

ФХМА (sol2019) / Учебная литература / Методички / Туркельтауб - Часть 1, Теория хроматографии, Газовая хроматография
.pdf56
миграции форма сконденсированной зоны станет
сильно |
асимметричной |
с вертикальным тылом и |
|
сильно |
растянутым |
фронтом |
(рис. 21). Если |
вместе |
с растворителем в |
колонку вводят |
|
вещества, летучесть которых ниже летучести растворителя, они будут задерживаться у вертикальной границы сконденсированной зоны. При-
чем полосы |
анализируемых |
веществ должны |
|||
сжиматься, так как |
их |
фронты |
надвигаются |
на |
|
очень толстую |
и |
все |
более увеличивающуюся |
по |
|
толщине пленку жидкого растворителя (фронт
тормозится), |
а тыльные |
участки |
двигаются |
с |
|||
гораздо |
более |
высокой |
скоростью |
по |
|||
относительно |
тонкой |
пленке неподвижной фазы |
|||||
(тыл |
ускоряется). |
В |
этом |
|
суть |
||
реконцентрирующего |
|
механизма |
эффекта |
||||
растворителя. |
|
|
|
|
|
|
|
Системы ввода пробы без деления потока получила широкое распространение благодаря тому, что вся
введенная проба попадает в колонку, что значительно повышает чувствительность. Это позволяет анализировать следовые количества, раз-
бавленные экстракты природных и промышленных смесей.
К недостаткам рассмотренной техники ввода следует
отнести: плохую воспроизводимость времен удерживания компонентов, выходящих сразу за
пиком растворителя; отсутствие эффекта
реконцентрирования для компонентов, элюируемых перед растворителем; разложение термолабильных соединений из-за длительного нахождения паров
образца в горячей зоне инжектора.
Указанные недостатки частично могут быть устранены в системах ввода, которые работают без
21
выделенных веществ будет составлять чуть более
98% при Rs = 1 и около 99,9% при Rs = 1,5.
На рис.6 представлены три хроматограммы, соответственно при Rs = 0,75; 1,0 и 1,5. Такое улучшение разделения достигается при увеличении эффективности или возрастании фактора разделения
.
Уравнение (16) можно представить в ином виде,
связав критерий разделения Rs c фактором емкости k, фактором разделения и числом теоретических тарелок n.
Примем 1 2= . Подставим в уравнение (16) ширину
пика у основания , представленную через число
теоретических тарелок n из соотношения (11):
RS |
tR |
2 |
tR1 |
|
n |
|
|
|
|
|
(17) |
||
|
|
tR2 |
4 |
|||
|
|
|
|
|
||
Подставим в полученное выражение величины
коэффициентов ёмкости из уравнения (6):
R |
|
k2 k1 |
|
n |
(18) |
|
4 |
||||
S |
|
k 1 |
|
||
Если в полученное выражение (18)
подставить из уравнения (7) величину коэффициента селективности , тогда можем записать:
R |
|
1 |
|
1 |
|
|
|
k |
|
|
|
|
|
|
(19) |
||
|
|
|
|
|
n |
|
|
||||||||||
|
|
|
k 1 |
|
|
||||||||||||
S |
4 |
|
|
|
|
|
|
|
|
||||||||
|
|
|
2 |
|
2 k 1 2 |
|
|||||||||||
n 16Rs |
|
|
|
|
|
|
|
|
|
|
|
(20) |
|||||
1 |
|
k |
' |
||||||||||||||
|
|
|
|
|
|
|
|
|
|
||||||||
Из уравнения (20) следует, что при заданном значении критерия разделения Rs и фактора
разделения (коэффициента селективности) , можно
вычислить необходимую эффективность колонки. Для
http://www.mitht.ru/e-library
22
разделения соединений с близкими свойствами надо
увеличить длину колонки или уменьшить ВЭТТ.
Последнее может быть достигнуто уменьшением
размера частиц, увеличением однородности насадки колонки, уменьшением толщины пленки жидкой фазы при одновременном снижении адсорбционной активности поверхности, на которую наносится эта неподвижная фаза. Помимо этого можно увеличить фактор разделения . Для этого необходимо
подобрать более селективную неподвижную, а для
жидкостной хроматографии и подвижную фазу. В газовой хроматографии снизить температуру колонки.
