

методички / 2347_EI
.pdfоказывает отрицательного влияния на работу детектора. Таким газом может быть аргон, азот, гелий, водород, а иногда даже водяной пар. К вспомогательным газам относится воздух, кислород, водород, метан, диоксид углерода и др. [4].
Дозаторы проб. Дозатор является составной частью хроматографа, которая позволяет вводить пробу в газ-носитель. Сначала отмеряют объем или массу пробы, а затем вводят ее в пространство, в котором поддерживается заданная температура и через которое проходит газ-носитель.
Конструкция дозатора должна решаться с таким расчетом, чтобы:
1)вносимая проба занимала минимальный объем колонки;
2)проба быстро переходила в газообразное состояние и поступала в колонку, не изменяя своего состава;
3)данные по временам удерживания воспроизводились с точностью до десятой доли процента, а площади пиков измерялись с отклонением менее 1 %;
4)разделяющая способность колонки не ухудшалась;
5)изменения условий работы колонки не влияли на правильность дозирования [4].
Колонки. Хроматографическая колонка представляет собой трубку с фиксированной неподвижной фазой, через которую протекает подвижная фаза.
Взависимости от расположения неподвижной фазы колонки делятся на
насадочные и капиллярные.
Насадочные колонки наполнены адсорбентом (система «газ – адсорбент») или инертным носителем, смоченным жидкой неподвижной фазой (система «газ – жидкость»). Капиллярные колонки имеют неподвижную фазу, твердую либо жидкую, нанесенную в виде тонкого слоя (толщиной максимум несколько мкм) на внутреннюю стенку капилляра, остальное пространство заполнено газом-носителем.
Насадочные колонки обычно представляют собой трубки длиной от 1 до 5 м и диаметром 2–3 мм. Трубки выполняются из нержавеющей стали, никеля, стекла или пластмасс. Эффективность этих колонок зависит от размеров зерен насадки, от качества микро- и макроповерхности насадки, от способа нанесения неподвижной фазы на носитель и от тщательности набивки трубки сорбентом. Капиллярные колонки имеют диаметр от 50 мкм до 1 мм и длину от единиц до сотен метров.
К адсорбентам относятся все твердые вещества с удельной поверхностью 10 – 103 м2/г. Наиболее подходящими для газовой хроматографии адсорбентами являются такие, у которых удельная поверхность колеблется в пределах 60 – 400 м2/г. К ним относятся активные угли, силикагель, активный оксид алюминия, молекулярные сита и пористые полимеры (порапак и др.).
Под термином носитель подразумевают мелкозернистый материал, на который можно нанести жидкую фазу, причем сам носитель химически почти нейтрален по отношению к разделяемым веществам. Он должен отличаться
11
термостойкостью и не изменять свои свойства даже после добавки кислот или щелочей. Зерна применяемого сорбента должны быть достаточно близки по диаметру, и из сорбента должны быть удалены все мелкие, особенно пылевидные, частицы. Товарные типы носителей поставляются уже просеянными и обработанными, и размер их зерен чаще всего составляет около 0,1 мм.
Капиллярные колонки изготавливают из нержавеющей стали, стекла или кварца и применяют в тех случаях, когда насадочные колонки не позволяют достаточно хорошо разделить компоненты, либо когда для хорошего разделения требуется слишком длительное время. Стеклянные капиллярные колонки получают вытягиванием капилляров из толстостенных трубок с последующей их обработкой; внутреннюю поверхность капилляра обрабатывают соляной кислотой, промывают, сушат, силилируют при высокой температуре (свыше 400 0С) и смачивают раствором неподвижной фазы. Кварцевые капилляры обрабатывают аналогичным способом. Преимущество последних перед стеклянными капиллярами заключается в инертности и гибкости [4].
Детекторы.
Детектор представляет собой устройство, которое реагирует на изменение состава протекающего газа и преобразует эту реакцию в электрически измеряемые величины. Критерии оценки детекторов общеприняты для всех систем детектирования; к ним относятся: чувствительность, отклик, шум, минимально детектируемая концентрация или массовая скорость потока, динамический линейный диапазон отклика, эффективный объем и время отклика.
