

лекции / ОФ_Фармакодинамика
.pdfФАРМАКОДИНАМИКА
От малых причин бывают великие последствия; так, отгрызение заусенца причинило моему знакомому рак. К. Прутков «Мысли и афоризмы», №79а.
Механизмы возникновенияпервичной фармакологической реакции. Природа рецепторов.
Как уже упоминалось, механизм действия лекарственных средств связан с их влиянием на биологические субстраты клетки, которое приводит к развитию фармакологического эффекта. Роль таких биологических субстратов могут выполнять:
•Молекулы неорганических веществ – ионы железа для дефероксамина, соляная кислота для гидроокиси алюминия, молекулы воды для наркотических газов;
•Структурные белки клетки – колхицин, мебендазол воздействуют на белок тубулин, который формирует микротрубочки;
•Белки-ферменты – каптоприл блокирует ангиотензин-превращающий фермент;
•Транспортные белки – сердечные гликозиды нарушают работу Na+/K+-АТФазы;
•Нуклеиновые кислоты – ряд противоопухолевых средств изменяет структуру ДНК клетки.
Наиболее важным является взаимодействие лекарственного вещества с особыми регуляторными молекулами клетки – рецепторами для нейромедиаторов, гормонов и других биологически активных веществ.
Рецептор (лат. recipere – получать) – специфические молекулярные компоненты клетки, которые при взаимодействии с лигандами подвергаются конформационным изменениям и за счет воздействия на эффекторные системы клетки изменяют функции ткани и органа в целом. От рецепторов следует отличать места инертного связывания – молекулярные компоненты с которыми могут взаимодействовать биологически активные вещества, не вызывая их конформационных изменений и передачи сигнала на эффекторные системы. Например, эстрогены взаимодействуя с эстрогеновыми рецепторами вызывают изменение транскрипции генов. В то же время, эстрогены могут связываться с секс-глобулином в плазме крови (транспортный белок) но это не приводит к какому-либо биологическому эффекту, поэтому в данном случае секс-глобулин – инертное место связывания для стероидов.
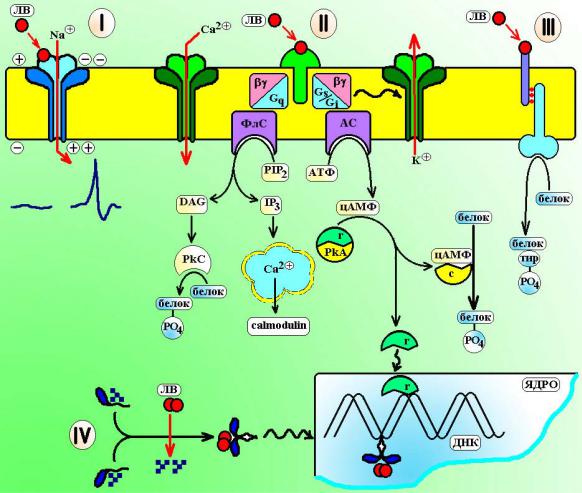
В клетке различают 4 типа рецепторов: три из них являются мембранными (т.е. встроены в мембраны клеток) и один тип – цитоплазматическим (см. схему 1).
• Трансмембранные рецепторы, связанные с ионными каналами. Представляют собой белки, которые формируют в мембране ионный канал. При взаимодействии лиганда с рецепторной субъединицей белка проницаемость ионного канала меняется. К данной группе рецепторов относят:
Н-холинорецепторы, связанные с Na+-каналами. При взаимодействии с ацетилхолином рецептор открывает натриевый канал и под влиянием тока ионов натрия в клетку возникает деполяризация мембраны и генерируется потенциал действия.
ГАМКА-рецепторы, которые связаны с Cl--каналами. При взаимодействии с γ- аминомасляной кислотой рецептор открывает канал и обеспечивает поступление в клетку ионов хлора. Возникает гиперполяризация мембраны и переход ее в состояние покоя.
• Трансмембранные рецепторы, связанные с G-белками. Эти рецепторы состоят из 3
субъединиц. Рецепторный белок располагается на наружной стороне мембраны. При взаимодействии с лигандом он передает сигнал на внутримембранный G-белок, который за счет энергии ГТФ перемещается к внутренней стороне мембраны и изменяет активность эффекторных белков. Эффекторные белки расположены на внутренней стороне мембраны и представляют собой ферменты, которые образуют «вторичные мессенджеры» – молекулы-посредники, которые передают сигнал в клетку и вызывают развитие ответной реакции. В качестве эффекторных белков выступают:
2
Аденилатциклаза – это фермент, который гидролизует АТФ с образованием циклического АМФ (цАМФ). Молекула цАМФ в цитоплазме клетки связывается с зависимой от нее протеинкиназой А (PkA). При этом молекула протеинкиназы распадается на 2 фрагмента: рецепторная субъединица PkA вместе с цАМф поступает в ядро клетки и влияет на транскрипцию генов, а каталитическая субъединица PkA остается в цитоплазме и обеспечивает фосфорилирование белков. Работу аденилатциклазы через Gs- белок стимулируют β-адренорецепторы, Н2-гистаминовые рецепторы, D1- дофаминовые рецепторы. Через Gi-белок работу аденилатциклазы тормозят α2- адренорецепторы, М2-холинорецепторы, µ-опиоидные рецепторы.