Помимо диффузии и сопротивления массопередачи размывание пика будет происходить в случае нелинейности изотермы распределения.
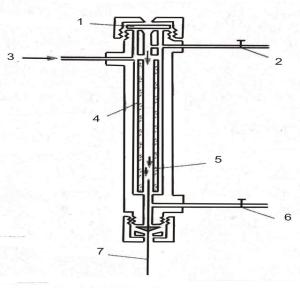
4. Общая схема хроматографического анализа.
Несмотря на большое многообразие вариантов хроматографии, процесс хроматографического анализа можно представить единой схемой,
Рисунок 7. Схема газового хроматографа.
1 – газ-носитель; 2 – регулятор потока газ-носителя; 3 – микрошприц для ввода пробы; 4 – испаритель; 5 – хроматографические колонки; 6 – термостат колонок; 7 – детектор; 8 – система регистрации.
55
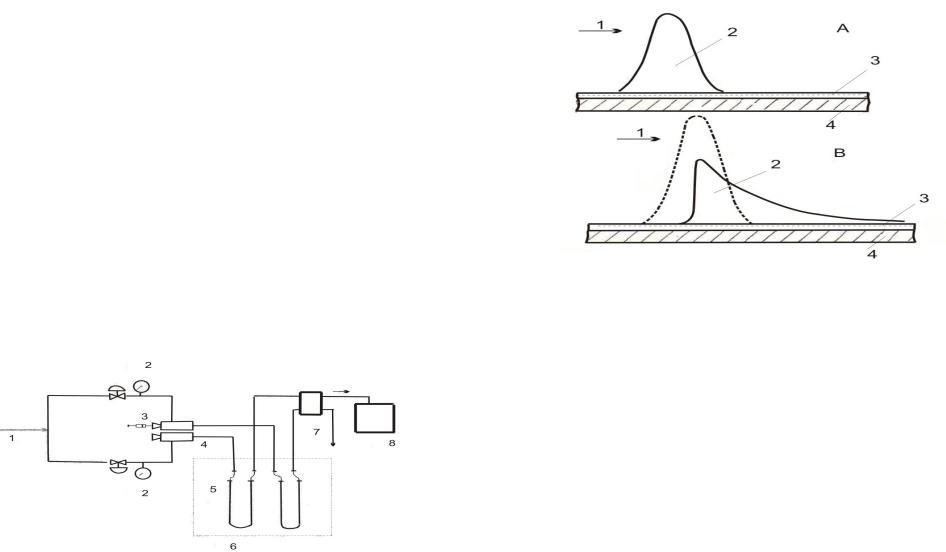
Рисунок 21. Схема формирования зоны
сконденсированного растворителя на начальном
участке капиллярной колонки: А – начальный момент
времени; В – начальная форма зоны. 1 – поток газаносителя; 2 – зона сконденсированного
растворителя; 3 – пленка неподвижной фазы; 4 –
внутренняя стенка капиллярной колонки.
Слой сконденсированного растворителя можно
рассматривать как пленку неподвижной фазы, поэтому относительная скорость миграции каждой
узкой полосы зоны будет определяться толщиной пленки растворителя на участке, над которым должна
двигаться данная узкая полоса. Поскольку толщина пленки жидкости в максимуме зоны в 100—300 раз
превышает толщину пленки |
неподвижной |
фазы |
||||
в |
остальной части колонки, скорость |
миграции |
||||
фронтальных полос зоны будет намного |
превышать |
|||||
скорость миграции |
тыльных |
полос |
зоны. |
|
||
В |
результате через |
короткое |
время |
после |
начала |
|
http://www.mitht.ru/e-library
54
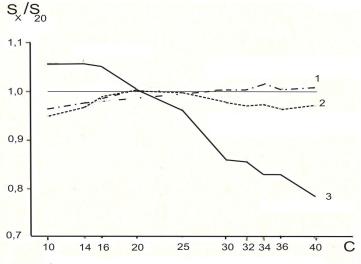
Рисунок 20. Фракционирование компонентов пробы при различных методах ввода.
