

Практикум ФизХим Кинетика. Катализ Михаленко 2018
.pdfГомогенный кислотно-основной катализ.
Кислотный катализ протекает в присутствии веществ, способных передавать и принимать протон Н+ (специфический кислотный катализ) или другой акцептор электронной пары (общий кислотный катализ). Схему последнего можно представить двумя стадиями
 S HA K1 SH A k2 P HA
S HA K1 SH A k2 P HA ,
,
где S – эсубстрат (исходное вещество), Р – продукт реакции. Первая стадия равновесная с константой равновесия K1, а вторая стадия необратимая с константой k2 определяет
скорость реакции в целом, то есть является лимитируюшей.
По закону действия масс w w2 k2CSH CA |
(1.21`) . Из |
|||
константы равновесия К1 |
|
СSH CA |
выразим |
CSH CA и |
|
||||
|
|
CS CHA |
|
|
подставим в 1.21`. Уравнение скорости реакции имеет вид
w K1k2CHACS k'эффСS |
(1.21) . Эффективная |
константа |
|
kэфф K1k2CHA зависит |
от природы кислоты |
(K1k2), что |
|
видно по разному |
тангенсу угла наклона линейной |
||
зависимости kэфф от |
CHA (рис.1.7а). |
|
|
Рис. 1.7. Влияние концентрации кислоты и рН на эффективную константу общего кислотного катализа (а) и специфического кислотно-основного катализа (б).
33
В специфическом кислотном катализе катализатор ион гидроксония Н3О+. Примером являются реакция
этерификации и реакция гидролиза сахарозы. |
|
|
|||||||
Для |
2х-стадийной |
схемы |
|
реакции |
|||||
S H3О |
|
K1 |
|
Н2О и |
SH |
|
k2 |
|
|
|
SH |
|
|
P H3O |
|
||||
получим уравнение типа 1.21 с эффективной константой |
||||
kэфф |
K1k2CH |
О |
и lg kэфф lg K1k2 pH |
(1.22) . Из |
|
3 |
|
|
|
уравнения 1.22 следует, что с увеличением рН значение логарифма lgkэфф линейно уменьшается, что отражает график на рис.1.7б.
Основной катализ. Для специфического основного катализа можно также записать две стадии реакции
SН |
|
ОН |
|
K1 |
k2 |
|
. |
|
|
S Н2О и |
S P ОН |
|
|||
В случае основного катализа lgkэфф |
линейно увеличивается |
||||||
с ростом рН, т.к. после проведения аналогичных выше
математических |
операций |
получается уравнение |
вида |
lg kэфф lg K1k2 |
* pH |
(1.22`) . Здесь, в эффективную |
|
константу включена константа ионизация субстрата S. |
|||
Для концентрированных кислот и щелочей |
смысл |
||
kэфф усложняется, вводятся коэффициенты активности и понятие кислотности среды h0 и функция кислотности Гаммета Н0= lgh0.
В заключении отметим, что кислотно-основной катализ включает перенос протона от одной молекулы к другой, значит, в реакционной среде должны быть доноры и акцепторы протонов. Кислотно-основной катализ с участием аминокислот (ферментов) связан с тем, что у аминокислот хорошими донорами протонов являются
группы СООН, NH3+, SH, а акцепторами протонов выступают группы СОО , NH2, S .
34
Ферментативный катализ
Ферментами (энзимами E) называются вещества белковой природы, которые ускоряют химические реакции, протекающие в клетках и тканях живого организма. Поэтому их еще называют биокатализаторами, а словами физиолога Павлова «возбудителями жизни». Активность ферментов выше активности других видов катализаторов.
Молекула фермента имеет чередующиеся полярные группы СООН, NH2, NH, OH, SH и др. и неполярные гидрофобные группы (первичная структура белка). Макромолекула фермента может изгибаться и свертываться в клубки с образованием внутримолекулярных водородных связей (вторичная структура белка). Сложные ферменты содержат ионы металлов с переменной степенью окисления (Fe,Cu), участвующих в окислительно-восстановительных реакциях.
