

учебник / Maydannik_V_G_Pediatria
.pdfВторичная гипероксалурия — патология обмена, обусловленная гастроинтестинальной гиперабсорбцией щавелевой кислоты из богатых ею пищевых продуктов, а также экзоили эндогенным дефицитом витаминов В, или В6, являющихся кофакторами ферментов, осуществляющих метаболизм глиоксилата. Локальное образование оксалатов в почках возможно в связи с разрушением мембранных фосфолипидов на фоне ишемии почек. Морфологически выявляют деструкцию щеточных каемок в канальцах нефрона, лимфогистиоцитарную инфильтрацию в интерстиции почек.
Основные клинические симптомы первичной гипероксалурии обнаруживаются в раннем возрасте. Заболевание обычно проявляется у двух третей детей в возрасте 3—5 лет. Первичная гипероксалурия составляет до 2% всех случаев уролитиаза у детей. Мальчики болеют несколько чаще девочек.
Выраженность клинических проявлений заболевания определяется активностью нарушенного метаболизма глиоксаловой кислоты и процессов кальцификации, наличием оксалатно-кальциевого уролитиаза и препятствия оттоку мочи.
Больные жалуются на боли внизу живота или в области поясницы, особенно во время мочеиспускания. Болевой синдром обусловлен раздражением нервных окончаний слизистой оболочки лоханок, мочеточников или мочевого пузыря оксалатными кристаллами или камнями. Боль в виде почечной колики является следствием острой задержки мочи (механическая закупорка или спазм лоханки и мочеточника). Почечная колика у детей часто сопровождается рефлекторным парезом кишок — метеоризмом, тошнотой, рвотой, напряжением передней брюшной стенки. Длительность болевого приступа зависит от времени передвижения оксалатного песка или камня по суженным участкам мочевых путей — от нескольких часов до суток. Постоянным симптомом является эритроцитурия, или гематурия, которая отмечается во всех фазах заболевания, за исключением полной закупорки мочеточника, и обычно усиливается при движениях. При наслоении мочевой инфекции, что часто бывает при этой патологии, отмечается лейкоцитурия, лихорадка, лейкоцитоз, ускоренная СОЭ.
Диагноз заболевания подтверждается повышенной экскрецией с мочой оксалатов, которая превышает 40 мг/сут и достигает до 100—400 мг/сут (или 1110—4440 мкмоль/сут на стандартную поверхность тела). Кроме того, при I типе первичной гипероксалурии, наряду с повышенной экскрецией оксалатов, выявляется избыточное количество глиоксилата до 100 мг/сут и более (при норме до 5 мг/сут) и повышенная экскреция гликолата до 100 мг/сут (при норме до 15 мг/сут). При II типе первичной гипероксалурии в моче появляется в избыточном количестве L-гли- церат (до 300—600 мг/сут), тогда как гликолат в моче практически отсутствует.
С помощью сонографии в чашечно-лоханочной системе почек выявляются скопления солей, а при сформированном камне обнаруживается локальное расширение лоханки или чашки с наличием эхопозитивного образования, что подтверждается рентгенологическим выявлением тени оксалата кальция в просвете лоханки почки или в почечной паренхиме.
691
Клинико-лабораторные критерии диагностики вторичной гипероксалурии те же, что и первичной гипероксалурии.
Лечить больных с оксалатной нефропатией довольно сложно. Прежде всего необходимо назначить картофельно-капустную диету. Ее использование уменьшает функциональную нагрузку на тубулярный аппарат. Предусматривается исключение из рациона питания продуктов, • богатых оксалатами, — какао, шоколада, щавеля, шпината, петрушки, свеклы и др. Вместо них целесообразно употреблять капусту, горох, тыкву, грибы, огурцы, в которых содержится мало оксалатов. Не показаны экстрактивные бульоны. Разрешается белый хлеб, свежее свиное сало, растительное и сливочное масло, сметана, мясо в отварном виде. Организм «подщелачивают» введением груш, чернослива и кураги. Соотношение белков, жиров, углеводов сохраняется в пределах возрастной нормы. Увеличивают жидкость до 2 л на стандартную поверхность тела. Важно обеспечить высокожидкостный режим в ночное время, когда моча более концентрирована и создаются благоприятные условия для кристаллизации солей. Две-три недели картофельно-капустная диета достоверно снижает экскрецию оксалатов.