Гуанилатциклаза – это фермент, который гидролизует ГТФ с образованием цГМФ. Молекула цГМФ активирует в клетке цГМФ-зависимые протеинкиназы, которые также фосфорилируют белки. С гуанилатциклазой связаны рецепторы предсердного натрийуретического пептида.
Фосфолипаза С – это фермент, который обеспечивает гидролиз фосфатидилинозитол бифосфата (PIP2) до инозитол-трифосфата (IP3) и диацилглицерола (DAG). IP3 воздействует на внутриклеточные кальций-депонирующие органелы, а DAG обеспечивает активацию протеинкиназы С (PkC), которая обеспечивает фосфорилирование внутриклеточных белков. С фосфолипазой С связаны через Gq-белок α1-адренорецепторы, М1 и М3-холинорецепторы, Н1-гистаминовые рецепторы.
Схема 1. Типы циторецепторов. I – трансмембранные рецепторы, связанные с ионным каналом, II – трансмембранные рецепторы, связанные с G-белками, III – трансмембранные рецепторы с ферментативной активностью, IV – цитозильныерецепторы.
ЛВ – лекарственное вещество, ФлС – фосфолипаза С, АС – аденилатциклаза, PIP2 – фосфатидил бифосфат, IP3 – инозитол трифосфат, DAG – диацилглицерол, PkC – протеинкиназа С, PkA – протеинкиназа А (r – рецепторная субъединица, с– каталитическая субъединица), тир– остаткитирозина вмолекулебелка.

3
•Трансмембранные рецепторы-ферменты. Данный вид рецепторов представлен молекулами, состоящими из 2 субъединиц. Рецепторная субъединица располагается с наружной стороны мембраны, а каталитическая (т.е. обладающая ферментативногй активностью) – прошивает мембрану клетки насквозь. При взаимодействии с лигандом рецепторная субъединица активирует каталитическую часть молекулы. В качестве каталитической субъединицы выступают:
Тирозинкиназы – ферменты, которые фосфорилируют остатки тирозина в молекулах белков. К такому типу рецепторов относится инсулиновый рецептор. Серин-треонинкиназы – ферменты, которые фосфорилируют остатки серина и треонина
вмолекулах белков. К такому типу относят некоторые из интерлейкиновых рецепторов.
•Цитоплазматические рецепторы. Находятся в цитозоле клетки. Лиганд рецептора (липофильное вещество) проникает через ее мембрану и связывается с рецептором. В покое эти рецепторы экранированы особым белком теплового шока (hsp-белок). При связывании с лигандом рецептор освобождает этот белок и образует пары с другими рецепторами данного семейства. Затем, активированный рецептор поступает в ядро клетки, где связывается с особыми рецепторными последовательностями нуклеотидов ДНК и регулирует экспрессию генов. К данному семейству относятся рецепторы для стероидных гормонов, витаминов А и D, тиреоидных гормонов.
Взаимодействие лекарственных веществ с рецепторами.
Способность вещества связываться с рецепторами клетки называется аффинностью1. Аффинность обусловлена тем, что пространственная конфигурация лекарства может напоминать конфигурацию эндогенного лиганда этого рецептора. Внутренней активностью называют способность лекарственного вещества вызывать активацию рецептора. В зависимости от величины внутренней активности все лекарственные вещества можно разделить на несколько групп:
Агонисты или миметики (от греч. agonistes – соперник; mimeomai - подражать) – вещества, которые связываясь с рецепторами способны их активировать, что вызывает развитие ответа, характерного для данного типа рецепторов. Считают, что внутренняя активность у агонистов равна 1,0 (т.е. они вызывают полный ответ ткани).
Антагонисты или блокаторы (от греч. antagonisma – соперничество, противоборство) – это лекарственные вещества, которые связываются с рецепторами, но не вызывают их активации (т.е. их внутренняя активность равна нулю). Антагонисты экранируют циторецепторы и препятствуют развитию ответа при действии эндогенных агонистов.
Парциальные агонисты – это вещества, которые связываются с рецепторами и вызывают их активацию, однако, даже если они займут все рецепторы, эти вещества не способны вызвать максимальный ответ для данного типа рецепторов. Т.е. внутренняя активность таких агонистов меньше 1,0 и составляет обычно 0,3-0,6.