Sx/S20 – отношение площадей н-алканов к площади пика С20Н42; C – число атомов углерода в молекуле н- алкана. 1 – непосредственный ввод; 2 –
автоматический ввод с делителем потока; 3 – ввод вручную.
Растворитель при этом свободно проходит через
колонку. После выхода зоны растворителя вклю-
чается нагрев колонки и начинается процесс разделения и анализа.
Механизм эффекта растворителя более сложен. Экспериментально показано, что при поступлении в
капиллярную колонку растворитель конденсируется
на ее начальном участке (рис.21). В первый момент зона сконденсированного растворителя имеет гауссово распределение. Под влиянием потока газаносителя зона мигрирует.
23
состоящей из следующих стадий: подача подвижной фазы, ввод анализируемой пробы, собственно
хроматографическое разделение, детектирование, обработка результатов разделения. Каждой из
указанной стадий соответствует определенный узел
или блок хроматографа. На рис.7 показана схема хроматографа.
Газ-носитель из газового баллона через редуктор
поступает в хроматограф (1). Регулятором расхода
устанавливается скорость газа-носителя (2). Ввод пробы обычно осуществляется с помощью шприца (3) или дозатора. Автоматические дозаторы позволяют работать в полностью автоматизированном режиме. В дозаторе в специальных гнездах располагаются склянки с образцами анализируемых смесей.
«Механическая рука» вводит шприц в склянку,
промывает его раствором анализируемой смеси и вводит в испаритель хроматографа. Затем берется
проба из другой склянки для следующего анализа. В
испарителе (4) проба переводится в газообразное
состояние. Затем она захватывается потоком подвижной фазы и попадает в колонку. В
хроматографической колонке (5) смесь разделяется на отдельные компоненты. Колонки обычно помещаются в воздушный термостат (6). Компоненты анализируемой пробы вместе с потоком подвижной
фазы поступают в детектор (7). В детекторе изменение концентрации компонента преобразуется в электрический сигнал, усиливаемый и затем
регистрируемый в виде выходной кривой – хроматограммы (8). В современных хроматографах
задание и поддержание температуры, скорости подвижной фазы и всех других параметров
http://www.mitht.ru/e-library
24
хроматографического опыта берет на себя
компьютер.
Схема высокоэффективного жидкостного хроматографа (ВЭЖХ) представлена на рис. 20 будет рассмотрена позже.
Основными техническими характеристиками
хроматографических детекторов являются:
- чувствительность, которую характеризуют
отношением сигнала детектора к количеству регистрируемого вещества;
- предел детектирования, или предел
обнаружения, - минимальное количество вещества,
соответствующее сигналу детектора, вдвое превышающему высоту пика, характерного для шума детектора;
-линейность, представляющая собой диапазон концентраций, в пределах которого зависимость
«сигнал – концентрация» имеет линейный характер.
-стабильность работы (низкая чувствительность к
колебаниям температуры;
-воспроизводимость. Под воспроизводимостью работы детектора понимается воспроизводимость его
показаний во времени при постоянных
чувствительности, фоновом токе и других условиях эксперимента.
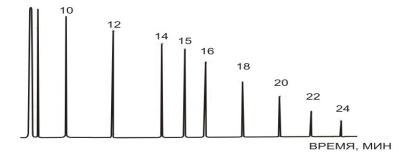
Анализ и методы расчета хроматограмм
Аналитическую хроматографию используют для качественного и количественного анализа простых и очень сложных смесей, в том числе, таких как
пищевые продукты, лекарства, продукты, загрязняющие окружающую среду. Препаративную хроматографию применяют для выделения некоторых веществ с близкими свойствами (например, лево- и
правовращающие изомеры), для очистки
53
капиллярную колонку, был предложен в 1969 году.
Сущность этого способа состоит в том, что
относительно большое количество (1—5 мкл)
разбавленного образца вводится в испаритель, испаряется и в виде пара переводится в колонку. Для исключения перегрузки колонки количество растворенных анализируемых компонентов в пробе не должно превышать 50 нг.
