

Гель - хроматография (эксклюзионная хроматография)
Это вид хроматографии, основанный на использовании различия в размерах молекул. Его называют также гель-фильтрацией или ситовой хроматографией. Неподвижной фазой является растворитель, находящийся в порах геля, а подвижной – сам растворитель, т.е. и подвижную, и неподвижную фазы составляет одно и то же вещество или одна и та же смесь вещества. Эксклюзионная хроматография представляет собой вариант жидкостной хроматографии, в котором разделение происходит за счет распределения молекул между растворителем, находящимся внутри пор сорбента, и растворителем, протекающим между его частицами. В отличие от остальных вариантов ВЭЖХ, где разделение идет за счет различного взаимодействия компонентов с поверхностью сорбента, роль твердого наполнителя в эксклюзионной хроматографии заключается только в формировании пор определенного размера, а неподвижной фазой является растворитель, заполняющий эти поры. Поэтому применение термина «сорбент» к данным наполнителям в определенной степени условно.
В процессе гель-хроматографии могут быть отделены крупные молекулы, которые гелем не сорбируются, т.к. их размеры превышают размеры пор, от мелких, которые проникают в поры, а затем могут быть элюированы. Модель разделения молекул по размеру в эксклюзионной хроматографии (рис.)


Объем эксклюзионной колонки можно выразить суммой трех слагаемых:
Vс = Vо+Vi+Vd
Где: Vо – мертвый объем – объем растворителя между частицами сорбента (объем подвижной фазы);
Vi – объем пор, занятый растворителем (объем неподвижной фазы);
Vd – объем матрицы сорбента без учета пор.
Полный объем растворителя в колонке Vt (его часто называют полным объемом колонки, так как Vd не принимает участия в хроматографическом процессе) представляет собой суммy объемов подвижной и неподвижной фаз:
Vt= Vо + Vi
Связь между удерживаемым объемом и молекулярной массой (или размером молекул) образца описывается калибровочной кривой (рис. вверху). Каждый сорбент характеризуется своей калибровочной кривой, по которой легко оценить область разделяемых на нем молекулярных масс. Точка А соответствует пределу эксклюзии, или мертвому объему колонки Vo. Все молекулы, масса которых больше, чем в точке А, будут элюироваться одним пиком с удерживаемым объемом Vo. Точка В отражает предел проникания, и все молекулы, масса которых меньше, чем в точке В, также будут выходить из колонки одним пиком с удерживаемым объемом Vt. Между точками А и В располагается диапазон селективного разделения. Соответствующий ему объем Vi = Vt - Vo часто называют рабочим объемом колонки. Отрезок CD представляет собой линейный участок калибровочной кривой, построенной в координатахVR – lgM. Этот участок описывается уравнением: Vr = C1 – C2 * lgM
Где: C1 – отрезок, отсекаемый на оси ординат продолжением отрезка СD,
C2 – тангенс угла наклона этого отрезка к оси ординат.
Всегда нужно стремиться выбирать колонку (или набор колонок) так, чтобы разделение анализируемого полимера протекало в пределах линейного участка калибровочной кривой. Удерживание молекул в эксклюзионной колонке определяется вероятностью их диффузии в поры и зависит от соотношения размеров молекул и пор, что схематически показано на рис. Коэффициент распределения Kd, как и в других вариантах хроматографии, представляет собой отношение концентраций вещества в неподвижной (Сi) и подвижной (Со) фазах:
Кd = Сi / Со Так как подвижная и неподвижная фазы имеют одинаковый состав, то Kd вещества, для которого обе фазы одинаково доступны, равен единице. Эта ситуация реализуется для молекул с самыми малыми размерами (в том числе и молекул растворителя), которые проникают во все поры (см. рис.) и поэтому движутся через колонку наиболее медленно. Их удерживаемый объем равен полному объему растворителя Vt. Все молекулы, размер которых больше размера пор сорбента, не могут попасть в них (полная эксклюзия) и проходят по каналам между частицами. Они элюируются из колонки с одним и тем же удерживаемым объемом, равным объему подвижной фазы Vo. Коэффициент распределения для этих молекул равен нулю. Молекулы промежуточного размера, способные проникать только в какую-то часть пор, удерживаются в колонке в соответствии с их размером. Коэффициент распределения этих молекул изменяется в пределах от нуля до единицы и характеризует долю объема пор, доступных для молекул данного размера. Их удерживаемый объем определяется суммой Vo и доступной части объема пор:
VR=Vo+Kd *Vi