Снижение синтеза и экскреции оксалатов достигается назначением небольших доз окиси магния (0,15—2 г/сут), витамина В6 в больших дозах (до 400 мг/сут), а также применением ортофосфата и синтетических аналогов неорганических пирофосфатов (оксиэтилиден-дифосфоновая кислота). Продолжительность курсов лечения определяется индивидуально с учетом динамики заболевания и достижения нормализации метаболизма щавелевой кислоты. В среднем она составляет 4—6 недель.
Показано применение мембраностабилизаторов и антиоксидантов, таких как димефосфон (30—50 мг на 1 кг в сутки 2—3 недели), эссенциале, унитиол, липоевая кислота, витамин А и Ε и др. Широко используется фитотерапия.
Нередко у детей встречается УРАТНАЯ НЕФРОПАТИЯ, которая характеризуется интерстициальным нефритом и развитием нефролииаза, возникающим при нарушении пуринового обмена с накоплением и избыточным выведением через почки мочевой кислоты и ее солей (уратов). По сводным данным, уратные нефропатии являются причиной формирования нефро- и уролитиаза у 1,3—7,6% детей с мочекаменной болезнью.
Взависимости от механизмов происхождения различают первичные
ивторичные уратные нефропатии. Первичные нефропатии возникают вследствие наследственно обусловленного дефекта метаболизма мочевой кислоты, а вторичные являются осложнением эритремии, миеломной болезни, хронической гемолитической анемии, лекарственной терапии тиазидовыми диуретиками, цитостатиками, салицилатами и др.
Нарушения пуринового обмена, приводящие к уратным нефропатиям, довольно сложны, и выявляется несколько механизмов, которые лежат в их основе. В частности, уратные нефропатии возникают в результате
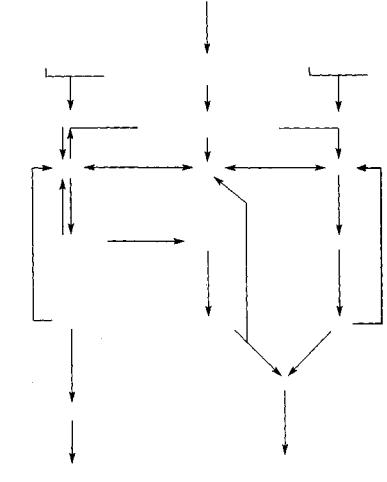
(рис. 85):
а) высокой активности пурин-фосфорибозил-пирофосфат синтетазы (ПФПС), обеспечивающей перенос пирофосфатной группы с АТФ на ри-
692
|
|
Рибозо-5-фосфат + АТФ |
|
|
|
|
ПФПС |
|
|
цАМФ |
РНК |
5-фосфорибозил |
цГМФ |
РНК |
|
|
|
|
|
|
|
1-пирофосфат |
|
|
АТФ днк
АМФ
5-НТ
АД
Аденозин
АФРТ
Аденин
КО
8-Гидроксиаденин КО
2,8-Дигидроксиаденин
5-фосфори- |
ДНК |
ГТФ |
|
бозиламин |
|
|
|
ИМФ |
|
ГМФ |
|
|
5-НТ |
|
|
Инозин |
|
Гуанозин |
|
НЗ |
ГФРТ |
НЗ |
ГФРТ |
Гипоксантин |
|
Гуанин |
|
КО |
|
ГД |
|
|
Ксантин |
|
|
|
КО |
|
|
Мочевая
кислота
Рис. 85. Схема метаболизма пуринов в норме и патологии:
КО — ксантиноксидаза, ГФРТ — гипоксантин-гуанин-фосфорибозил трансфераза, АФРТ— аденин-фосфорибозил трансфераза,
ПФПС — пурин-фосфорибозил-пирофосфат синтетаза, ГД — гуанин-дезаминаза, НЗ — нуклеозидаза,
5-НТ — 5-нуклеотидаза, АД — аденозин-дезаминаза
бозо-5-фосфат с образованием 5-фосфорибозил-I-пирофосфата, который служит основой образования инозинмонофосфата и в последующем — мочевой кислоты. Этот механизм является наследственно обусловленным (описано более 20 семей) и связан с дефектом генов, локализованных в Х-хро- мосоме;
693
б) низкой активности фермента аденин-фосфорибозил трансферазы (АФРТ), который обеспечивает превращение аденина в аденозинмонофосфат, что способствует накоплению 2,8-дигидроксиаденина, обладающего нефротоксичностью и формирующего мочевые камни. Указанный дефект наследуется по аутосомно-рецессивному типу и обусловлен дефектными генами, которые находятся в 16-й хромосоме;
в) отсутствия фермента гипоксантин-гуанин-фосфорибозил трансферазы (ГФРТ), который катализирует превращение гипоксантина в инозинмонофосфат и гуанина — в гуанозинмонофосфат. Это ведет к гиперпродукции ксантина (из гипоксантина и гуанина) и мочевой кислоты (синдром Леша—Нихана, по имени авторов, описавших его в 1964 году). Указанный генетический дефект сцеплен с полом (болеют только мальчики), поскольку дефектные гены локализуются в Х-хромосоме.