Ферменты классифицируют по типу катализируемой реакции: гидролазы (реакции гидролиза), изомеразы (изомерные превращения), оксиредуктазы (окислительновосстановительные реакции). Выделяют шесть классов ферментов: I – оксидоредуктазы, II – трасферазы, III – гидролазы, IV – лиазы, V – изомеразы, VI – лигазы. Классы делятся на подклассы. Ферменты I класса катализируют окислительно-восстановительные реакции, II – реакции переноса различных функциональных групп от одного субстрата (донор) к другому (акцептор), III – разрыв в субстрате внутримолекулярных связей, IV – разрыв связей С-О, С-С, С-N связей, V – изомерные превращения в одной молекуле, VI – присоединение друг к другу двух молекул.
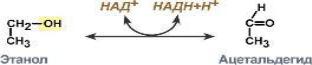
Пример 1. Фермент I класса – алкогольдегидрогеназа катализирует образование ацетальдегида из этанола.
35
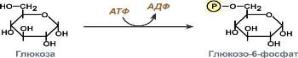
Пример 2. Фермент II класса – гексокиназа катализирует перенос фосфорсодержащих групп.
Активные центры фермента способны избирательно связывать молекулу реагирующего вещества субстрата S, образую с ней единый фермент-субстратный комплекс ES. Важно геометрическое соответствие структур активного центра фермента и субстрата (принцип «ключ-замок»).
Ферментативный катализ занимает промежуточное положение между гомогенным и гетерогенным катализом. Его отличительные черты: малые количества фермента и высокие скорости, специфичность или селективность действия (один фермент ускоряет определенную реакцию), низкие температуры и сильное влияние рН среды и присутствия других веществ.
Ингибиторами (I) называют вещества, осложняющие течение ферментативной реакции вследствие образования комплексов с ферментом или фермент-субстратным комплексом.
В фармацевтической продукции ферментные препараты занимают большую часть ассортимента. При инфекционных заболеваниях используются препараты, разрушающие оболочку болезнетворных бактерий. Широко известны препараты, применяемые при нарушениях функций желудочно-кишечного тракта, для рассасывания гнойных скоплений и тромбов, например, стрептокиназа.
Ферменты пищеварения незаменимы для людей, страдающих хроническими заболеваниями или периодическими воспалениями поджелудочной железы. Широко представлены ферменты растительного происхождения – альфа-амилаза, лактаза, протеаза, целлюлаза, липаза.
36
Кинетика ферментативных реакций. Уравнение Михаэлиса-Ментена
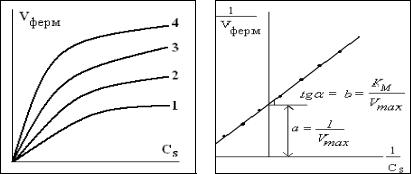
Скорость ферментативной (энзимной) реакции Vферм зависит от концентрации фермента СE и концентрации субстрата CS. Наблюдается прямо пропорциональная зависимость Vферм от начальной концентрации С0,Е (первый порядок по ферменту). Зависимость Vферм от концентрации субстрата имеет нелинейный вид (рис.1.7), однако, при низких концентрациях субстрата порядок по субстрату первый, а при высоких – нулевой.
Рис. 1.7. Зависимость скорости ферментативной реакции от концентрации субстрата (увеличение константы равновесия K1 в ряду 1-4) и способ определения константы Михаэлиса КМ графическим методом
Простейшая схема ферментативного катализа включает обратимое образование фермент-субстратного комплекса (ES) , который необратимо превращается в продукт:
E S K k1 / k 1 ES k2 E P .
Константа равновесия первой стадии выражена через константы скорости прямой (k1) и обратной (k 1) реакций, а константа скорости второй стадии k2 << k 1 , т.е. вторая
37

стадия является лимитирующей. Тогда, скорость
ферментативной реакции равна Vферм k2CES |
(1.23) . |
Константу k2 называют каталитической константой или
числом оборотов фермента.
В соответствии со схемой приведѐм вывод уравнения Михаэлиса-Ментена (1.24), используя закон действия масс и метод стационарных концентраций условие постоянства концентрации промежуточного вещества
dСES |
Vферм |
k1CS CE k 1CES |
k2CES |
0 (1.23`) . |
|
dt |
|||||
|
|
|
|
Текущую концентрацию фермента выразим через его
начальную концентрацию С0.Е и концентрацию комплекса (материальный баланс) CE C0,E CES и тогда получим
k1CS C0,E CES CS k 1CES k2CES 0 (1.23``) .
