

Учебники / Pediatric Sinusitis and Sinus Surgery Younis 2006
.pdfCystic Fibrosis and Sinusitis |
125 |
The genetic alteration results in a deficiency in this transmembrane regulator that causes a decrease in chloride permeability across apical membranes of epithelial cells. Researchers have identified over 1000 CFTR mutations, but the exact phenotypical manifestations of the genetic alterations to 7q31 are not well understood. The most common mutation is DF508, which causes a deletion of phenylalanine on the CFTR gene. In the United Kingdom, this accounts for approximately 70% of the mutations and is one of the most common mutations in Sweden (4,7). The majority of genetic screening programs use the DF508 mutation for their screening, which can detect about 87% of the CF mutations (6). However, some recommend testing a panel of genetic mutations based on the local prevalence of different mutations (8). DNA analysis is helpful in those patients in whom prior CF testing is equivocal or in couples with a family history of CF. This can be particularly important in preconceptual planning for such couples. In addition, in utero amniocentesis or chorionic villi sampling is available for couples desiring prenatal diagnosis (6).
Five broad categories of CFTR mutations have been described (4,9,10): class-I mutations cause CFTR synthesis defects resulting in absent production; class-II, which includes DF508, results in abnormal production of CFTR; class-III is characterized by normal production and trafficking of protein, but disrupted regulation at the cell membrane; class-IV has expression at the cell membrane but abnormal chloride conductance; and class-V has decreased splicing of normal CFTR. Classes I–III are associated with more severe phenotypic disease compared with classes IV and V.
In addition to the CFTR mutations, several other molecular events have been identified. Increased transmembrane sodium conductance due to a greater number of sodium channels has been described. In addition, there are an increased number of ATPase-dependent sodium/potassium pumps at the basal lateral surface of the respiratory epithelial cell. These alterations cause an influx of sodium from the extracellular matrix across the apical membrane of epithelial cells (11). This results in passive movement of water from the extracellular matrix into the cell, causing increased mucous viscosity and impaired mucociliary clearance due to desiccation of extracellular fluid.
PATHOPHYSIOLOGY
Epithelial cells of numerous organs, such as the lung, pancreas, and gastrointestinal tract, are affected in CF. While the lung is the primary organ affected, chronic sinusitis and nasal polyposis are the most common otolaryngological manifestations of CF. Of the paranasal sinuses, the ethmoid and maxillary sinuses are most commonly involved. There is a higher incidence of hypoplastic or aplastic sinuses in CF patients compared with non-CF patients (12–14). The frontal and sphenoid sinuses are not present at birth and frequently fail to develop in CF patients (15). It is believed that chronic
126 |
Derkay and Schraff |
inflammation and obstruction prevent pneumatization of these developing sinus cavities (16).
Similar to the tracheobroncial tree, the nasal cavity and paranasal sinuses are lined by pseudostratified ciliated epithelium. This epithelial layer acts as a barrier to inhaled microscopic foreign debris. It is believed that the cilia are embedded in two separate mucous layers that lie superficial to the respiratory epithelium (17). The more superficial of the layers is the gel layer, which is a rather viscous, mucous layer that helps entrap inhaled foreign particles. Beneath the gel layer but above the epithelium lies the sol layer, a more fluid layer that bathes the cilia. Ionic composition between these two layers and the epithelium is crucial for normal ciliary motion and mucociliary clearance.
In CF, mucociliary clearance is decreased despite normal ciliary structure and function. As described earlier, a deficiency in CFTR causes a decrease in chloride permeability across apical membranes of epithelial cells. This leads to an influx of sodium and water into cells. As a result, the extracellular matrix of exocrine secretions becomes more viscous. Although ciliary beat frequency does not change, the abnormal chloride conduction and sodium exchange result in impaired mucous clearance. The abnormal ionic composition also destroys the delicate balance between the gel, sol, and epithelial layers leading to mucous thickening (18).