К наиболее распространенным типам принадлежит универсальный детектор по теплопроводности (катарометр), пламенно-ионизационный детектор, а из селективных детекторов – пламенный фотометрический и термоионизационный, а также детектор электронного захвата. Встречается и фотоионизационный детектор, а в последние годы все чаще встречается комбинация хроматографа с простым масс-спектрометром (mass selective detection).
Детектор по теплопроводности является универсальным недеструктирующим детектором, реагирующим на изменение теплопроводности, возникающее в результате изменения состава газа. В качестве датчиков применяются платиновые, вольфрамовые или позолоченные вольфрамовые волокна в виде спиралек, помещенных в каналы металлического блока, через которые проходит газ-носитель.
12
Таблица 2
Детекторы, применяемые в экологическом анализе [3]
Детекторы |
Аббревиатура |
Селективность |
Область |
|
|
|
|
|
применения |
|
|
Пламенно- |
ПИД |
Универсальный |
Все органические |
||
ионизационный |
|
|
соединения |
|
|
Фотоионизационный |
ФИД |
То же |
То |
же |
(за |
|
|
|
некоторыми |
|
|
|
|
|
исключениями) |
|
|
Электронозахватный |
ЭЗД |
Специфичный |
Галогенсодер- |
|
|
|
|
|
жащие |
|
|
|
|
|
соединения |
|
|
Термоионный |
ТИД |
Селективный |
Соединения |
|
|
|
|
|
азота и фосфора |
||
Пламенно- |
ПФД |
То же |
Соединения серы |
||
фотометрический |
|
|
|
|
|
Хемилюминесцентный- |
ХЛД |
То же |
То же |
|
|
серный |
|
|
|
|
|
Атомно-эмиссионный |
АЭД |
Элемент- |
Соединения |
с |
|
|
|
специфический |
различными |
|
|
|
|
|
функциональным |
||
|
|
|
и группами |
|
|
Электролитический |
ЭЛКД |
Селективный |
Соединения |
|
|
кондуктометрический |
|
|
азота, |
серы |
и |
(Холла) |
|
|
галогенов |
|
|
Масс- |
МСД |
То же |
Все органические |
||
спектрометрический |
|
|
соединения |
|
|
Пламенно-ионизационный детектор – универсальный, применяющийся для анализа почти всех органических соединений. Детектор имеет селективные варианты: пламенный термоионизационный и пламенный фотометрический детекторы. Они имеют незначительный эффективный объем (0,06 – 4 мкл), низкую постоянную времени (единицы миллисекунд), высокую чувствительность ко всем органическим углеродсодержащим соединениям (предел обнаружения до пикограммов) и широкий линейный диапазон (шесть и более порядков, в зависимости от геометрии детектора).
Пламенный термоионизационный детектор отличается от описанного выше типа лишь тем, что в непосредственной близости от пламени помещается соль щелочного металла в виде силикатной чешуйки, укрепленной на платиновой проволочке, либо керамическое колечко, пропитанное раствором соли и закрепленное около горелки. Этот детектор обладает высокой селективностью по отношению к веществу, содержащему в молекуле азот или фосфор и достаточно чувствителен к этим элементам.
13
Пламенный фотометрический детектор – используется как селективный детектор для веществ, содержащих в молекуле серу или фосфор. Его работа основана на измерении излучения пламени при характеристической длине волн.
Детектор электронного захвата селективно обнаруживает все вещества, которые захватывают свободные электроны с образованием стабильных ионов. Чаще всего такой детектор применяют для обнаружения соединений, содержащих галогены, фосфор и серу, но он реагирует также и на NO2 , кислород, озон и водяные пары. Поэтому необходима тщательная предварительная очистка газа-носителя и вспомогательного газа.
Термостат. Все детали и составные части хроматографа, через которые проходит проба, должны термостатироваться.