Одним из проявлений нарушения пуринового обмена является ксантинурия, которая также часто ведет к нефро- и уролитиазу. Это патологическое состояние является одним из патогенетических вариантов уратных нефропатий. Ксантинурия может иметь наследственное и приобретенное происхождение. Наследственная ксантинурия обусловлена врожденным дефектом активности фермента ксантиноксидазы, который катализирует превращение гипоксантина в ксантин и далее — в мочевую кислоту, а также аденина — в 8-гидроксиаденин и 2,8-дигидрокси- аденин. Дефектные гены, контролирующие активность ксантиноксидазы, локализуются во 2-й хромосоме. Приобретенная низкая активность ксантиноксидазы может быть обусловлена длительным приемом аллопуринола, например у больным синдромом Леша—Нихана.
Выраженный и стойкий дефицит указанных выше энзимов приводит к повышенной продукции пуриновых оснований и гиперурикемии. Повышению биосинтеза пуриновых оснований способствует также избыточное питание. Накопление кристаллов мочевой кислоты в организме ведет к их отложению в интерстициальной ткани мозгового слоя, канальцевой системе, особенно в области петель нефрона, чашечно-лоханочном аппарате. При этом стенка канальцев повреждается, и выпавшие кристаллы проникают в интерстициальную ткань, накапливаются в ней и вызывают развитие интерстициального нефрита. При кислой реакции мочи могут формироваться уратные камни. Следствием высокой степени насыщения пуринами является уменьшение почек в размерах.
Макроскопически субкапсулярная поверхность почек бугристая и покрыта мелкими рубцами. Световая микроскопия выявляет гиалиноз клубочков, явления атрофии, дилятации или регенерации в канальцах, фиброз и лимфогистиоцитарную инфильтрацию в интерстициальной ткани.
Энзиматические дефекты пуринового обмена выявляются в раннем возрасте, но нередко уратная нефропатия длительное время может протекать бессимптомно. Больные с уратной нефропатией предъявляют жалобы на дизурию, боли в животе (по ходу мочеточников, над лобком), которые могут носить приступообразный характер. В моче выявляется кристаллурия, небольшая протеинурия с цилиндрурией, эритроцитурия, иногда — до степени гематурии и лейкоцитурия.
694
ta Синдром Леша—Нихана выявляется исключительно у мальчиков с первых недель жизни. Заболевание проявляется значительной кристаллурией (ураты), повышением уровня мочевой кислоты в крови и ее экскреции с мочой в 2—4 раза по сравнению с нормой. Дети с таким генетическим дефектом по мере их роста страдают умственной отсталостью и неврологической симптоматикой (агрессивность, атетоз, судороги и др.). Очень часто течение заболевания осложнется инфекцией верхних дыхательных и мочевых путей.
Ксантинурия у 40% детей клинически проявляется классическими симптомами — болезненностью, гематурией, кристаллурией (кристаллы ксантина в виде бесцветных ромбов), почечной коликой, инфекцией мочевых путей. У части больных наблюдается рвота, диарея или рецидивирующая инфекция.