In addition, it is believed that factors related to bacterial toxins also play a role in reducing clearance. A study on the bacterial content of the maxillary sinus found bacterial colonization in 95% of CF patients (19). Colonization is multimicrobial; however, CF patients suffer from Pseudomonas infection more often than non-CF patients. Bacterial toxins such as homolysin and pyocyanin produced by Pseudomonas contribute to impaired mucociliary clearance. In vitro studies have shown pyocyanin to slow ciliary beating and cause epithelial disruption (20).
Bacterial infection also causes inflammation resulting in goblet cell hyperplasia, squamous cell-metaplasia, and loss of ciliated cells (21). Individuals with CF have higher concentrations of interleukin-8 (IL-8) and other pro-inflammatory mediators in their sinuses and lungs (4,22). Mucous stasis results in retention of secretions and impaired gas exchange within the sinuses. As the partial pressure of carbon dioxide (CO2) rises, a hypoxic environment is created in which infection can flourish. Subsequent inflammation causes a decrease in sinus ostial patency. This results in a vicious cycle causing further damage to the sinus cilia and epithelium (15).
In addition to thickening of secretions and sinonasal mucosa, nasal polyposis is common in CF patients. Although some believe allergy may play a role in the development of nasal polyps in CF patients (23), most agree the incidence of allergy is not greater in CF patients compared with non-CF patients (24,25). The exact etiology of nasal polyps is unclear; however, polyps found in CF patients differ histologically compared with

Cystic Fibrosis and Sinusitis |
127 |
those found in non-CF, atopic patients. CF polyps have a thin basement membrane and lack submucosal hyalinization and eosinophils. In addition, the mucous glands contain an abundance of acid mucin. Atopic polyps, in contrast, demonstrate a thick basement membrane, eosinophilia, and neutral mucin (21).
There are several theories used to explain the pathophysiology of nasal polyp development in CF patients. One theory, proposed by Rulon et al. (26) and supported by histological studies (27), suggests that inspissated nasal mucosa secretions cause dilation of the mucous glands. This, in turn, causes capillary and venous congestion that leads to stromal edema and mucosal prolapse with subsequent polyp formation. Another theory is based on infection or allergy-induced chronic inflammation that causes vasodilation, epithelial damage, and lamina propria prolapse (28).
CLINICAL MANIFESTATIONS
CF affects many organ systems (Table 1) and patients tend to present with a variety of symptoms including salty skin, persistent cough, wheezing or shortness of breath, failure to thrive despite an excessive appetite, and greasy, bulky stools. Overall, the number of CF patients who complain of sinus symptoms is relatively small at about 10% (29,30). This may be due to patient acclimation to the numerous symptoms that accompany CF and an unhealthy baseline status (31).
The clinical symptoms of sinusitis in the CF patient are similar to those found in non-CF patients; however, CF patients tend to have more severe disease. Symptomatic patients tend to be older children and teenagers, which
Table 1 Systemic Effects of Cystic Fibrosis
Organ system |
Clinical manifestations |
|
|
Head and neck |
Sinusitis, nasal polyposis, and obstruction |
Pulmonary |
Impaired mucociliary clearance, |
|
hemoptysis, recurrent infections, |
|
bronchiectasis, bronchospasm, and |
|
poor pulmonary reserve |
Cardiovascular |
Cor pulmonale and clubbing |
Gastrointestinal |
Cirrhosis, colelithiasis, pancreatitis |
|
and pancreatic dysfunction, bowel |
|
obstruction, portal hypertension, |
|
meconium ileus, rectal prolapse, |
|
and malabsorption |
Genito-urinary |
Infertility |
Nutritional |
Vitamin deficiencies (A, D, E, and K) |
|
|
128 |
Derkay and Schraff |
may be due to a young child’s inability to verbalize complaints (23,30). Postnasal drainage, purulent rhinorrhea, facial pain, halitosis, headache, and snoring are typical, with the most common complaint being nasal obstruction (25,32). Headache is usually reported more often in older patients and anosmia is reported in 12–71% (31,32). These symptoms are caused by the CF-related mucous stasis and nasal polyposis. However, anatomical abnormalities of the nose and paranasal sinuses, such as a deviated septum or inferior turbinate hypertrophy, can exacerbate the disease process.