В отличие от системы с делением потока, где проба
при испарении должна гомогенно смешаться с газом-
носителем, в данном варианте обеспечивают минимальное смешение паров пробы с газом-
носителем.
Главным моментом в технике ввода без деления потока является создание условий для реконцентрирования анализируемых компонентов в виде узких полос на входе в капиллярную колонку. Без стадии реконцентрирования работа системы теряет смысл. Получили распространение
два способа реконцентрирования: низкотемпературное улавливание и так называемый эффект растворителя. Первый из них состоит в
конденсации и задержке при низкой температуре (приблизительно на 150°С ниже температуры
кипения компонентов) анализируемых соединений на
начальном участке колонки.
http://www.mitht.ru/e-library
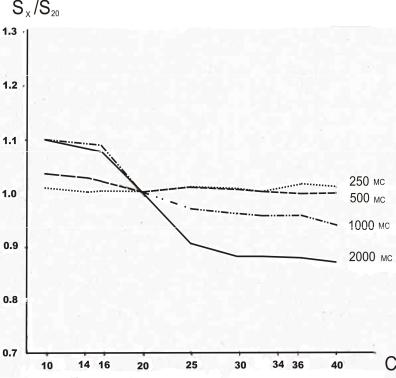
Рисунок 19. Влияние продолжительности
нахождения иглы в узле ввода пробы на фракционирование пробы углеводородов С10 - С40 в гексане. C – число атомов углерода в молекуле н- алкана.
Условия эксперимента: газовый хроматограф фирмы Hewlett-Packard с автоматическим устройством ввода пробы; колонка 10 м х 0,53 мм, НФ OV-1;
программирование температуры от 60°С до 350°С со скоростью 15 град/мин; температура испарителя
350°С.
потока не позволяет проводить количественный анализ с высокой точностью.
II. Метод ввода пробы без делителя потока
(splitless injection),
Метод ввода пробы без делителя потока, при котором весь образец целиком попадает в
25
лекарственных препаратов. Промышленную
хроматографию используют для контроля и
регулирования различных технологических
процессов.
Качественный анализ основывается на сопоставлении времен удерживания компонентов
смеси с веществами, выделенными из данной смеси
(или полученными другим путем), которые затем были идентифицированы иными независимыми методами.
Успешная идентификация проводится при сочетании хроматографии с некоторыми спектральными методами. Большое значение
приобретает хромато-масс-спектрометрия,
соединение хроматографии с УФ и ИК спектроскопией и некоторыми другими методами.
Широко используется идентификация, основанная
на:
-применении специальных детектирующих систем;
-применении корреляционных зависимостей параметров удерживания от физических или физикохимических свойств веществ.
Наиболее полно такие зависимости изучены, например, для температуры кипения, показателя преломления, молекулярной рефракции, числа
функциональных групп.
Количественный анализ.
Количественный анализ основывается на измерении площади хроматографического пика S или
его высоты h, которые пропорциональны
концентрации определяемого вещества.
Обычно используют один из трех методов обработки результатов анализа. Это методы нормировки, внешнего и внутреннего стандарта.
Метод нормировки. Нормировка в
http://www.mitht.ru/e-library

26
хроматографическом анализе представляет собой расчет массовой доли каждого компонента смеси xi путем деления площади соответствующего пика Si на
сумму площадей всех пиков. При этом считают, что из хроматографа выходят все компоненты смеси и чувствительность детектора к каждому их них одинакова:
xi% nSi 100%
Si ,
i 1
где n – число компонентов в смеси.
Часто чувствительность детектора к различным
соединениям неодинакова. Эту особенность
учитывают вводя, поправочные коэффициенты i. В этом случае массовую долю рассчитывают по формуле:
xi% nSi i 100Si i
i 1
При анализе многокомпонентных смесей обычно возникает ситуация, когда провести градуировку по всем компонентам смеси невозможно. Поэтому
приобретает актуальность априорная оценка поправочных коэффициентов. Такая оценка основана
на анализе механизма детектирования и учета особенностей химического строения анализируемых
веществ. Так, экспериментально установлено, что сигнал пламенно-ионизационного детектора при анализе углеводородов пропорционален числу атомов углерода в молекуле.