Раскрыв скобки, найдем текущую концентрацию комплекса
CES |
|
|
k1 |
CS C0,E |
|
|
CS C0,E |
|
|
CS C0,S |
|
|
(1.24`) |
||||||
|
k1CS k 1 k2 |
k2 |
k 1 |
|
|
KM CS |
|||||||||||||
|
|
|
|
|
CS |
|
|
||||||||||||
|
|
|
|
|
|
k1 |
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
и с учетом 1.23 получим |
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
V |
k2 C0,E CS |
|
(1.24а) |
|
V |
|
|
Vmax CS |
|
(1.24б) |
|||||||||
|
|
|
|
|
|
||||||||||||||
ферм |
|
KM |
CS |
|
|
|
|
ферм |
|
KM |
CS |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||
где Vmax |
k2 |
C0,S максимальная скорость при бесконечно |
|||||||||||||||||
большой концентрации субстрата (нулевой порядок по СS). |
|||||||||||||||||||
Константа |
Михаэлиса |
КМ |
– важный |
|
кинетический |
||||||||||||||
параметр ферментативной реакции. |
|
Она численно равна |
|||||||||||||||||
концентрация субстрата, при которой достигается половина максимальной скорости реакции. КМ характеризует специфичность фермента к субстрату (чем меньше КМ, тем больше специфичность), а еѐ значения КМ, лежат в интервале 10 6 (1 мкМ) 0,1М (примеры см. ниже).
38

Фермент |
Cубстрат |
КМ, мкМ |
KM |
|
(k2 k 1) |
|
Карбоангидраза |
СО2 |
8000 |
||||
k1 |
||||||
Пенициллаза |
Бензилпеницилин |
50 |
|
|
Определение КМ и Vmax по экспериментальным данным.
Уравнение (1.24а) преобразуем в линейную форму
1 |
|
1 |
|
KM |
|
1 |
(1.25) . |
|
|
|
|
||||
Vферм |
|
Vmax |
|
Vmax |
CS |
|
|
Представляя зависимости начальной скорости реакции V0 от начальной концентрации субстрата СS,0 в обратных координатах (рис.1.6) находим тангенс угла наклона b и отрезок, отсекаемый по оси ординат, равный коэффициенту а линейного уравнения y=а+bx. По значению а рассчитывается максимальная скорость реакции, а константа Михаэлиса равна КМ =b/a.
Можно использовать и другие координаты спрямления при обработке экспериментальных данных – V0 и V0/СS,0 , полученных для другой линейной формы уравнения
Михаэлиса-Ментена V V |
|
KM V0 |
. |
0 max |
|
CS ,0 |
|
|
|
||
Ингибирование. |
|
|
|
Протекание ферментативной реакции усложняется присутствием веществ, образующих комплексы с ферментом или фермент-субстратным комплексом. Конкурентный ингибитор конкурирует с субстратом за активные центры фермента и к указанной выше схеме
добавляется стадия E I EI .
При конкурентном ингибировании Vmax остается неизменной, а эффективная KM увеличивается
|
|
|
|
|
|
С |
|
|
К1 называют константой ингибирования |
K |
|
К |
|
1 |
|
|
0,I |
|
K I СE CI / CEI , |
|
|
|
|
||||||
|
M .эфф |
|
М |
|
|
K1 |
|
||
|
|
|
|
|
|
|
С0,I – исходная концентрация ингибитора |
||
|
|
|
|
|
|
|
|
|
|
39
Неконкурентный ингибитор обратимо связывает как фермент, так и фермент-субстратный комплекс – стадии
E I EI и ES I ESI . При неконкурентном ингибировании KM не изменяется, а Vmax уменьшается.
Смешанное ингибирование описывается сложными кинетическими схемами (механизмами) и изменяются как константа КМ, так и Vmax. Такой тип ингибирования можно наблюдать и в случае двухсубстратных реакций.
Особенности ферментативных реакций.
1.Для ферментных катализаторов высокая активность (ускорение реакции в 108-1014 раз), которая на порядки превышает активность кислот, оснований и гетерогенных
катализаторов. Так, разложение Н2О2, катализируемое кислотой и уреазой, отличаются по значениям константы скорости в 1013 раз (710-7 и 5 106 с-1). Энергии активации реакции равны соответственно 103 и 28 кДж/моль.