Колонка помещается в выполненном в виде шкафа термостате, обогреваемом горячим воздухом. Поскольку скорость движения компонентов смеси в колонке экспоненциально зависит от температуры, крайне необходимо, чтобы колонка была хорошо термостатирована по всей своей длине. Термостат с колонкой должен отвечать следующим требованиям:
1)температура должна быть одинаковой по всему пространству термостата и главным образом вдоль всей колонки;
2)температура должна регулироваться в интервале 20 – 400 0С, а в некоторых случаях от – 50 до 400 0С;
3)подвод или съем тепла дозатором и детектором должны быть минимальными;
4)теплопотери должны быть наименьшими;
5)должен обеспечиваться хороший доступ ко всем соединениям, применяемым при установке или замене колонок;
6)термостат должен иметь минимальную общую массу, чтобы любое изменение температуры происходило достаточно быстро [4].
Количественная обработка результатов хроматографического анализа
Сигнал с детектора поступает на регистрирующий прибор. В качестве регистратора могут использоваться самопишущие потенциометры.
Запись сигнала детектора на диаграммной ленте называется хроматограммой. Каждый пик хроматограммы соответствует индивидуаьному веществу или смеси неразделенных веществ.
Параметром хроматограммы, который непосредственно отражает количество данного вещества в анализируемой пробе и наиболее точно воспроизводится при изменении условий хроматографирования, является площадь пика Q, ограниченная контуром пика и продолжением нулевой линии
(рис. 5).
14
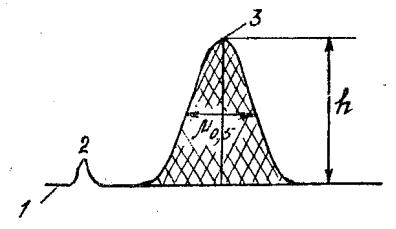
Рис. 5. Основные параметры хроматограммы: 1 – нулевая линия;
2 – пик несорбирующегося вещества; 3 – пик определяемого компонента
Обычно измерение пика проводят в автоматическом режиме с помощью интеграторов, однако, ручная обработка хроматограмм тоже используется. Для этого используют формулу:
Q = h * μ0,5,
где h – высота пика, мм;
h * μ0,5 – ширина пика на уровне половины высоты, мм.
Основными методами количественной обработки хроматограмм являются метод абсолютной градуировки, метод внутреннего стандарта и метод внутренней нормализации [1].
Метод абсолютной градуировки основан на использовании зависимости площади пика Q от количества соответствующего сорбата в смеси:
Сi = Ka * Q * 100/ q,
где Ka – градуировочный коэффициент; q – величина пробы;
Сi – концентрация данного сорбата в анализируемой пробе, %.
Значение градуировочного коэффициента зависит от условий проведения хроматографического процесса, поэтому градуировку проводят в тех же условиях, что и разделение анализируемой смеси. Для получения точной градуировки исследуют не менее 10 эталонных смесей с известной концентрацией сорбата.
Метод абсолютной градуировки прост, но довольно трудоемок, его точность зависит от постоянства режима хроматографирования и тщательности приготовления эталонных градуировочных смесей, а также воспроизводимости размера вводимой пробы. Этот метод является основным при определении микропримесей.
Метод внутреннего стандарта основан на введении в анализируемую смесь определенного количества стандартного вещества (вещества сравнения). Содержание исследуемого сорбата в смеси Сi , % масс вычисляют по формуле:
15
Сi =100 r Кi Qi /КстQст,
где Кi и Кст – относительные поправочные коэффициенты к площади пиков i- компонента и внутреннего стандарта;
Qi и Qст – площади соответствующих пиков;
r – отношение массы внутреннего стандарта к массе анализируемой смеси. Относительные поправочные коэффициенты определяют на основании анализа смеси стандарта с данным компонентом с известной концентрацией. Для стандартного вещества поправочный коэффициент обычно принимают
равным 1. в этом случае расчет Сi выполняют по формуле:
Сi =100 r Кi Qi /Qст.
Относительные поправочные коэффициенты к площадям пиков мало зависят от параметров процесса, поэтому имеются многочисленные справочные данные по относительным поправочным коэффициентам различных классов соединений для наиболее широко применяемых в ГХ детекторов. Обычно стандартным веществом выбирается бензол.