Действие парциального агониста будет зависеть от того, с какими рецепторами они взаимодействуют – с покоящимися или с рецепторами, которые уже активированы полными агонистами. Если парциальный агонист взаимодействует с покоящимися рецепторами, то он вызывает их активацию и ответ ткани возрастает с нуля до некоторой величины (≈30-60%), т.е. это типичное агонистическое воздействие. Если парциальный агонист действует на ткань, рецепторы которой уже активированы полным агонистом, то он начинает вытеснять агонист из связи с рецептором и занимать его место. Поскольку эффект парциального агониста уступает полному – ответ ткани снижается со 100% до более низкой величины (≈30-60%), т.е. это типичное блокирующее действие (см. схему 2).
Иногда выделяют также понятие агонистов-антагонистов. Агонисты-антагонисты – это вещества с низкой аффинностью, которые могут взаимодействовать не с одним, а с не-
1 Мерой аффинности служит константа диссоциации (Kd) вещества от рецептора, т.е. такая его концентрация при которой вещество связывает 50% рецепторов. Чем меньше Kd, тем более аффинным к рецептору является лекарственное вещество.
4
сколькими типами рецепторов, при этом они одни рецепторы активируют, а другие блокируют.
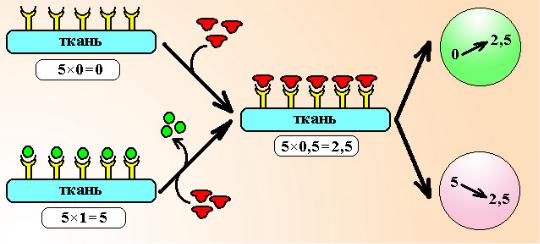
Схема 2. Действие парциальных агонистов. Предположим, что имеется тест-ткань, несущая всего 5 рецепторов. Если рецепторы свободны, ответ ткани равен нулю. После добавления парциального агониста с внутренней активностью 0,5 он будет активировать рецепторы и общий ответ ткани составит 5×0,5=2,5 ЕД. Если рецепторы уже заняты полным агонистом, ответ ткани максимальный (5×1,0=5 ЕД), при добавлении к системе парциального агониста он начинает вытеснять лиганд из связи с рецептором и будет активировать их слабее полного агониста. Общий ответ ткани при этом уменьшится и составит 5×0,5=2,5 ЕД, т.е. возникнет эффект блокады.
Можно показать, что чем больше изначальное число активированных рецепторов в ткани, тем будет более выражен блокирующий эффект парциального агониста.
Инверсные агонисты – это вещества, которые связываются с рецепторами и вызывают эффект обратный тому, который возникает при действии обычного агониста. Т.е. внутренняя активность реверсных агонистов меньше нуля (-1,0). Инверсные агонисты не следует путать с антагонистами. Антагонист предупреждает эффект агониста, блокируя рецептор, инверсный агонист – вызывает при взаимодействии с рецептором зеркально противоположный эффект. Примером реверсных агонистов могут служить β-карболины. Диазепам является агонистом бензодиазепин-ГАМК рецепторного комплекса и вызывает при активации рецептора развитие седативного и снотворного эффектов. Флумазенил – антагонист этих рецепторов, если его ввести в организм пациента, то воспроизвести снотворное действие диазепама не удается. β-карболины при взаимодействии с бензодиазепиновым рецептором будут вызывать судороги (качественно противоположный снотворному действию диазепама эффект).
Теории взаимодействия лекарственных веществ с рецепторами.
Впервые теория взаимодействия вещества и рецепторов была разработана A.J. Clark в 1926-1937 гг. Она получила название классической оккупационной теории. Ее основные положения включают:
•Взаимодействие вещества и рецептора аналогично взаимодействию двух химических веществ путем абсорбции свободного вещества (лиганд) на поверхности фиксированной молекулы (рецептор) и описывается зависимостью Langmuir, которая была предложена в 1917 г.
•Имеется прямая пропорциональная зависмость между числом занятых рецепторов и ответом ткани: чем больше рецепторов занято, тем больше ответ ткани.
•Каждая молекула рецептора при взаимодействии с лигандом изменяет свою конформацию и передает сиг-
нал на эффектор. При диссоциации лиганда от рецептора он вновь переходит в состояние покоя.
Однако, оккупационная теория не могла объяснить действие антагонистов на рецепторы – почему при связывании с лигандом-антагонистом рецепторы не переходят в активное состояние? Кроме того, нельзя было объяснить почему некоторые агонисты при активации всех рецепторов не способны вызвать максимальный эффект.