Метод внешнего стандарта (абсолютной калибровки) заключается в построении графической
51
При вводе пробы «холодной» иглой ее охлаждают во
время ввода пробы. Предложено специальное
устройство, в котором игла шприца, находясь во
входной трубке, охлаждается холодным воздухом или газообразным диоксидом углерода. В горячей камере испарителя находится только кончик иглы 2-3 мм. Стеклянный вкладыш не охлаждается, так как узел охлаждения тщательно теплоизолирован. Этот метод позволяет избежать селективного испарения
легкокипящих компонентов пробы из иглы.
Быстрый ввод пробы позволяет получить воспроизводимые результаты. Чтобы избежать
селективного испарения легкокипящих компонентов
пробы из иглы, необходимо очень быстро ввести пробу. Это следует провести так, чтобы ввод иглы, впрыскивание пробы и удаление иглы, было осуществлено за время недостаточное для нагрева иглы. При этом испарения легкокипящих компонентов пробы из иглы не происходит.
Продолжительность нахождения иглы в испарителе не должна превышать 500 мс (рис. 19). Это условие практически неосуществимо вручную.
Это следует из результатов сравнения автоматического и ручного ввода пробы в
капиллярную колонку. Все полученные данные
соотнесены с результатами холодного ввода этой же пробы непосредственно в колонку, так как в этом слу-
чае не наблюдается фракционирования ни в игле, ни
в устройстве ввода пробы.
Из приведенных результатов следует, что при использовании систем с делителем потока
проведение достоверного количественного анализа возможно, хотя и сопряжено с рядом трудностей. Однако в некоторых случаях ввод пробы с делителем
http://www.mitht.ru/e-library
16
и дисперсию 2. При рассмотрении рис.1 мы
воспользовались теорией теоретических тарелок,
предложенной Мартином и Сингом. По этой теории (смотри рис.1) движение вещества вдоль колонки можно представить как последовательный его перенос c одной теоретической тарелки на другую.
Число теоретических тарелок n равно отношению длины колонки L к высоте, эквивалентной
теоретической тарелке Н:
n = L/H |
(8) |
Высота, эквивалентная теоретической |
тарелке |
(ВЭТТ), связана с дисперсией пика в колонке 2 ,
которая выражена в единицах длины 2L или времени
2t..
|
|
2 |
|
|
2 |
|
H |
L |
или H |
t |
( 9) |
||
|
|
2 |
||||
|
L |
t |
||||
|
|
|
R |
|
ВЭТТ имеет размерность длины. Уменьшение ВЭТТ (8) позволяет, повысить эффективность
колонки.
Для пика гауссовой формы (рис.2)
справедливо равенство |
4 t . Подставляя |
в |
||||||
уравнение (9), получим: |
|
|
||||||
|
|
|
2 L |
|
|
|||
H |
|
|
|
|
|
. |
(10) |
|
1 6 t |
2 |
|||||||
|
R |
|
|
|||||
Число теоретических тарелок равно |
|
|||||||
|
t |
R |
2 |
|
|
|||
n 16 |
|
|
|
. |
|
(11) |
||
|
|
|
||||||
|
|
|
|
|
||||
Эта величина может быть рассчитана
непосредственно из хроматограммы. Однако в этом
случае легче измерить ширину пика на половине
27
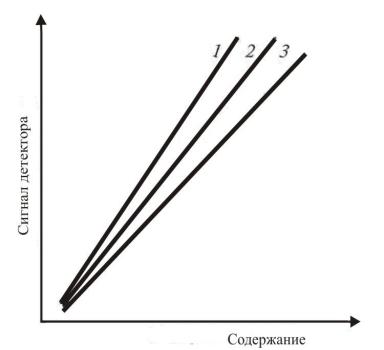
зависимости площади (высоты) пиков от количества
(концентрации) вещества в смеси (рис. 8). При
анализе смеси из i компонентов строят число
графиков, равное их числу. Метод является наиболее точным, надежным при анализе малых концентраций. При его использовании необходимо точно вводить пробу и строго соблюдать условия хроматографирования при калибровке и определении анализируемого вещества.
Рисунок 8. Зависимость сигнала (площади пика) S от содержания компонента % (метод внешнего стандарта) для соединений 1, 2 и 3.