Отсюда следует еще одно существенное отличие эксклюзионной хроматографии: в данном методе разделение заканчивается до выхода пика растворителя, в то время как в других вариантах ВЭЖХ компоненты смеси элюируются после пика растворителя. Если какое-либо вещество элюируется с удерживаемым объемом больше Vt, то это указывает на проявление других механизмов разделения (чаще всего адсорбционного). Адсорбционные эффекты обычно проявляются на жестких сорбентах, но иногда наблюдаются и на полужестких гелях, видимо, из-за повышенного сродства к матрице геля. Примером может служить адсорбция ароматических соединений на стирол-дивинилбензольных гелях. Исследователи предложили теорию единого механизма жидкостной хроматографии полимеров на жестких гелях, из которой следует, что изменением параметров взаимодействия в системе полимер – сорбент – растворитель можно переходить от адсорбционного механизма к эксклюзионному, и наоборот. В общем случае в эксклюзионной хроматографии нужно стремиться полностью подавить адсорбционные и другие побочные эффекты, так как они, особенно при исследовании молекулярно-массового распределения (ММР) полимеров, могут существенно исказить результаты анализа. Принципиальными отличиями эксклюзионной хроматографии от других вариантов являются заранее известная продолжительность анализа в конкретной используемой системе, возможность предсказания порядка элюирования компонентов по размеру их молекул, примерно одинаковая ширина пиков во всем диапазоне селективного разделения и уверенность в выходе всех компонентов пробы за достаточно короткий промежуток времени, соответствующий объему Vt.
Размеры пор могут быть регулированы изменением состава растворителя и набухания геля. Гели готовят на основе полиакриламида, крахмала, декстрана, агар-агара или других природных и синтетических соединений. Применяемые гели подразделяют на мягкие, полужесткие и жесткие. Мягкими являются высокомолекулярные органические соединения с незначительным числом поперечных связей (декстраны, крахмал) Полужесткими являются гели полученные путем полимеризации (стирогели – продукты сополимеризации стирола и дивинилбензола), на них осуществляется гель-проникающая хроматография. К жестким гелям относят силикагели и часто пористые стекла, хотя они и не являются собственно гелями. Они имеют фиксированные поры и используются при высоком давлении. Практическое применение гель-хроматографии в нефтехимии связано, в основном, с разделением смеси ВМС, например смол и асфальтенов.
Принципиальной особенностью метода является возможность разделения молекул по их размеру в растворе в диапазоне практически любых молекулярных масс – от 102 до108, что делает его незаменимым для исследования синтетических веществ и биополимеров. Хотя данный метод применяют, главным образом, для исследования ММР полимеров и анализа макромолекул биологического происхождения (белки, нуклеиновые кислоты и т.д.), указанные особенности делают его чрезвычайно перспективным для анализа низкомолекулярных примесей в полимерах и предварительного разделения проб неизвестного состава. Получаемая при этом информация существенно облегчает выбор наилучшего варианта ВЭЖХ для анализа данной пробы. Кроме того, микропрепаративное эксклюзионное разделение часто используют в качестве первого этапа при разделении сложных смесей путем комбинации различных видов ВЭЖХ.
По традиции процесс, проводимый в органических растворителях, все еще часто называют гель-проникающей, а в водных системах – гель-фильтрационной хроматографией. Принят единый термин, который происходит от английского «Size Exclusion» – исключение по размеру – и в наиболее полной степени отражает механизм процесса. Ограниченный диапазон коэффициентов распределения определяет и главный недостаток эксклюзионной хроматографии – заметно меньшее, чем в других вариантах ВЭЖХ, число пиков, которые могут быть полностью разделены на колонке заданной эффективности.
Лекция 14
ГАЗО-ЖИДКОСТНАЯ РАСПРЕДЕЛИТЕЛЬНАЯ ХРОМАТОГРАФИЯ (ГЖХ)
В 1941 г. американские ученые А. Мартин и Р. Синг впервые предложили метод хроматографии в жидкостно-жидкостном варианте и указали на возможность осуществления газо-жидкостной хроматографии. В 1952 г. А. Мартин и А.Джеймс создали теорию процесса и разработали конкретную методику анализа углеводородов ГЖХ. В основу было положено различие в коэффициентах распределения анализируемых веществ между 2-мя несмешивающимися жидкой неподвижной и подвижной газообразной фазами.
Неподвижной фазой является жидкость, подвижной фазой – газ-носитель.
Принципиальная схема ГЖ хроматографа (рис):
загрузка разделяемой смеси