Нередко течение уратной нефропатии осложняется возникновением инфекции мочевых путей, а также могут обнаруживаться мочевые камни, что приводит к постренальной обструкции, острой олигурической почечной недостаточности. По мере прогрессирования склеротических изменений в почках снижается их концентрационная способность, появляются признаки хронической почечной недостаточности.
Диагноз уратной нефропатии подтверждают исследованием содержания в крови и моче мочевой кислоты и других продуктов пуринового обмена. У детей на фоне выраженной уратной нефропатии всегда имеет место гиперурикемия (более 0,350 ммоль/л) и гиперурикозурия (более 4 ммоль/л). При ксантинурии у больных повышен уровень ксантина в плазме крови (более 1 мкмоль/л).
При лечении больным с уратными нефропатиями назначается мо- лочно-растительная диета, исключающая продукты, богатые пуриновыми основаниями (печенка, почки, мозги, мясные бульоны, сельдь, паштет, шпроты, горох, бобы, фасоль, орехи, какао). Рекомендуется вводить в
рацион питания фрукты, крупы, овощи (картофель), молоко, рис, яйца, в которых нет пуринов. Важно включать в диету лимоны, цитратные смеси, бикарбонат натрия. Такая диета способствует предупреждению образования солей уратов и уратных камней. При тяжелом мочекислом кризе назначают разгрузочную диету — фруктовую, картофельно-овощ- ную, молочнокислую.
Важно обеспечить достаточное употребление жидкости, ее объем должен составлять до 1—2 л/сут (в зависимости от возраста). Для поддержания рН мочи в пределах 6,2—6,6 следует использовать цитратные препараты (солимок, уралит, магурлит, блемарен и др.), которые обладают значительной буферной емкостью.
При гиперурикемии назначают аллопуринол (ингибитор ксантиноксидазы), который блокирует превращение пуринов в мочевую кислоту. При этом отмечается снижение уровня уратов в крови и моче уже через 24—48 ч после его приема и повышение уровня предшественников уратов (ксантина, гипоксантина). Суточная доза — 5 мг на 1 кг массы тела (но не более 200—300 мг) в 2—3 приема в течение 3—6 месяцев и более. Назначают также колхицин — по 0,5—2 мг в день непрерывными прие-
695
мами в течение многих месяцев (свыше 18 месяцев), который стимулирует продукцию коллагеназы в клетках, секрецию стероидных гормонов, подавляет транспорт пуриновых оснований, что приводит к снижению скорости обмена пуринов и содержания их в организме. Урикозурическим и урикостатическим действием обладает также бензобромарон, который назначают по 50—100 мг/сут на стандартную поверхность тела 2—3 раза в день в сочетании с салуретиками и цитратом натрия для поддержания рН мочи в пределах 6,2—6,6. В комплексном лечении используют оротовую кислоту в дозах 2—6 мг/сут, которая снижает уровень мочевой кислоты в крови за счет урикозурического эффекта.
В заключение следует подчеркнуть, что за детьми из семей, у которых имеется наследственная предрасположенность к мочекаменной болезни, в целях профилактики кальциевого и уратного нефролитиаза рекомендуется диспансерное наблюдение.
ТУБУЛОПАТИИ
Тубулопатии — это заболевания, которые обусловлены стойкими нарушениями мембранного транспорта различных веществ в почечных канальцах. В зависимости от причин, приводящих к развитию тубулопатии, последние делят на первичные (наследственные) и вторичные (табл. 105). Первичные тубулопатии могут быть вызваны: наследственно обусловленной недостаточностью ферментных систем, обеспечивающих активный мембранный транспорт; снижением чувствительности клеток эпителия канальцев к действию гормонов; изменением структуры мембранных белков-носителей; нарушением структуры цитомембран клеток. Вторичные тубулопатии развиваются при воспалительных заболеваниях почек, при наследственных и приобретенных болезнях обмена, медикаментозном повреждении.
Рассмотрим наиболее часто встречающиеся тубулопатии в детском возрасте.