Метод внутреннего стандарта (относительной
http://www.mitht.ru/e-library
28
калибровки). По хроматограммам специально
приготовленных смесей с известным соотношением
анализируемого и стандартного веществ находят
площади пиков и рассчитывают их отношение. Для каждого вещества строят график зависимости этого отношения (S/Sст) от величины массового соотношения компонента и стандартного вещества в смеси. При анализе смеси неизвестного состава к ней добавляют точное количество стандартного вещества и хроматографируют.
Измеряют площади. Находят отношение площадей пиков и рассчитывают массу определяемого
вещества. Этот метод позволяет получить
правильные и воспроизводимые результаты. Он применим, если на хроматограмме отсутствует
значительная часть пиков компонентов
анализируемой смеси.
5. Газовая хроматография
Бурное развитие газовой хроматографии началось с 1952 года, после того как Мартин и Джеймс
осуществили газожидкостной вариант, использовав
смешанную неподвижную фазу, состоящую из стеариновой кислоты и полиметилфенилсилоксана.
Это позволило сразу включить широкий набор жидкостей вместо ограниченного круга адсорбентов.
Кроме того, линейность изотермы распределения анализируемых веществ (постоянство времени
удерживания) позволила проводить надежную
идентификацию даже очень сложных смесей.
Примерно в это же время целый ряд фирм начал серийный выпуск газовых хроматографов. Принципиальная схема хроматографической
установки приведена на рис.7.
5.1. Газожидкостная хроматография.
49
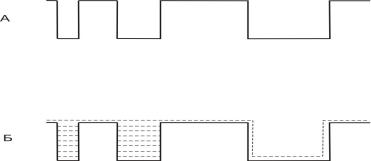
меньший поступает в капиллярную колонку, а
больший — сбрасывается. Если гомогенизация
полная, то образец будет делиться в отношении,
определяемом скоростями двух указанных потоков. Соотношение этих потоков называют отношением деления. На практике используют делители с отношением деления от 1:10 до 1:1000. Конструкция делителя должна обеспечивать в процессе ввода строгое постоянство отношения деления.
Однако на практике происходит фракционирование
компонентов образца. Делитель разделяет компоненты смеси не в одной и той же пропорции.
Происходит селективное испарение молекул
различного размера, что приводит к дискриминации компонентов (рис.18).
Рисунок 18. Дискриминация смеси н-алканов в н- гексане при вводе пробы c делением потока. Коэффициент деления потока 1:40.
Дискриминация пробы при вводе с делителем потока
обусловлена как характеристиками устройства вода,
так и условиями ввода, например работой шприца. Нелинейность делителя потока обусловлена
различной летучестью, полярностью и концентрацией компонентов смеси.
Для преодоления дискриминации пробы при вводе с
http://www.mitht.ru/e-library
48
капиллярную колонку нежелательно вводить пробу бо-
лее 1-10-5 г, иначе эффективность ее сильно
уменьшится.
Обычно это достигается при вводе пробы с делением потока.
I.Ввод пробы с делением потока (split injection).
Проба в количестве (0,1 —1,0 мкл) через мембрану подается в камеру испарения (рис.17),
Рисунок 17. Схема устройства ввода пробы с делителем потока.
1 - мембрана; 2,6 – вентиль тонкой регулировки; 3 - газ-носитель; 4 – камера испарения; 5 – деление потока; 7– капиллярная колонка.
представляющую собой вкладыш из стекла или
кварца. В камере испарения парообразная проба смешивается с газом-носителем.
Затем гомогенная смесь паров пробы с газомносителем разделяется на два неравных потока:
29
Разделение анализируемой смеси в ГХ
происходит |
за |
счет |
различия |
констант |
распределения K1 |
и K2, как это показано, например, |
|||
на рис.1. |
При этом приведенный объем |
|||
удерживания, V’R |
прямо |
зависит от |
количества |
|
жидкой фазы Vs в колонке.
Приведенный объем удерживания получается, если из объем удерживания VR вычесть мертвый объем VM.