1. баллон с газом-носителем
2. дозатор - устройство для ввода пробы в хроматографическую колонку
3. хроматографическая колонка с жидкой неподвижной фазой
4. термостат
5. детектор
6. блок - преобразует параметры веществ в разделенной смеси в электрический сигнал, усиливает сигналы
7. регистрирующее устройство
ПАРАМЕТРЫ, ХАРАКТЕРИЗУЮЩИЕ ХРОМАТОГРАММЫ ГЖХ Регистрируемая самописцем кривая изменения сигнала детектора называется хроматограммой.

Типичная выходная кривая (хроматограмма) проявительного (элюентного) анализа приведена на рис.:


А” А B C
где: точка «А”» соответствует вводу анализируемой пробы;
т. «А» – появлению на выходе несорбирующегося (слабосорбирующегося) компонента;
т. «В», «С» - появлению на выходе компонентов анализируемого вещества.
Линия А”ABC – называют нулевой линией
Кривая – называется хроматографическим пиком, характеризуется высотой h, шириной w и площадью.
1. Время удерживания tR - это время, прошедшее от момента ввода пробы в колонку до выхода максимума соответствующего пика, оно складывается из 2 величин:
– пребывание в пустотах сорбента (мертвый объем Vo),
– t0 - время удерживания несорбирующегося (слабосорбирующегося) вещества, например, воздуха (при этом появляется маленький пик).
Исправленное время удерживания – время, прошедшее с момента появления максимума пика несорбирующегося (слабосорбирующегося) компонента до пика соответствующего соединения:
tR'(1) = tR(1) - t0
Время удерживания зависит от ТоС, скорости газа-носителя и размера образца.
2. Относительное время удерживания tотн, определяется как отношение времени удерживания неизвестного вещества tх к времени удерживания эталонного вещества tэтал:
t отн = tх / tэтал
Относительное время удерживания ни от чего не зависит.
3. Умножив время удерживания на объемную скорость элюента F или среднюю скорость газа-носителя (мл/мин), прошедшего через колонку с момента ввода пробы до момента появления максимального пика, получим удерживаемый объем VR:
VR = tR . F
Удерживаемый объем пропорционален времени удерживания.
Приведенный или исправленный объем удерживания VR' – это объем удерживания с поправкой на мертвый объем колонки V0, т. е. на объем удерживания несорбируемого (слабосорбируемого) компонента:
VR' = VR - V0
VR' пропорционален отрезку АА” (рис.), характеризует удерживаемый объем несорбирующегося газа, или мертвый объем колонки. Характеристикой удерживания является также коэффициент емкости K', определяемый как отношение массы вещества в неподвижной фазе к массе вещества в подвижной фазе:
K' = mн / mп
Величину K' легко определить по хроматограмме: K'= tR'- t0 / t0
4. В ГЖХ при анализе соединений одного гомологического ряда для качественной идентификации отдельных компонентов смеси пользуются индексами удерживания – индексами Ковача Ix. В качестве стандарта выбирают нормальный алкан. Далее берут два соседних алкана, один из которых элюируется до, а другой – после исследуемого соединения.
lg t’x - lg t’n
Ix = 100 ---------------- + 100n
lg t’(n+1) - lg t’n
где: t’n < t’x < t’(n+1) n – число атомов углерода алкана
Для качественного анализа и идентификации какого-либо неизвестного соединения снимают хроматограмму этого соединения, а также хроматограммы двух нормальных парафинов (алканов) с известным числом атомов углерода в молекуле, время удерживания одного из которых меньше, а другого больше, чем время удерживания исследуемого соединения в тех же условиях. Индексы удерживания позволяют не только идентифицировать по их значениям неизвестные вещества, но и предсказывать, каким должен быть индекс удерживания того или иного вещества на определенной фазе и при определенной температуре.
КРИТЕРИИ ЭФФЕКТИВНОСТИ ХРОМАТОГРАФИЧЕСКОГО РАЗДЕЛЕНИЯ
Для полной характеристики хроматографической колонки необходимо знание того, насколько избирательно действие выбранного адсорбента по отношению к компонентам разделяемой смеси, т.е. необходимо знание селективности адсорбента. Важнейшими параметрами хроматографического разделения являются его эффективность и селективность.