НАСЛЕДСТВЕННЫЙ ФОСФАТ-ДИАБЕТ (ВИТАМИН-О-РЕЗИС- ТЕНТНЫЙ РАХИТ). Это гетерогенная группа наследственно обусловленных заболеваний с нарушением метаболизма витамина D. В основе заболевания лежит врожденный дефект фосфатного гомеостазиса, проявляющегося нарушением всасывания фосфатов в проксимальных канальцах почек. В патогенезе данной патологии придают значение нарушениям структуры белков, участвующих в активном транспорте фосфатов, повышенной чувствительности эпителия канальцев почек к действию паратгормона, первичному дефекту транспорта неорганических фосфатов в почках, а также синтезу в организме метаболитов витамина D.
ПАТОГЕНЕЗ. Обсуждается несколько механизмов развития: наследственное нарушение метаболизма витамина D в печени и почках; нару-
696

Таблица 105
Классификация тубулопатий по локализации дефекта
|
(Ю.Е. Вельтищев, |
1982) |
|
|
|
|
|
Локализация |
Тубулопатий |
||
|
|
|
|
поражения |
первичные |
|
вторичные |
|
|
||
|
|
|
|
ПроксимальБолезнь де Тони—Дебре— |
|
Цистиноз, синдром Лоу, |
|
ные каналь- |
Фанкони, глюкозаминовый |
|
тирозинемия, галактоземия, |
цы |
диабет, глюкозурия, фосфат- |
|
гликогенозы, наследствен- |
|
диабет, аминоацидурия |
|
ная непереносимость |
|
(цистинурия, аминоглицину- |
|
фруктозы, при отравлениях |
|
рия, болезнь Гартнапа, гли- |
|
солями тяжелых металлов, |
|
цинурия), почечный тубуляр- |
|
лизолом, крезолом, тетра- |
|
ный ацидоз, тип II |
|
циклином и др., болезнь |
|
|
|
Вильсона—Коновалова, пер- |
|
|
|
вичный гиперпаратиреои- |
|
|
|
дизм, гипофосфатазия, |
|
|
|
целиакия, синдром Олпор- |
|
|
|
та, первичная гипероксалу- |
|
|
|
рия, сахарный диабет, |
|
|
|
ксантинурия |
Дистальные |
Почечный несахарный диабет, |
|
Пиелонефрит |
извитые ка- |
почечный тубулярный ацидоз, |
|
|
нальцы и |
тип 1, псевдогипоаль- |
|
|
собиратель- |
достеронизм |
|
|
ные протоки |
|
|
|
Общее по- |
— |
|
ХПН, нефронофтиз Фанкони |
вреждение |
|
|
|
канальцевого аппарата
шение структуры белков, участвующих в транспорте фосфатов в канальцах; первичный дефект транспорта фосфатов в кишечнике и проксимальных канальцах почек. Предполагается, что при Х-сцепленном гипофосфатемическом рахите нарушается регуляция активности 1-аль- фа-гидроксилазы фосфатом, что свидетельствует о дефекте синтеза 1,25-(OH)2-D3 в дополнение к уже известному дефекту в транспорте фосфата. Вследствие дефицита фосфора у ребенка с раннего возраста развиваются рахитоподобные изменения скелета.
При первых двух формах фосфат-диабета имеется изолированное нарушение реабсорбции фосфатов в проксимальных канальцах. При третьем типе — гипофосфатемия, гипокальциемия и дефицит фосфора и кальция в экстрацеллюлярной жидкости кости, по-видимому, обусловлены снижением абсорбции в кишечнике или дефектом механизма прямого влияния витамина D на специфические рецепторы кости вследствие нарушения метаболизма витамина D.
697
Фосфат-диабет (гипофосфатемическая тубулонатия, семейная гипофосфатемия, наследственный фосфатный почечный диабет, почечный фосфатный диабет, семейный персистирующий фосфатный диабет, ренальный тубулярный рахит, синдром Олбрайта—Батлера—Блумберга) — наследственное заболевание, обусловленное снижением реабсорбции фосфатов в проксимальном отделе канальцев почки, проявляющееся гиперфосфатурией, гипофосфатемией и развитием рахитоподобных изменений, резистентных к обычным дозам витамина D.