Чтобы решить эти противоречия в 1954 г E.J. Ariens ввел поправки в теорию A.J. Clark и предложил характеризовать каждый лиганд как по аффинности (сродству к рецептору), так и по его внутренней активности, о которой уже упоминалось выше. Однако, поправки E.J. Ariens не могла решить всех противоречий: не была ясна природа внутренней активности – почему один лиганд рецептор активирует полностью, а другой лишь на 40%. Что определяет меру внутренней активности? В 1956 г R.B. Stephenson обнаружил еще одно противоречие в теории A.J. Clark: он показал, что между числом занятых рецепторов и
5
в теории A.J. Clark: он показал, что между числом занятых рецепторов и ответом ткани может быть непропорциональная зависимость – некоторые лиганды могут вызывать полный ответ, активируя менее 100% рецепторов.
Для того, чтобы решить эти противоречия в 1961 г. W.D. Paton предложил теорию скорости взаимодействия. Согласно этой теории характер действия веществ на рецепторы определяется скоростью ассоциации и диссоциации вещества от рецептора. Согласно теории W.D. Paton агонисты – это вещества, которые имеют высокую скорость ассоциации и диссоциации от рецептора. Чем выше эта скорость, тем более активен агонист, тем меньше молекул рецептора ему требуется занять, чтобы вызвать максимальный ответ. Антагонисты – это вещества, которые связываются с рецептором, но крайне медленно освобождают его. Таким образом, сила антагониста будет пропорциональна числу занятых рецепторов, как и в теории A.J. Clark.
Иногда, очень образно, теорию A.J. Clark сравнивают с игрой на органе – музыка извлекается лишь в тот момент, когда пальцы музыканта нажимают на клавиши органа. Если клавишу отпустить звучание тотчас же исчезает. Теорию W.D. Paton сравнивают с игрой на рояле – звучание инструмента сохраняется и после того, как палец музыканта уже нанес удар по клавише. Чем быстрее двигаются пальцы пианиста, тем мощнее мелодия и звучание инструмента. К сожалению, теория W.D. Paton не смогла решить всех противоречий. Вскоре после ее создания были обнаружены агонисты с медленной кинетикой связывания и натагонисты с быстрой кинетикой диссоциации.
В 1967 г. A.T. Karlin создает аллостерическую теорию рецепции. Согласно этой теории рецептор находится в двух состояниях: активном и неактивном, причем между состояниями существует динамический переход: Ri Ra. Если вещество взаимодействует преимущественно с активной формой рецептора и стабилизирует его в этом состоянии, то оно является агонистом, если вещество взаимодействует с неактивной формой рецептора – это антагонист. Однако, в теории A.T. Karlin вскоре был обнаружен изъян – установили, что агонисты способны вытеснять антагонисты из связи с рецепторами, т.е. они заведомо способны связываться и с неактивной формой рецептора.
В 1979-1982 гг. E.J. Ariens предложил объединить все теории рецепции. Согласно этой теории при взаимодействии лиганда с рецептором образуется неактивный комплекс лиганд-рецептор (LR), который может обратимо переходить в активированное состояние (LR*). При этом скорость активации комплекса с агонистом значительно выше, чем скорость активации комплекса с антагонистом. Биологический ответ ткани при этом пропорционален не числу связанных рецепторов, а числу активных комплексов. Таким образом, E.J. Ariens предложил систему перехода: L+R LR LR* L+R* L+R. Решением этой системы переходов служит сово-
− dR |
= k R2 |
|
||||
|
|
dt |
|
|
1 |
|
|
|
dLR |
|
|
|
|
|
− |
= k2 LR |
|
|||
|
dt |
|
|
|||
|
|
|
|
. До настоящего времени нет достаточно убеди- |
||
купность дифференциальных уравнений: |
|
|
|
|
|
|
− dLR* |
= k3 LR* |
|||||
|
|
dt |
|
|
|
|
|
|
dR* |
= k4 R* |
2 |
||
− |
dt |
|
|
|||
|
|
|
|
|
|
|
тельных экспериментальных данных, которые смогли бы подтвердить или опровергнуть данную теорию.
Соотношение между концентрацией лекарственного вещества и его фармакологическим эффектом
Градуальная и квантовая кривые зависимости «доза-эффект»
Как правило, по мере увеличения дозы (концентрации) лекарственного вещества его эффект возрастает. Вначале процесс прироста эффекта происходит пропорционально дозе лекарства, однако, процесс возрастания эффекта не носит бесконечного характера – обычно при достижении некоторого уровня прирост эффекта вначале уменьшается, а затем прекращается совсем и наступает фаза плато (т.е. дальнейшее увеличение дозы не приводит к возрастанию эффекта лекарства).
Если представить эту зависимость изменения эффекта лекарства от его дозы графически, то она будет иметь вид экспоненциальной кривой. Предел эффекта, к которому стремиться кривая при бесконечном увеличении дозы лекарства называют максимальным эффектом (Emax). Зная Emax можно рассчитать, при какой дозе (концентрации) лекарства возникает эффект, равный половине максимального (½Emax или E50). Величину дозы или концентрации, вызывающей полумаксимальный эффект обозначают ED50 или ЕС50 соответственно. Таким образом:
6
•Еmax – максимальный эффект лекарства, который удается воспроизвести при введении в
организм бесконечно больших количеств лекарства.
