюмерное и некоторые другие. Несмотря на то, что нефтяные масла эксплуатируются в разнообразных условиях и используются в различных целях, многие требования к ним имеют общий характер. Поэтому качество большинства масел оценивается одинаковыми физико-химическими показателями, а различие заключается в основном в их абсолютных значениях. Остановимся вкратце на этих общих и специальных требованиях к нефтяным маслам.
Требования к качеству смазочных масел и нормируемые показатели
32
vk.com/club152685050 | vk.com/id446425943
Основной физико-химической характеристикой нефтяных масел является их вязкость, или коэффициент внутреннего трения. От вязкости зависит способность данного масла при температуре, характерной для определённого узла трения, выполнять свои функции, т.е. обеспечивать замену сухого трения жидкостным и предотвращать износ материала, так как коэффициент жидкостного трения в десятки и сотни раз меньше коэффициента сухого трения. Ввиду исключительно большого разнообразия конструкций узлов трения, в характере и скорости движения трущихся поверхностей, а также в возникающих удельных нагрузках различные группы масел, а внутри групп отдельные сорта и марки должны отличаться друг от друга по вязкости в широком интервале.
Для всей группы моторных масел важное эксплуатационное значение имеет вязкостнотемпературная характеристика, гарантирующая достаточную пологость температурной кривой вязкости. Действительно, при низких температурах вязкость масла не должна быть слишком высокой, чтобы не затруднять запуск двигателя. Наоборот, при характерной для поршневой группы высокой температуре, масло должно обеспечить гидродинамический режим смазки, т.е. вязкость его должна быть достаточно высокой.
Смазочные масла не должны вызывать коррозии металлов. Это общее требование оценивается прежде всего кислотным числом, которое для всех масел нормируется в очень узких пределах: 0,05…0,35 мг КОН на 1 г масла. Кроме того, для многих трансмиссионных масел, для масел, применяемых в холодильных машинах, и для сульфофрезола установлено специальное ускоренное испытание на коррозию стальных и медных пластинок при 100°С в течение 3 ч, которое все эти масла должны выдерживать. Очень серьёзное эксплуатационное значение для многих групп нефтяных масел (моторных, турбинных, компрессорных, для холодильных машин и трансформаторных) имеет химическая стабильность, т.е. способность масла противостоять окислению кислородом воздуха.
Большинство смазочных масел должны прокачиваться при низких температурах, что связано с условиями их эксплуатации и транспортировки. В технических нормах это требование контролируется температурой застывания, хотя должного соответствия между температурой застывания, определённой по стандартной методике, и температурой потери подвижности в реальных условиях, как уже указывалось, часто не наблюдается. Для отдельных сортов масел низкотемпературные свойства оцениваются значением вязкости при низких температурах.
Все смазочные масла контролируются по температуре вспышки. Нормируемый низший предел температуры вспышки характеризует пожарную безопасность масла и лимитирует примесь низкокипящих компонентов, которые при эксплуатации масел будут испаряться.
Все смазочные масла должны быть в достаточной мере очищены от минеральных примесей, обладать невысоким коксовым числом, не содержать селективных растворителей, т.е. быть чистыми и однородными. Поэтому в технические условия на нефтяные масла включены также следующие показатели: содержание селективных растворителей (отсутствие или следы); зольность — не более тысячных (реже сотых) долей процента, для масел с присадками содержание золы повышается до десятых долей процента; содержание механических примесей — отсутствие или тысячные и сотые доли процента, для масел с присадками десятые доли процента; содержание воды — отсутствие или следы;
содержание водорастворимых кислот и щёлочей — отсутствие; коксуемость — от 0,1 до 3,0 % для тяжёлых масел.
Определение показателя преломления
При переходе из одной среды в другую падающий луч света отклоняется от своего первоначального направления, т.е. преломляется, вследствие изменения скорости распространения света. С изменением угла падения α меняется и угол преломления β, но отношение синуса к синусу угла преломления охраняется постоянным. При переходе луча света из пустоты или воздуха в оптически более плотную среду имеем:
(sin α/sin β)=n
Это отношение называется показателем преломления или коэффициентом рефракции. Его значение характеризует оптическую плотность среды. Значение показателя преломления зависит также
33
vk.com/club152685050 | vk.com/id446425943
от длины волны падающего света и от температуры определения. Поэтому в символе показателя преломления необходимо указывать температуру и условное буквенное обозначение длины волны источника света. В обычных анализах чаще всего определение проводят при температуре 20°С на солнечном свету при длине волны жёлтой линии спектра паров натрия λ= 589 нм, что условно
обозначается индексом
Определение показателя преломления проводят на приборах - рефрактометрах при температуре 20°С. Основная оптическая часть рефрактометра состоит из двух призм (рис. 44): осветительной 2 и измерительной 1. Обе призмы сложены по гипотенузе, и между ними находится тонкий слой испытуемой жидкости 3. Лучи света, пройдя через призму 2 и преломившись на границе раздела сред «жидкость – стекло», войдут в призму 1. При полном внутреннем отражении лучи света попадут в зрительную трубу 4. При этом одна половина светового поля 5 будет освещена, а вторая останется темной. Это явление легко наблюдается в окуляре рефрактометра.