КЛИНИКА И ДИАГНОСТИКА. Заболевание проявляется в возрасте первых двух лет. Наиболее характерными признаками являются: возбудимость, гипотония, искривление ног с началом ходьбы, задержка роста, грубые деформации костей, особенно нижних конечностей, алопеция, аномалия зубов, снижение неорганического фосфора в сыворотке крови до 0,64 ммоль/л и ниже (при норме у детей 1-го года жизни 1,29—2,26 ммоль/л), повышенное выделение фосфора с мочой до 5 г/сут и активности щелочной фосфатазы в 2—4 раза по сравнению с нормой. Содержание кальция в сыворотке крови нормальное. Гипераминоацидурия и глюкозурия не характерны. При рентгенографии костей обнаруживаются широкие диафизы с утолщением кортикального слоя, грубый рисунок трабекул, остеопороз, вагусная деформация нижних конечностей, запаздывание скелетного возраста и другие признаки рахита.
Лабораторные данные указывают на выраженную гипофосфатемию, достигающую 0,6—0,9 ммоль/л, повышение щелочной фосфатазы при обычно нормальном уровне кальция сыворотки крови. Экскреция фосфора с мочой значительно повышена, кальция — не изменена. При I— Π типах фосфат-диабета снижается реабсорбция фосфатов в проксимальных канальцах, что обуславливает значительную фосфатурию (более 20—30 ммоль/л). При III типе отсутствует фосфатурия, экскреция фосфора в пределах нормы, но выражена гипераминоаиидурия. При рентгенологическом исследовании выявляются рахитоподобные изменения в метафизарной зоне трубчатых костей с блюдцеобразным ее расширением. Бокаловидное изменение метафизов происходит в проксимальной и дистальной частях большой берцовой кости, в дистальной частях малой берцовой, лучевой и локтевой костях. При первой форме отмечается утолщение кортикального слоя и отсутствие признаков остеопороза. При второй форме отсутствуют признаки остеомаляции. При третьей форме — тяжелые рахитические изменения с истончением кортикального слоя, остеопороз.
Диагноз ставится на основании характерных клинических симптомов, наличия подобных заболеваний у других членов семьи, гиперфосфатурии, гипофосфатемии, повышенной активности щелочной фосфатазы и отсутствии лечебного эффекта от умеренных доз витамина D. Дифференцировать необходимо с витамин-О-дефицитным рахитом, который хорошо поддается комплексному лечению, синдромом де Тони—Дебре—Фанко- ни, остеопатией при хронической почечной недостаточности.
698
ЛЕЧЕНИЕ. Рекомендуется начинать с препаратов фосфора (1—2 г в сутки), а затем приступить к использованию витамина D. Такая методика позволяет добиться лечебного эффекта при введении витамина D в умеренных дозах. Начальные дозы витамина D должны составлять 20 000— 30 000 ME вдень, через 4—6 недель ее увеличивают на 10 000—15 000 ME ежедневно, пока не будут достигнуты нормализация уровня фосфора в крови, снижение активности щелочной фосфатазы, исчезновение болей в костях нижних конечностей, восстановление структуры костной ткани. Обязательным является контроль за выделением кальция с мочой по пробе Сулковича. Отсутствие симптомов интоксикации, небольшое выделение кальция с мочой являются показаниями к увеличению дозы витамина D. В большинстве случаев эффективной дозой витамина D является 100 000—150 000 ME в сутки. Показаны сочетания витамина D с дифосфонатом (ксидифон) или со смесью Олбрайта (80 мл смеси-ра- створа в сутки в 5—6 приемов внутрь). Наличие грубых деформаций костной системы служит показанием к ортопедическому лечению (используется иммобилизация конечностей).
БОЛЕЗНЬ ДЕ ТОНИ-ДЕБРЕ-ФАНКОНИ наследуется по аутосом- но-рецессивному типу, характеризуется преимущественным поражением проксимальных канальцев нефронов (истончение, дегенеративные изменения). В процесс вовлекаются клубочки и интерстиций паренхимы почек с постепенным развитием фиброза и склероза. Возникают тубулярные дисфункции, проявляющиеся снижением реабсорбции воды, фосфатов, натрия, калия, глюкозы, аминокислот, гидрокарбонатов. Впервые описано де Тони (1933), Дебре (1934), Фанкони (1936).