Despite the low number of symptomatic patients, the prevalence of sinus disease in CF patients is close to 100%, and imaging studies reveal opacification of the maxillary and ethmoid sinuses in virtually all patients. Mucoceles have also been reported in pediatric CF patients (31,33,34). Given the rarity of mucocele findings in children, the presence of mucocele in a child should raise suspicion for CF (33). Anterior rhinoscopy usually shows edematous, and erythematous nasal mucosa with abundant secretions and polyposis also is fairly common, typically multiple and bilateral. Literature reports of polyposis incidence vary from 6% to 86% of patients; however, most report an incidence of about 10% (25,27,30,35–37). Most patients will develop polyps between the ages of 5 and 20 (25,30,38). Undiagnosed children with milder forms of the disease may first present to the otolaryngologist with polyp disease sequelae such as nasal obstruction, septal deviations, proptosis, or hypertelorism. Nasal polyps in a pediatric patient should raise suspicion for CF. It is recommended that all children with polyp disease should be screened for CF with a sweat chloride test, especially if there is a history of repeated lung infections.
Studies have shown that CF patients with nasal polyposis are more likely to have Pseudomonas colonization of their lower airways than patients without polyps (7,19). Despite colonization with more virulent bacteria, few CF patients develop serious sinus-related complications such as orbital and brain abscesses or osteomyelitis (39).
The increased viscosity of mucous in CF patients leads to both pulmonary and non-pulmonary disease. CF patients suffer life-long recurrences of pulmonary disease. Tenacious secretions and mucous plugging cause atelectasis resulting in bronchitis, pneumonia, abscess, empyema, pneumothorax, pulmonary hypertension, or respiratory failure. CF-related exocrine dysfunction also affects the gastrointestinal tract. Pancreatitis and pancreatic insufficiency are not uncommon. Pancreatic dysfunction can also lead to diabetes and nutritional deficiencies due to malabsorption of fat-soluble vitamins. Stasis of secretions can result in colelithiasis, biliary cirrhosis, and eventual portal hypertension. Meconium ileus, fecal impaction, volvulus, and rectal prolapse are intestinal manifestations. Infertility due to failure of vas deferens development in men and tubal stasis of secretions in women has been reported (5) and more than 95% of men with CF are sterile (2).
Cystic Fibrosis and Sinusitis |
129 |
DIAGNOSIS
The diagnosis of CF can be confirmed by both laboratory test and clinical disease. Newborn screening has markedly improved identification and is based on elevated levels of serum immunoreactive trypsinogen. Newborn screening has also shown other benefits, as those identified early in life are less likely to suffer from malnutrition and growth retardation (40). Genetic testing for DF508 can also be performed. However, in many instances, family history may not be revealing and the diagnosis is not made until clinical symptoms arise. Usually, repeated pulmonary infections or sino-nasal disease lead to a suspicion of CF. The diagnosis is usually made with an abnormally high sweat chloride value (3). Sweat, an exocrine secretion of the skin, has an abnormally high chloride concentration (greater than 60 mEq/L) in CF patients and is the basis of the sweat test. It is recommended that patients with a borderline sweat test should undergo genetic testing (41).
MEDICAL MANAGEMENT
Medical management of CF patients continues to evolve and provides many challenges for the clinician. From an otolaryngologic standpoint, management of sinus disease often parallels treatment for pulmonary disease. The primary treatment modalities revolve around anti-inflammatory and antimicrobial medications.
Ideally, medical management involves controlling sinus disease and polyposis. It is presumed that control of sinus disease in these patients will improve pulmonary function and, hopefully, reduce pulmonary disease. Saline and nasal steroid sprays used for daily nasal hygiene have typically been the backbone of medical management, with oral steroids and antibiotics reserved for acute exacerbations.
Saline sprays are used to help mechanically debride the nasal cavities and decrease congestion. Buffered (3%) saline is preferred to physiologic (0.9%) saline due to its hypertonicity. It is felt that this hypertonic solution may help decongest nasal mucosa osmotically and has been shown to improve mucociliary clearance (42,43).