Метод внутреннего стандарта особенно удобен в тех случаях, когда отсутствуют чистые вещества для приготовления эталонных смесей. Использование табличных значений относительных поправочных коэффициентов также значительно сокращает трудоемкость анализа. Основная трудоемкость данного метода заключается в выборе и точной дозировке стандартного вещества.
Метод внутренней нормализации основан на определении соотношений между концентрациями компонентов смеси. Поэтому необходимым условием определения содержания какого-либо вещества в смеси является регистрация всех компонентов. Расчет состоит в приведении к 100 % суммы произведений площадей пиков Q на относительные поправочные коэффициенты K, обусловленные чувствительностью детектора к данному компоненту:
n
Ci = 100% Ki Qi / ∑ (Ki Qi).
i=1
Метод внутренней нормализации также часто осуществляют без специальной градуировки с использованием литературных данных по относительным поправочным коэффициентам.
Достоинство метода внутренней нормализации заключается в том, что искажения, имеющиеся в одинаковой степени у всех пиков, в конечном счете не влияют на точность результатов. Ошибки метода связаны с постепенным изменением режима в процессе анализа [1].
16

ПРОВЕДЕНИЕ АНАЛИЗА СМЕСИ УГЛЕВОДОРОДОВ
Для проведения опыта берут смесь известных газов. В данном случае, поскольку колонку подбирается так, чтобы произошло разделение всех веществ, можно воспользоваться методом внутренней нормализации.
Анализируемую смесь нормальных парафинов и ароматических углеводородов вводят с помощью микрошприца в нагретый испаритель хроматографа, соединенный с рабочей колонкой. Результаты хроматографического разделения регистрируют на диаграммной ленте самописца. Одновременно с помощью секундомера измеряют время от момента ввода пробы до момента выхода максимума каждого пика. Хроматографирование анализируемой смеси проводят до 10 раз. Выпадающие результаты выбраковываются, оставшиеся заносят в таблицу журнала опыта.
Затем в хроматографическую колонку вводят 3–5 раз пробу чистого бензола и пробу несорбирующего газа (воздух), каждый раз фиксируя по секундомеру время удерживания.
Обработку результатов начинают с идентификации всех хроматографических пиков. Первым идентифицируется бензол. Ему соответствует тот пик, для которого время удерживания соответствует времени удерживания чистого бензола. Бензол является стандартным веществом, относительно которого для всех остальных пиков рассчитывается относительное удерживание.
Идентификацию остальных пиков хроматограммы проводят путем сравнения рассчитанных значений с табличными [1].
Риc. 6. Хроматограмма смеси н-парафинов С6 – С10 и ароматических углеводородов C6 – C8 :
1 – н-гексан, 2 – н-гептан, 3 – н-октан, 4 – н-нонан, 5 – н-декан, 6 – бензол, 7 – толуол, 8 – м-ксилол
17
После того, как будет установлено соответствие пиков индивидуальным соединениям, проводят расчет площадей пиков для каждого компонента смеси. Для этого линейкой измеряют высоту пика от нулевой линии до максимума пика h, затем делят данный отрезок пополам и измеряют ширину пика на половине
его высоты μ0,5 .
Площадь пика рассчитывается как произведение
Q = h * μ0,5,
где h – высота пика, мм;
h * μ0,5 – ширина пика на уровне половины высоты, мм.
Расчет площадей пиков проводят для всех хроматограмм, отобранных в ходе анализа (прил. 1). Все рассчитанные площади пиков заносят в табл. 4.
Пользуясь литературными данными по относительным поправочным коэффициентам, приведенным в табл. 3, для каждого пика рассчитывают произведение KiQi и затем для каждой хроматограммы находят сумму произведений ∑ (Ki Qi) для всех пиков. Полученные данные заносят в табл. 4.
Концентрацию каждого компонента, % масс, находят для каждой повторности по формуле:
n
Ci = 100% Ki Qi / ∑ (Ki Qi).
i=1
Итоговую концентрацию каждого компонента в анализируемой смеси находят как среднее арифметическое значений повторностей.