ПАТОГЕНЕЗ. Различают два варианта этого заболевания: первичный и вторичный. Первый возникает как самостоятельное заболевание, второй — при метаболических нарушениях (цистинозе, гликогенозе, галактоземии, фруктоземии, окулоцеребраренальном синдроме), при приобретенных заболеваниях (множественной миеломе), при отравлении солями тяжелых металлов, лизолом и др.
Заболевание наследуется по аутосомно-рецессивному типу. Однако указывают и на возможность аутосомно-доминантного пути передачи. Описаны и спорадические случаи. В основе данной патологии лежит снижение реабсорбции аминокислот, глюкозы, фосфатов в проксимальном отделе канальцев.
Вместе с тем страдает реабсорбция бикарбонатов натрия и калия. Потеря бикарбонатов с мочой ведет к почечному канальцевому ацидозу. В результате чрезмерного выделения с мочой бикарбонатов и глюкозы теряется большое количество калия. Потеря натрия объясняется следствием выделения значительного количества анионов. Кальциурия и нарушение всасывания в кишечнике обуславливают значительные вариации уровня его в сыворотке крови. Нарушается реабсорбция воды.
Рахитоподобная остеопатия развивается в результате сочетанного действия метаболического ацидоза и гипофосфатемии, дефицита кальция. Понижение чувствительности к витамину D, по-видимому, обу-
699
словлено нарушением превращения его в активные формы в условиях метаболического ацидоза.
Первичная форма заболевания — генерализованная проксимальная тубулопатия. Первичная форма заболевания рассматривается как канальцевая полиэнзимопатия, характеризующаяся нарушением синтеза ферментов, ответственных за транспорт аминокислот, фосфатов, глюкозы, реабсорбцию бикарбонатов.
При морфологическом исследовании на ранних этапах заболевания паренхима почек производит впечатление неизменной. В дальнейшем выявляются изменения в канальцах, особенно в проксимальном отделе, расширение их в области петли Генле и собирательных трубок. В интерстиции обнаруживают лимфогистиоцитарную инфильтрацию и нередко — фиброз. Клубочки интактны до развития ХПН.
КЛИНИКА И ДИАГНОСТИКА. Заболевание проявляется в первые два года жизни. Обращает на себя внимание отставание в физическом и умственном развитии — задержка в росте, гипотрофия, рахитоподобные изменения скелета, обусловленные потерей фосфатов с мочой и др. Дети часто болеют в связи со сниженными защитными свойствами организма. Отмечается полиурия и чувство жажды, возникают признаки обезвоживания — результат нарушения реабсорбции воды. Большие потери калия с мочой ведут к гипокалиемии, клинически проявляющейся слабостью, тошнотой, мышечной гипотонией, гипорефлексией, гипотензией, расширением границ сердца, тахикардией, изменениями на ЭКГ. У всех больных возникает метаболический ацидоз в связи со сниженной реабсорбцией гидрокарбонатов, признаками которого являются вялость, раздражительность, бледность и др.
При лабораторном исследовании для полного синдрома де Тони— Дебре—Фанкони характерны: глюкозурия, фосфатурия, гипофосфатемия, ацидоз и аминоацидурия; для неполного — отсутствие одного из них. Гипераминоацидурия генерализованная (аланин, глицин, аргинин, пролил, глутаминовая кислота и др.). Характерным признаком является выраженная фосфатурия, приводящая к значительной гипофосфатемии. Уровень кальция в плазме крови, как правило, снижен. Экскреция его с мочой нормальная или увеличена. В плазме повышена активность щелочнойфосфатазы. В большинстве случаев выявляется гипокалиемия. Метаболический ацидоз на ранних этапах заболевания обусловлен снижением реабсорбции бикарбонатов. Постепенно развивается гипоизостенурия, протеинурия. Клубочковая фильтрация снижается лишь при хронической почечной недостаточности.
Рентгенологическая картина характеризуется общим остеопорозом, истончением и разрыхленностью кортикального слоя, бокаловидным расширением дистальных и проксимальных отделов трубчатых костей, искривлением конечностей, возможны переломы.
Выделяют наследственный синдром де Тони—Дебре—Фанкони. Он обусловлен накоплением кристаллов цистина в клетках различных органов, в том числе в почках. Проявляется задержкой физического разви-
700