Nasal steroids have been shown to be clearly beneficial in CF patients. While polyp response rates to nasal steroid sprays have been variable in the past, a recent prospective, double-blinded, and randomized trial demonstrated that betamethasone nasal drops result in a statistically significant reduction in nasal polyp size compared to a placebo (44). Oral glucocorticoids also are typically used, but their use is tempered by the side effects, such as hypothalamic-pituitary-adrenal axis suppression, growth retardation, Cushing’s disease, glucose intolerance, water retention, increased gastric acid secretion, and impaired wound healing. Unpleasant side effects led to the
130 |
Derkay and Schraff |
discontinuation of the largest controlled trial on their use. In addition, the benefits of oral steroids are short-lived and do not persist once the medication is discontinued (4).
Antibacterial nasal sprays have also been used with mixed results. In theory, such sprays should help decrease bacterial load, and inhaled tobramycin has been used successfully to treat Pseudomonas pulmonary disease. Clinical trial results show that this reformulated version of the common antibiotic improved lung function in people with CF and reduced the number of hospital days (45,46). A recent clinical trial on azithromycin showed that patients with CF who took the antibiotic experienced an almost 50% reduction in hospitalizations, a significant improvement in lung function, and weight gain (2). However, a study using tobramycin and saline administered by a large-particle nebulizer reduced pain but increased nasal congestion (47). There also is the concern that daily use of antimicrobial nasal sprays will select for resistant bacteria.
However, a promising topical preparation called dornase alfa has been shown to improve nasal symptoms in CF patients. Dornase alfa, recombinant human deoxyribonuclease I, acts by decreasing inflammation and mucous viscosity, thus increasing mucociliary clearance. When administered to CF patients by jet nebulizer, it has been shown to improve forced expiratory volume in one second. A recent study (48) demonstrated a reduction in nasal mucosal edema, polyps, and need for revision surgery in a small cohort of CF patients treated after functional endoscopic sinus surgery.
While Streptococcus pneumoniae, Haemophilus influenza, and Moraxella catarrhalis are the typical bacteria found in non-CF patients, Staphylococcus aureus and Pseudomonas aeruginosa have a particular affinity for the respiratory mucosa in CF patients. One study (37) showed that 89% of CF patients had cultures positive for Pseudomonas while none of the non-CF patients demonstrated its presence. Pseudomonas is particularly problematic due to its biofilm production, which enhances its antibiotic resistance. To prevent the development of biofilms and reduce bacterial load, many clinicians recommend extended courses of high dose anti-pseudomonal antibiotics (49).
Macrolides have recently been discovered to possibly have anti-inflam- matory effects that are beneficial to CF patients. IL-8 is a potent chemotactic and neutrophil-activating factor that is believed to play an important role in inflammation. It is highly expressed in nasal polyps and the nasal mucosa of patients with chronic rhinosinusitis. Several studies have demonstrated that macrolides reduce IL-8 and neutrophil concentration as well as modulate immune-complex reaction against Pseudomonas biofilm. They have also been shown to reduce nasal polyp size (50–53).
Sputum, middle meatus, and tracheal aspirate cultures can be used to guide antimicrobial therapy; however, these usually do not accurately correlate with the organism colonizing the sinuses (19,54). Despite lack of U.S. Food and Drug Administration approval for use in the pediatric population
Cystic Fibrosis and Sinusitis |
131 |
due to potential joint maldevelopment, quinolones are being used due to the prevalence of Psuedomonas (5). Aminoglycoside and other broad-spectrum antibiotics, such as piperacillin, ceftazidime, cefsulodin, and imipenem, have been frequently used in the past; however, renal, cochlear, and vestibular toxicities must be closely monitored.
We recommend aggressive, individually tailored medical treatment when CF patients present with sinusitis. In addition, maintenance nasal care is mandatory during times of quiescent disease. Hypertonic saline irrigation (3–7%), used two to three times a day, and a nasal steroid spray should be used daily. During flare-ups of acute disease, oral antibiotics with antipseudomonal coverage should be added to the nasal hygiene regimen for a minimum of three to four weeks. A short course of oral steroids can be given. Antihistamines and decongestants should be avoided in CF patients because these can cause mucous thickening and worsen disease. Broad-spectrum intravenous or oral fluoroquinolone antibiotics may be needed for those with persistent disease, polyposis, or sinus-induced pulmonary complications. Typically, CF patients harbor resistant organisms due to frequent antibiotic use to treat airway disease, and surgical management should be considered in patients with recalcitrant disease.