VR’ = VR - VM = VS K , |
(21) |
где K - коэффициентом распределения. |
|
Объем удерживания VR определяется: |
|
VR = uср tR , |
(22) |
uср – объемная скорость газа-носителя.
Для сорбентов насадочных колонок количество жидкой фазы в колонке получило название степени пропитки и задается как доля жидкой фазы от массы
всего сорбента. Выбор степени пропитки зависит от
особенностей решаемых задач и твердого носителя. Наибольшее распространение получили твердые
носители на основе «диатомитов», которые
образовались в результате отмирания
одноклеточных диатомитовых водорослей. Для
анализа высококипящих соединений используется малый процент неподвижной фазы - 5% (а иногда 3 и
даже 1%). Часто на упаковках с таким сорбентом
ставится надпись: «для высокотемпературной хроматографии».
Вообще, можно нанести на твердый носитель практически любое количество жидкой фазы, но обычно степень пропитки составляет от 3 до 25%.
Сам процесс приготовления сорбента для газожидкостной хроматографии предельно прост. Сначала взвешивают требуемое количество жидкой фазы и твердого носителя. Взвешенное количество
http://www.mitht.ru/e-library
30
жидкой фазы (обычно несколько грамм) растворяют в
50-100 мл низкокипящего растворителя. Готовый
раствор выливают в «выпаривательную» чашку. Туда
же помещают взвешенный твердый носитель так, чтобы полученный раствор полностью покрывал его поверхность. Затем при слабом нагревании и медленном перемешивании ждут полного испарения растворителя. Спустя некоторое время, после того как приготовленный сорбент вновь приобрел
«сыпучесть», его можно засыпать в колонку.
Поверхность твердого носителя представляет
собой набор узких и широких пор, заполненных
жидкостью и участков поверхности, где жидкая фаза вовсе отсутствует (рис. 9).
Твердый носитель должен быть инертным,
сохраняя при этом большую удельную поверхность. К
сожалению, диатомитовые твердые носители содержат ряд примесей, которые приводят к
образованию «хвостов» (смотри рис. 3) и сдвигу
времен удерживания, характерному для выпуклой
изотермы адсорбции.
Рисунок 9. Модель заполнения твердого носителя неподвижной жидкой фазой. А – поверхность твердого носителя без неподвижной жидкой фазы. Б – поверхность твердого носителя, покрытая неподвижной жидкой фазой.
47
диэтиленгликольсукцианатом определялась для пары
компонентов метилстеарат/метилолеат. Температура
колонки 180°С. Газ-носитель – гелий.
Таблица 7. Сравнение параметров трех типов колонок
Параметр |
|
Тип колонки |
|
||
|
|
|
|
|
|
|
Насадочные |
|
SCOT |
|
WCOT |
|
|
|
|
|
|
L, м |
2,4 |
|
15 |
|
100 |
|
|
|
|
|
|
dс, мм |
2,2 |
|
0,5 |
|
0,27 |
|
|
|
|
|
|
|
10 |
|
54 |
|
225 |
|
|
|
|
|
|
k |
58,6 |
|
11,2 |
|
2,7 |
|
|
|
|
|
|
u, см/с |
15,9 |
|
20,3 |
|
41,1 |
|
|
|
|
|
|
Rs |
1,2 |
|
3,8 |
|
6,3 |
|
|
|
|
|
|
t,мин |
15 |
|
15 |
|
15 |
|
|
|
|
|
|
Zt,мин -1/2 |
0,31 |
|
1,0 |
|
1,6 |
|
|
|
|
|
|
Сравнение проводилось при заданном времени для метилолеата равном 15 мин. (k найдено для метилолеата).
Ввод пробы в капиллярную колонку
Уменьшение объема неподвижной фазы в
капиллярной колонке заставляет снизить величину пробы, чтобы избежать перегрузки колонки. Величина
максимальной пробы qmax , которая еще не вызывает
перегрузки колонки, равна: |
|
qmax 0,02 (VM + K VS )/n |
(21) |
где VM и VS - объем газовой и жидкой фаз в объеме
одной тарелки; К – коэффициент распределения; n - число теоретических тарелок колонки.
Приближенный расчет показывает, что в
http://www.mitht.ru/e-library