•ED50 или ЕС50 – доза или концентрация лекарства, при которой развивается эффект, равный половине от максимального.
Однако, проводить анализ экспоненциальной кривой сложно, поэтому обычно прибегают к логарифмическому преобразованию координат. Если построить этот же график в полулогарифмических координатах, то он принимает вид S-образной кривой. Причем часть кривой на участке 20-80% эффекта имеет вид прямой линии и может быть легко преобразована в аналитическую форму (форму уравнения) для расчета Emax и ЕС50.
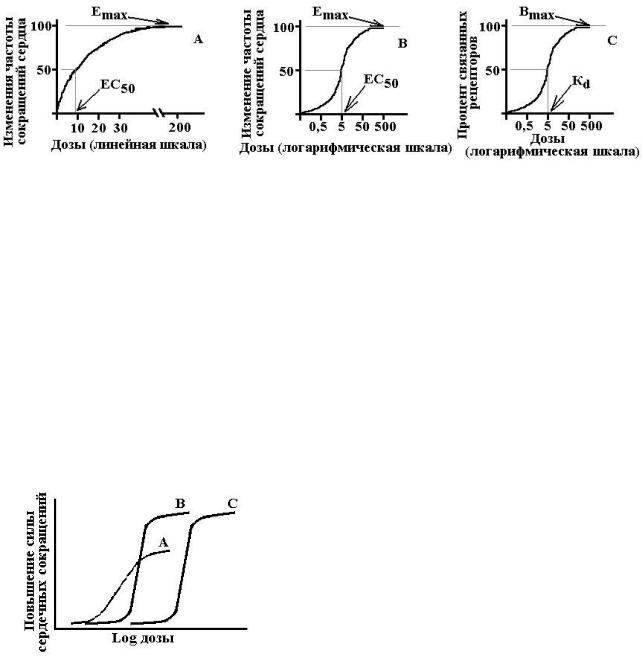
Схема 3. Градуальные кривые зависимости «доза-эффект». А – зависимость изменения частоты сердечных сокращений от дозы лекарства в нормальных координатах, В – та же кривая в полулогарифмических координатах (одна из шкал переведена в форму десятичных логарифмов), С – кривая связывания того же вещества с рецепторами миокарда.
Emax – максимальный эффект, Bmax – максимальное число связанных рецепторов, ЕС50 – концентрация лекарства при которой возникает эффект равный половине максимального, Kd – константа диссоциации вещества от рецептора, при которой связано 50% рецепторов.
На основании построения таких кривых можно определить два важнейших показателя, характеризующих каждое лекарство: активность и эффективность.
•Активность (сила лекарства) – параметр, который показывает какая доза (концентрация) лекарства способна вызвать развитие стандартного эффекта, равного 50% от максимально возможного для этого лекарства. Т.е. активность лекарства численно характеризуется ве-
личиной ЕС50 или ED50. Чем выше активность лекарства, тем меньшая его доза требуется для воспроизведения терапевтического эффекта.
•Эффективность – это способность лекарства оказывать максимально возможное для него действие. Т.е. фактически это максимальная величина эффекта, которую можно достигнуть при введении данного лекарства. Эффективность численно характеризуется величиной Еmax. Чем выше Еmax, тем выше эффективность лекарства.
|
На схеме 3С представлена кривая связывания |
|
лекарства с рецепторами, которую часто исполь- |
|
зуют в молекулярной биологии. Как нетрудно за- |
|
метить, эта кривая практически идентична зависи- |
|
мости «доза-эффект», при этом величина макси- |
|
мального связывания вещества с рецептором (Вmax) |
|
выступает в роли аналога максимального эффекта, |
|
а показатель Kd (константа диссоциации) – как |
|
аналог ЕС50. |
Схема 4. Сравнительная характеристика кар- |
Согласно теории A.J. Clark очевидно, что |
диотонических средств. Как следует из пред- |
максимальный эффект лекарства должен возникать |
ставленных графиков EC50,A<EC50,B<EC50,C – сред- |
в тот момент, когда заняты все рецепторы, а 50% |
ство А самое сильное, но Emax,A<Emax,B=Emax,C – |
эффект – если занята их половина. Однако, имеют- |
средстваВиСсамыеэффективные. |
ся лекарства, которые могут вызвать максималь- |
|
ный эффект даже в том случае, если они оккупировали менее 100% рецепторов. Такая ситуация, когда часть рецепторов оказывается избыточной и не нужна для достижения макси-
7
мального эффекта получила название «молчащих» рецепторов ткани. Очевидно, что и величина ЕС50 у таких лекарств проявится при оккупации не 50%, а значительно меньшего числа рецепторов. Если изобразить кривые «доза-эффект» и «доза-оккупация рецепторов» в одних координатах, то величина ЕС50 окажется меньше, чем показатель Kd для этого лекарства. Таким образом, о наличии молчащих рецепторов к лекарству говорят в том случае, если у него
Kd>>EC50 (см. схему5).