SURGICAL MANAGEMENT
Surgical management of sinusitis in CF patients is reserved for those who have failed medical management, have polyposis, or are awaiting lung transplantation. CF patient comorbidities related to lung and hematological abnormalities, as well as anatomical variances, present challenges for the sinus surgeon. Fortunately, the advent of endoscopic sinus surgery and power instruments has greatly facilitated surgical management of sinus disease and polyposis.
While timing, indications, and extent of surgery can be controversial in non-CF pediatric patients, it is generally agreed that CF patients deserve special consideration, especially regarding preoperative work-up (Table 2). CF patients who present with either frequent exacerbation of lung disease, polyposis, or radiological evidence of sinusitis along with clinical symptoms (nasal obstruction, facial pain, headache, and fevers) refractory to medical management merit surgical intervention. Nasal obstruction and the resulting increase in pulmonary effort is particularly concerning given the lack of pulmonary reserve in CF patients (55). It should be noted, however, that abnormal radiographic findings are common in CF patients, but are not always accompanied by symptoms.
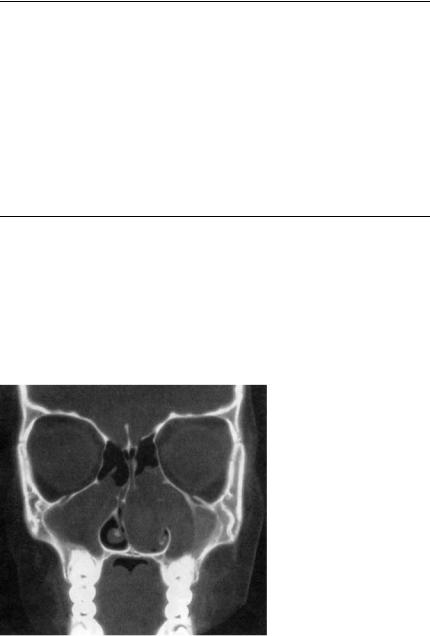
The preoperative imaging for CF patients is similar to non-CF patients; however, particular attention should be focused on CT findings (Fig. 2). Paranasal sinus development in CF patients has been shown to be characteristically different compared with controls who have non-CF
132 |
Derkay and Schraff |
Table 2 Preoperative Work-Up
All cases
CT scan (image-guidance system protocol, if available)
Serum electrolyte analysis
Complete blood count
Coagulopathy panel
Blood glucose level
Liver function test
Chest x-ray
Pulmonary function test
Sputum culture and sensitivity
When indicated
Arterial blood gas analysis
Electrocardiogram
Echocardiogram
Source: From Refs. 6, 59.
inflammatory sinonasal disease (13). CF patients have a higher incidence of hypoplastic or aplastic sinuses, especially the frontal and sphenoid sinuses, and have fewer pneumatization variants, such as Haller or Agger Nasi cells. Typically, these patients have medial bowing of the medial wall of the maxillary sinus as well as resorption of the uncinate process (32). It is hypothesized that these bony changes are caused by osteitis or a pressure-induced phenomenon caused by polyps and inspissated mucous (32,56).
Figure 2 Coronal CT scan of CF patient with sinusitis. Abbreviations: CT, computed
tomography; CF, cystic fibrosis.