2.Высокая специфичность. Так, амилаза не катализирует гидролиз сахарозы, но катализирует расщепление крахмала. Различают абсолютную специфичность (1 фермент1 субстрат), групповую специфичность (1 фермент несколько похожих субстратов) и стереоспецифичность (ферменты работают с оптически активными субстратами левовращающими L или правовращающими D).
3.Ферменты «работают» только в мягких условиях (36-37 0С, рН 7-8, атмосферное давление), действуют в малых количествах и не расходуются в реакции.
4.Ферменты ускоряют только термодинамически возможные реакции.
40

Гетерогенный катализ
Гетерогенно-каталитические реакции протекают на границу раздела фаз, образуемых на поверхности катализатора и реактантами. Механизм гетерогенного катализа более сложен, чем гомогенного катализа. Роль катализатора состоит в том, что увеличивается вероятность встречи и взаимодействия молекул за счет концентрирования веществ на твердой поверхности катализатора, т.е. адсорбции. Поэтому в основе механизма гетерогенного катализа лежит адсорбционная теория.
Рассматривают пять основных стадий в реакциях, протекающих на твердой поверхности: диффузия вещества к поверхности катализатора (1), адсорбция вещества (2), реакция в адсорбционном слое (3), десорбция продуктов с поверхности (4), диффузия продуктов в объем (5). Простейший механизм гетерогенного катализа можно записать двухстадийной схемой, для субстрата S(газ), находящегося в газовой фазе
S(газ) К1адс S(адс) k2 P(адс) .
Первая стадия обратимая с константой адсорбционного
равновесия K1адс= kaдс/kдес , где kадс константа скорости прямой реакции (адсорбции), а kдес – константа скорости
десорбции вещества.
Скорость гетерогенной реакции (W) прямо пропорциональна поверхностной концентрации реагентов (степени заполнения поверхности данными веществами S,i) и количеством свободных центров Z на поверхности
катализатора ( |
): W k 1 |
.... n |
(1.26) . |
|
|
|
|
0 |
1 S |
0 |
|
|
|
|
|
1 |
|
|
|
|
|
Уравнение 1.26 отражает закон действующих масс для |
|||||
необратимой |
гетерогенной |
реакции |
в |
виде |
||
S (адс) ... nZ(центры) *P (адс) ... |
(1.26`) . |
|||||
1 |
1 |
|
1 |
1 |
|
|
Степени заполнения поверхности веществами, находящимися в реакционном объеме в виде газов, можно
41
выразить через парциальные давления. Заметим, что поверхность реального твердофазного катализатора неоднородна, что означает присутствие различных центров, из которых выделяют каталитически активные центры. На этих центрах адсорбция приводит к ослаблению связей между атомами превращаемой молекулы, и она становиться более реакционно-способной. При участии двухатомных молекул Н2, О2 (реакции гидрирования и окисления) присходит их диссоциация, например, реакции C2H4 + H2 = C2H6 (Ni), N2+3H2=2NH3 (Fe), 2SO2+O2= 2SO3 (Pt), CO + ½ O2 = CO2 (Rh) и другие. Приведенные выше реакции являются крупнотоннажными каталитическими процессами. Преимуществом этих процессов является простота разделения продуктов реакции и катализатора, который после регенерации можно использовать повторно.
Для увеличения эффективности гетерогенных катализаторов и снижения стоимости за счет уменьшения содержания благородных металлов (платина, родий) каталитически активную фазу наносят на твердые вещества (носители) с развитой поверхность. Простыми и инертными носителями являются уголь, силикагель, алюмосиликаты, цеолиты). Высокой активностью обладают оксиды металлов - меди, кобальта, цинка, хрома, железа, титана и др.
Гетерогенные катализаторы очень чувствительны к присутствию каталитических ядов, представляющих собой вещества (сера, и еѐ соединения, ртуть, свинец, молекулы с кратной связью, например СО и HCN), блокирующие активные центры поверхности и дезактивирующие еѐ из-за образования устойчивых соединений с катализатором. В реакциях с участием углеводородов наблюдается снижение активности катализатора из-за зауглероживания.
Вещества, повышающие активность и стабильность работы катализатора называют промоторами. Например, в алюмосиликатный катализатор вводят ионы Cl или F для регулирования кислотно-основных свойств поверхности.
42