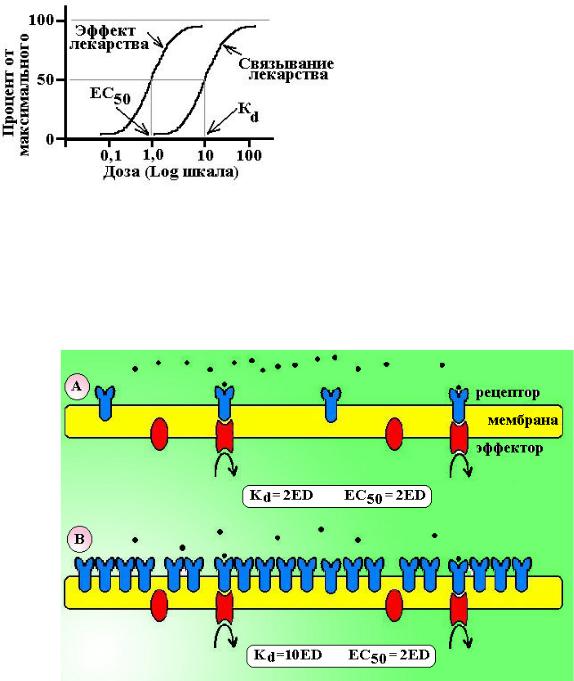
Схема 5. Обнаружение молчащих рецепторов в ткани. У
данного лекарства полумаксимальный эффект возникает при концентрации 1,0 ЕД, а половина рецепторов оккупируется лишь в дозе 10 ЕД.
Почему в ткани возникают молчащие рецепторы? Это можно объяснить двумя причинами:
• Эффект некоторых лекарств сохраняется значительно дольше, чем взаимодействие лекарства с самим рецептором. Такой механизм появления молчащих рецепторов получил название «time»-механизма. Он имеет место при действии сердечных гликозидов на фермент Na+/K+-АТФазу. В этом случае изменение Са2+ гомеостаза клетки сохраняется значительно дольше, чем взаимодействие самого гликозида с молекулой АТФазы.
• Наличие рецепторов, не связанных с эффекторными молекулами. Данная ситуация изображена на схеме 6В. Для развития полумаксимального фармакологического эффекта лекарству достаточно занять половину рецепторов связанных с эффекторами, но оккупация 50% рецепторов будет рассчитываться исходя из их общего числа.
Схема 6. Появление молчащих рецепторов в ткани.
А. Изначально ткань содержит 4 рецептора и 4 эффектора, поэтому величина ЕС50 для нее равна 2 ЕД лекарства и доза при которой лекарство занимает 50% рецепторов также составляет 2 ЕД.
В. При увеличении числа рецепторных молекул в 5 раз (20 молекул) с прежним числом эффекторов (4 молекулы) возникает ситуация, когда лекарству по-прежнему, чтобы вызвать 50% эффект достаточно 2 ЕД, но чтобы занять 50% рецепторов требуется 10 ЕД.
Наличие молчащих рецепторов имеет место в миокарде, где число β1-адренорецепторов в 10 раз превосходит число эффекторных молекул, связанных с ними. Избыточность β1- адренорецепторов имеет важное значение и позволяет миокарду в полной мере реагировать
8
на медиаторы (норадреналин и адреналин) даже после того, как большая часть рецепторов будет утрачена.
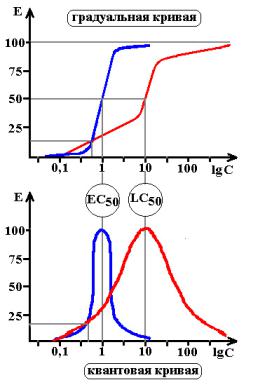
Описывая и разбирая процедуру построения кривой «доза-эффект», мы исходили из предположения о том, что эффект лекарства является непрерывной величиной, которая может быть измерена количественно. Например, гипотензивный эффект лекарства может быть измерен по уровню артериального давления, жаропонижающий эффект – по степени снижения температуры тела. Такие эффекты носят название градуальных (непрерывных, интегральных), а кривые, которые описывают такой эффект получили название градуальных кривых «доза-эффект».