Cystic Fibrosis and Sinusitis |
133 |
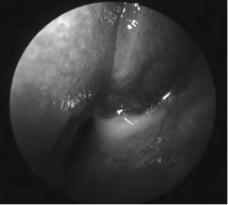
Other anatomical abnormalities, such as maxillary and ethmoid sinus hypoplasia, place the orbit at risk during surgery. In maxillary sinus hypoplasia, the orbit is enlarged and the lateral nasal wall is more laterally located. In ethmoid sinus hypoplasia, the lamina papyracea is more medial and often found medial to the lateral wall of the maxillary ostium. Furthermore, the fovea ethmoidalis is often lower in CF children compared with non-CF children and, when combined with other CF-related abnormalities, exposes these patients to a greater risk of intracranial injury or cerebrospinal fluid leak (13). However, recent advances in image-guided technology and the use of nasal endoscopes (Fig. 3) have greatly reduced the risks and complications of sinus surgery in CF patients. Endoscopic sinus surgery combined with an image-guidance system allows surgical precision to less than 2 mm (57) and should be considered if the equipment is readily available.
In addition to CT variances, the sinus surgeon must be wary of hematological abnormalities in CF patients. Nutritional deficiencies and coagulopathies are common in these patients. Frequently, CF can involve the liver and lead to impaired processing of Vitamin K as well as elevated prothrombin (PT) and partial thromboplastin times (PTT). These deficiencies need to be corrected preoperatively and monitored closely in the perioperative period. Pancreatic and intestinal involvement can greatly diminish the absorption of fat-soluble vitamins, which can have nutritional and hematological consequences.
These patients also deserve special consideration from an anesthesia standpoint. In children and teenagers with CF, the most common reason for anesthesia is sinus surgery (58). In the past, CF patients, due to poor lung function, had tolerated anesthesia poorly. However, as medical management of pulmonary disease has improved, the anesthetic risks have subsequently declined. Only about 5% of patients undergoing routine types of ear, nose,
Figure 3 Nasal endoscopy of CF patient with sinusitis.
134 |
Derkay and Schraff |
or throat surgery suffer minor postoperative complications (58). However, in patients with moderate to severe pulmonary disease, preoperative pulmonary function studies should be obtained. CF patients typically demonstrate an obstructive pattern with an increased functional residual capacity and decreased vital capacity, FEV1, and peak expiratory flow rate. Fibrosis, secretions and bronchospasm can cause ventilation problems. An indicator of advanced disease is increased levels of arterial CO2, which are usually low to normal in CF patients (58,59).
Preoperative patients should be given anti-reflux medications to prevent pulmonary irritation due to gastroesophageal reflux. Opioids should be avoided due to problems with constipation. It is recommended that all routine pulmonary medications, such as inhalers and corticosteroids, should be continued and given on the morning of surgery (59). Preoperative consultation with the patient’s pulmonologist is recommended since some patients will need ‘‘tuning-up’’ from a pulmonary standpoint prior to undergoing general anesthesia.
Intraoperatively, the choice of anesthetic agent is very important. Sevoflurane should be used given its bronchodilatory properties, and ketamine, isoflurane, and desflurane should be avoided. These agents have little bronchodilatory effect and can cause increased pulmonary secretions. Muscle relaxants can be used, but sparingly. It should be noted that aminoglycosides can potentiate the effect of neuromuscular blockers (6). Propofol is a good choice as a short-acting sedative. Stress dose steroids should also be considered in patients who take daily maintenance oral steroids.
Postoperatively, the head of bed should be elevated to 30 and the patient admitted to the post-anesthetic care unit for aggressive respiratory therapy. The extent of surgery and CF severity will determine whether the patient should be admitted to the hospital. These patients are at risk for respiratory depression, obstruction, pneumonia, and pneumothorax following endotracheal intubation (58). Stool softeners and laxatives can be used to alleviate constipation.
Sinus disease can be particularly problematic for the CF patient awaiting lung transplantation. The sinuses can act as a pathogen reserve that can seed the lungs in the post-transplant patient. To avoid this, all CF transplant patients need to be aggressively managed, both medically and sometimes surgically. There is controversy regarding prophylactic sinus surgery in pretransplant patients. Those in favor believe surgery will lower the sinus bacterial load, thus decreasing the chance of infection in the post-transplanted, immunocompromised patient. Others, however, have shown that pretransplant surgery confers no benefit and does not affect transplant success rates (3). A recent consensus statement stated that the benefit of sinus surgery toward preventing or decreasing the morbidity and mortality in the post-transplant patient is unknown (60).