Таблица 3
Значения относительного удерживания r и относительных поправочных
коэффициентов Кв |
для нормальных парафинов С6 – С10 |
||
и ароматических углеводородов C6 – C8 (стандарт - бензол) |
|||
|
|
|
|
Сорбат |
|
r |
Квi |
н-гексан |
|
0,06 |
0,89 |
н-гептан |
|
0,11 |
0,91 |
н-октан |
|
0,19 |
0,94 |
н-нонан |
|
0,31 |
0,95 |
н-декан |
|
0,52 |
0,95 |
бензол |
|
1,00 |
1,00 |
толуол |
|
1,62 |
0,99 |
м-ксилол |
|
2,55 |
1,03 |
18
Таблица 4
Количественное содержание компонентов в анализируемой смеси
Сорбаты |
н- |
н- |
н-октан |
н- |
н- |
бензол |
толуол |
м- |
||
|
|
|
гексан |
гептан |
|
нонан |
декан |
|
|
ксилол |
|
|
|
|
|
|
Повторность 1 |
|
|
|
|
Qi |
|
|
|
|
|
|
|
|
|
|
Ki |
Qi |
|
|
|
|
|
|
|
|
|
∑ Ki Qi |
|
|
|
|
|
|
|
|
|
|
Ci |
, |
% |
|
|
|
|
|
|
|
|
масс |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Повторность 2 |
|
|
|
|
Qi |
|
|
|
|
|
|
|
|
|
|
Ki |
Qi |
|
|
|
|
|
|
|
|
|
∑ Ki Qi |
|
|
|
|
|
|
|
|
|
|
Ci |
, |
% |
|
|
|
|
|
|
|
|
масс |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Повторность 3 |
|
|
|
|
Qi |
|
|
|
|
|
|
|
|
|
|
Ki |
Qi |
|
|
|
|
|
|
|
|
|
∑ Ki Qi |
|
|
|
|
|
|
|
|
|
|
Ci |
, |
% |
|
|
|
|
|
|
|
|
масс |
|
|
|
|
|
|
|
|
|
|
Ci |
ср. |
|
|
|
|
|
|
|
|
|
Вопросы для самоподготовки
1.Перечислите источники химического загрязнения воздушной среды железнодорожным транспортом.
2.В чем заключается хроматографический метод анализа смесей? Каковы его преимущества?
3.Опишите принципиальную схему газового хроматографа.
4.Какие типы хроматографических детекторов вам известны? Какие загрязнители можно определять с их помощью?
5.Какие вещества называются сорбентами и сорбатами? Что такое хроматограмма? Как рассчитать площадь хроматографического пика?
6.Какие приемы используют при определении качественного и количественного состава загрязнителей в анализируемой пробе?
19
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1.Анфилофьев Б.А., Сеницкая Г.Б. Методические указания к выполнению лабораторной работы «Исследование загрязнений воздушной среды газохроматографическим методом» по дисциплине «Безопасность жизнедеятельности». – Самара, 1993. – 21 с.
2.Гольберт К.А., Вигдергауз М.С. Введение в газовую хроматографию. –
М. : Химия, 1990. – 352 с.
3.Дугов Ю.С., Родин А.А. Газохроматографический анализ загрязненного воздуха : Практическое руководство. – М. : БИНОМ. Лаборатория знаний, 2006. – 628 с.
4.Крейчи М., Паюрек Я., Комерс Р. и др. Вычисления и величины в сорбционной колончатой хроматографии : Пер. с чешск. – М.: Мир, 1993. – 208 с.
5.Крупенио Н.Н. Экологический мониторинг и контроль транспортных систем : Учеб.пособие для вузов ж.д. транспорта. – М.: Маршрут, 2006. – 133 с.
6.Лозановская И.Н., Орлов Д.С., Садовникова Л.К. Экология и охрана биосферы при химическом загрязнении : Учеб. пособие. – М.: Высш.шк. – 1998. – 287 с.
7.Суворов С.В., Штеренгарц Р.Я. Вредные вещества на железнодорожном транспорте. – М.: Транспорт, 1986. – 176 с.
20