К сожалению, некоторые эффекты лекарств являются дискретной величиной или качественным признаком, т.е. они описываются всего лишь несколькими вариантами состояния. Например, головная боль может либо быть, либо отсутствовать у конкретного пациента, контрацептивный эффект лекарства либо проявляется, либо нет. Такие эффекты лекарств называют квантовыми или дискретными. Для квантового эффекта не возможно построить кривую зависимости величины эффекта от дозы для каждого конкретного индивидуума. Чтобы избежать возникшего затруднения прибегают к построению квантовой кривой «доза-эффект», где отмечают зависимость проявления эффекта в популяции от величины принимаемой дозы лекарства.
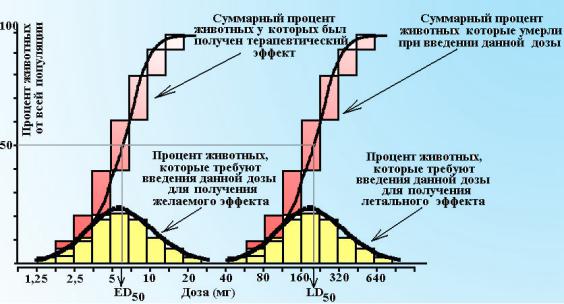
Вначале при введении небольших доз эффект развивается у малого числа объектов. По мере повышения дозы на лекарство начинает реагировать все большее число испытуемых и наконец остается лишь небольшая группа объектов у которой реакцию удается вызвать применяя высокие дозы лекарства. График зависимости «доза-эффект» при этом имеет куполообразный вид и идентичен Гауссовой кривой нормального распределения (см. схему 7). Величина соответствующая куполу кривой позволяет определить активность лекарства – т.е. ту его дозу, которая вызывает развитие эффекта у 50% лиц в популяции.
Схема 7. Квантовые кривые «доза-эффект». Левая кривая представляет построение зависимости для терапевтического эффекта лекарства, а правая – развитие токсического эффекта (смерти) после введения летальных доз лекарства. Как следует из графика доза в 1,25 и 2,5 мг вызывают эффект у незначительного количества животных, однако введение лекарства в дозе 5 мг позволяет вызвать эффект у наибольшего числа особей, оставшееся небольшое число животных реагируют на высокие доза лекарства. Аналогичные рассуждения справедливы и для введения летальных доз лекарства.
Таким образом, квантовая кривая «доза эффект» может помочь установить величину ЕС50, но ничего не говорит об эффективности лекарства.
Если на графике зависимости отмечать не то число объектов, у которого удалось получить ответ на каждую дозу в отдельности, а накопленное число объектов – т.е. всех лиц у ко-
9
торых к моменту введения дозы уже удалось добиться эффекта и пациентов прореагировавших на данную дозу, то кривая может быть трансформирована в стандартную S-образную форму, напоминающую градуальную кривую. Однако, эта кривая не будет нести никакой дополнительной информации.
Таблица1. Различиямеждуградуальнойиквантовойкривыми«доза-эффект»
Параметр |
Градуальная кривая |
Квантовая кривая |
Характер эффекта |
Количественный |
Качественный |
Возможность построения |
У индивидуума |
В популяции |
Сила лекарства |
Определяется величиной ED50 |
Определяется величиной ED50 |
|
(доза, которая создает полу- |
(доза, которая вызывает разви- |
|
максимальный эффект) |
тие эффекта у 50% популяции) |
Эффективность |
Определяется величиной Emax |
Неможетбытьопределенабез |
|
|
специальногоанализа |
Вид кривой |
Экспоненциальнаязависимость |
Кривая Гауссова распределения |
|
(S-образнаявполулогарифми- |
|
|
ческихкоординатах) |
|
Широтатерапевтическогодействия. Терапевтическийиндекс. Терапевтическийкоридор.