Рис. 44. Оптическая схема рефрактометра: 1 и 2 - призмы; 3 - анализируемая жидкость; 4 - зрительная труба; 5 - световое поле.
Устройство прибора
Во внешней части корпуса 3 рефрактометра ИРФ-22 (рис. 45) имеется маховичок 11, вращением которого можно наклонить измерительную головку 7 до нужного положения. При этом, наблюдая в окуляр зрительной трубы, находят границу раздела света и тени. Здесь же имеется зеркало 9 для подсвечивания шкалы показателя преломления и стопорный болт 16, с помощью которого укрепляется осветительное зеркало 17, служащее для подсвечивания исследуемого вещества.
Измерительная головка 7 прикреплена к корпусу 3 и состоит из двух полушарий, в которые вставлены измерительная и осветительная призмы. Верхнее полушарие откидывается для удобства нанесения на поверхность измерительной призмы капли испытуемого вещества. Через окно 15 наблюдают, равномерно ли заполняется исследуемым веществом пространство между призмами. Для поддержания постоянной температуры в полушариях имеются камеры, через которые пропускается вода с помощью шлангов 6, надеваемых на штуцеры 5. Температура воды контролируется термометром 8.
Рис. 45. Внешний вид рефрактометра ИРФ-22:
1 - зрительная труба с отсчётным устройством; 2 - фланец; 3 - корпус; 4 - барабан со шкалой; 5 - штуцеры; 6 - шланг; 7 - измерительная головка; 8 - термометр; 9, 17 - зеркала для подсвечивания жидкости и шкалы; 10, 18 -
34
vk.com/club152685050 | vk.com/id446425943
крышки; 11, 19 - маховички; 12 - диафрагма; 13 - ключ; 14 - котировочная пластина; 15 - окно; 16 - стопорный винт зеркала.
Зрительная труба 1 с отсчётным устройством соединена с измерительной головкой 7, внутри корпуса которой расположена шкала отсчёта показателя преломления. На отсчётном устройстве имеется маховичок 11, вращением которого можно устранить окраску наблюдаемой границы раздела.
Для измерения показателя преломления окрашенных или мутных проб имеется диафрагма 12; которая вставляется в отверстие после открытия крышки 10. В этом случае луч света, пройдя через диафрагму и измерительную призму, равномерно осветит поле зрения.
Подготовка прибора
Для поддержания постоянной температуры из термостата ТС-15 подают воду через шланг 6 в рубашку рефрактометра. Скорость по дачи воды регулируют винтовым зажимом.
Перед работой необходимо убедиться в правильности показаний рефрактометра. Для проверки прибора используют дистиллированную воду при температуре 15, 20 и 30°С или стеклянную пластинку с известным показателем преломления, причём перед каждым измерением пластинку переставляют в приборе. Определение показателя преломления пластинки и дистиллированной воды проводят так же, как описано ниже в «Методике определения».
Если отсчёты по шкале прибора совпадают с показателем преломления воды или пластинки или отличаются от них не более, чем на 0,000l, то прибор пригоден к работе. В противном случае устанавливают по шкале прибора необходимый отсчёт и, поворачивая ключом 13 винт с четырёхгранной головкой, подводят границу раздела к точке пересечений нитей. После этого прибор подготовлен к работе.
Методика определения
Откидывая верхнее полушарие, протирают поверхности призмы мягкой неворсистой тканью, смоченной лёгким бензином или спиртом, и наносят на нижнюю призму 1-2 капли испытуемого масла. Опускают полушарие и наблюдают в окне 15 равномерное распределение жидкости по поверхности призмы. Зеркало 9 устанавливают так, чтобы источник света через окно 15 равномерно освещал верхнюю призму. Открывают ставню зеркала 17, освещают шкалу показателя преломления и наблюдают её в окуляре. При этом, вращая маховичок 19, находят границу раздела света и тени. Затем, вращая маховичок 11, добиваются исчезновения окрашенности. Снова осторожно поворачивая маховичок 19, более точно совмещают границу раздела с перекрестием.
В этих условиях в окуляре трубы 1 отчётливо видна линия границы, крест, деления шкалы и её штрихи (рис. 46). Целые, десятые, сотые и тысячные доли значения показателя преломления отсчитывают по шкале, а десятитысячные доли отсчитывают на глаз. Отсчёт проводят 2—3 раза с точностью до 0,0002 и каждый раз отмечают температуру воды. Из полученных отсчётов выводят среднее значение показателя преломления и температуру воды.
Рис. 46. Поле зрения трубы рефрактометра.
Если температура воды во время определения отличалась от 20°С, то в найденный показатель преломления 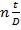 следует внести поправку по формуле:
следует внести поправку по формуле:
где t — температура воды; 0,00035 — средняя поправка для показателя преломления масел при изменении температуры на 1°С.
35
vk.com/club152685050 | vk.com/id446425943
По окончании работы призмы следует протереть мягкой неворсистой тканью, смоченной в бензине, просушить, проложить бумагой и закрыть.
36