Если после того, как достигается плато эффекта доза лекарства будет продолжать расти, то через определенный промежуток времени начнет проявляться токсическое действие лекарства. Зависимость токсического действия от дозы (концентрации) лекарства носит такой же характер, как и его полезный эффект и может быть описана градуальной или квантовой кривыми. На этих кривых также может быть определена величина TD50 или ТС50 – токсиче-
ской дозы (концентрации) лекарства, которая вызыва-
|
ет токсический эффект, равный 50% от максимально- |
|
|
го (для квантовой кривой – токсический эффект у |
|
|
50% лиц в популяции). Иногда, вместо TD50 пользу- |
|
|
ются показателем LD50 – летальная доза, которая вы- |
|
|
зывает гибель 50% объектов в популяции. |
|
|
Информация относительного токсического по- |
|
|
тенциала лекарства позволяет дать характеристику |
|
|
его безопасности. Основными критериями безопасно- |
|
|
сти являются: |
|
|
Терапевтический индекс – это соотношение меж- |
|
|
ду токсической и эффективной дозами лекарства, |
|
|
которые вызывают появление полумаксимального |
|
|
эффекта. ТИ=TD50/ED50. Чем больше величина те- |
|
|
рапевтического индекса, тем более безопасным |
|
|
является лекарство. Например, на схеме 6 величи- |
|
|
на ED50≈6 мг, а LD50≈210 мг и ТИ=210/6=35. Пе- |
|
|
нициллин является лекарством у которого величи- |
|
|
на терапевтического индекса составляет более 100, |
|
|
а дигоксин имеет терапевтический индекс равный |
|
|
всего лишь 2. Таким образом, фактически тера- |
|
Схема 8. Терапевтический индекс и ши- |
певтический индекс определяет расстояние между |
|
рота действия. Терапевтический эффект |
точками ED50 и TD50 на кривых «доза-эффект». |
|
лекарства проявляется с высокой скоро- |
При этом мы исходим из допущения о том, что |
|
стью и EC50=1, токсические эффекты на- |
сами кривые имеют одинаковый наклон и харак- |
|
растают медленно, при этом LC50=10. Не- |
тер нарастания эффекта, а также, что токсическое |
|
смотря на то, что ТИ=10/1=10, терапев- |
||
действие лекарства проявляется позже его тера- |
||
тическая широта равна нулю, т.к. |
||
TD15=LD15=0,6 |
певтического эффекта. К сожалению, достаточно |
10
часто характер кривых терапевтического и токсического действия лекарства будет различаться.
Терапевтическая широта (терапевтическое окно) – это диапазон доз между минимальной терапевтической и минимальной токсической дозами лекарства (ТШ=TD10/ED10). Терапевтическая широта более корректный показатель безопасности лекарства, поскольку он позволяет учитывать степень нарастания нежелательных эффектов на кривой «дозаэффект».
Например, на схеме 8 показано лекарственное средство, у которого фармакологическое действие проявляется в виде быстро нарастающего эффекта в ответ на незначительное повышение дозы. Токсическое действие у него, напротив, имеет вид плавно нарастающей кривой. В данном случае величина терапевтического индекса составляет около 10, но благодаря тому, что токсические эффекты нарастают медленно и одновременно с терапевтическим действием 15% эффект лекарства возникает одновременно с развитием отравления у 15% пациентов. Т.е. терапевтическая широта у данного средства отсутствует и его полезное действие возникает одновременно с токсическим.
Фактор надежной безопасности – это отношение минимальной токсической дозы к максимальной эффективной (ФНБ=TD1/ED99). Фактически, данный критерий представляет собой несколько модифицированную форму терапевтической широты и показывает: во сколько раз может быть превышена терапевтическая доза лекарства без риска развития интоксикации (нежелательных эффектов).
Терапевтический коридор – это диапазон эффективных концентраций лекарственного вещества в крови, которые необходимо создать и поддерживать в организме, чтобы обеспечить достижение желаемого терапевтического действия.
Зависимость действия лекарств от их структуры, физико-химических свойств, лекарственной формы и путей введения.
Химическая структура лекарства определяет следующие особенности его действия:
• Пространственную конфигурацию молекул лекарства и его способность активировать или блокировать рецепторы. Так, например, l-энантиомер пропранолола способен блокировать β1 и β2-адренорецепторы, тогда как его d-энантиомер в несколько раз более слабый адреноблокатор.
•Тип биосубстрата, с которым вещество способно взаимодействовать. Например, стероид-
ные молекулы с ароматизированным кольцом из класса С18-стероидов активируют эстрогеновые рецепторы, а при насыщении кольца приобретают способность стимулировать андрогеновые рецепторы.
•Характер устанавливаемых с биосубстратом связей и продолжительность действия. Например, ацетилсалициловая кислота образует ковалентную связь с циклооксигеназой, ацетилирует активный центр фермента и необратимо лишает его активности. Напротив салицилат натрия образует с активным центром фермента ионную связь и лишь временно лишает его активности.
Физико-химические свойства лекарства. Данная группа свойств определяет, главным образом, кинетику лекарства и его концентрацию в области биологического субстрата. Ведущую роль здесь играет степень полярности молекулы вещества, сочетание липофильных и гидрофильных свойств. Все эти факторы были уже рассмотрены ранее.
Лекарственная форма. Лекарственная форма определяет скорость поступления лекарства в системный кровоток и продолжительность его действия. Так, в ряду водный раствор > суспензия > порошок > таблетка скорость поступления в кровоток падает. Данный эффект связан, отчасти, с площадью поверхности лекарственной формы – чем она больше, тем быстрее происходит всасывание, т.к. большая часть лекарства контактирует с биологической мембраной. Данную зависимость можно проиллюстрировать следующим примером: площадь поверхности куба с ребром 1 см составляет 6 см2, а если этот куб разделить на меньшие кубики с ребром 1 мм, то площадь поверхности составит 60 см2 при том же общем объеме.