Существенное значение в теории хроматографического процесса имеет метод теоретических тарелок (ТТ). В этом методе (ТТ) впервые предложенном А.Мартином и Р.Сингхом, хроматографическая колонка мысленно делится на ряд элементарных участков –«тарелок» и предполагается, что на каждой тарелке очень быстро устанавливается равновесие между сорбентом и подвижной фазой. Каждая новая порция газа-носителя вызывает смещение этого равновесия, вследствие чего часть вещества переносится на следующую тарелку, на которой, в свою очередь, устанавливается новое равновесное распределение и происходит перенос вещества на следующую тарелку. В результате этих процессов хроматографируемое вещество распределяется на нескольких тарелках, причем на средних тарелках его концентрация оказывается максимальной по сравнению с соседними тарелками. ТТ – условный участок колонки, на котором происходит единичный акт разделения смеси.
L L
Число ТТ N = 5,54 * (------)2 N= ------ H= L / N
w 1/2 H
где: H - высота, эквивалентная теоретической тарелке (ВЭTТ)
L - длина слоя сорбента, на которой произведено поглощение и размещено N теоретических тарелок
w 1/2 - ширина пика на половине высоты
Эффективность колонки, измеряемая высотой теоретических тарелок (ВЭТТ) и обратно пропорциональная их числу (N) тем выше, чем уже пик вещества, выходящего при том же времени удерживания. Значение эффективности может быть вычислено по хроматограмме по следующей формуле:
tR
Число ТТ N = 5,54 * (-------)2
w 1/2
где: tR - время удерживания,
w 1/2 - ширина пика на половине высоты
В набивных колонках N = 5-20 тыс В капиллярных N =106
Задачей хроматографии является разделение веществ за счет разности в их удерживании. Степень разделения двух веществ называется разрешением (resolution). Термин «разрешение» необходим для того, чтобы выразить степень разделения двух пиков количественно. Непосредственно из хроматограммы разрешение двух пиков вычисляется как отношение разницы во временах удерживания к половине суммы ширин пиков на уровне базовой линии. При рассмотрении разделения смеси двух компонентов важным параметром служит также степень (коэффициент) разделения RS:
RS = t R .
w 1/2 (1)+ w 1/2 (2)
Где: w1/2 - ширина 1 и 2 пиков на половине высоты
Пики считаются разрешенными, если величина RS больше или равна 1,5.
Для современных аналитических колонок размера 250х4.6 (длиной 250 мм и внутренним диаметром 4.6 мм) при скорости подачи элюента 2 мл/мин нулевое время составляет величину порядка t0 ≈ 1.3 мин., а оптимальное время элюирования целевого соединения изменяется от 4 до 7 мин. Если вещества не разделяются, то разрешение равно нулю. При разрешении, равном единице, перекрываются лишь около 2% площадей двух пиков – при условии, что пики идеально симметричные, имеющие форму Гауссовой кривой.
2. Селективность разделения (коэффициент) двух веществ Kc может быть выражен с помощью времен удерживания, по уравнению:
tR(2) – tR(1)
Kc= 2 -------------------
tR(2) + tR(1)
Коэффициент селективности может быть выражен также соотношением:
Г2 – Г1
Kc = 2 -------------- Г – коэффициент Генри
Г2 + Г1
Концентрация вещества в адсорбенте (неподвижной фазе), г/см3
Г = ----------------------------------------------------------------------
Концентрация вещества в газовой фазе (подвижной фазе), г/см3
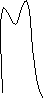
Селективность адсорбента связана с различием его адсорбционной способности по отношению к компонентам анализируемой смеси. Разделительная способность хроматографической колонки определяется как ее эффективностью, так и селективностью. Достаточно полное разделение можно осуществить лишь при сочетании высокой эффективности колонки с хорошей селективностью фазы: (рис. )
высокая эффективность высокая эффективность низкая эффективность
высокая селективность низкая селективность высокая селективность

 1
2 1 2
1 2
1
2 1 2
1 2

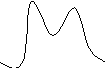

Если t R = w 1/2 (1)+ w 1/2 (2), тогда RS =1 – происходит еще недостаточно полное разделение
RS ≥1 - можно говорить об эффективном разделении, это основное условие эффективного разделения (рис)





 1
2 1 2
1+2
1
2 1 2
1+2



0,6 < RS <1 0,4 < RS <0,6 RS <0,4
RS ≤ 1 - неэффективное разделение
Г рафически
уравнение Генри выглядит так: (рис
рафически
уравнение Генри выглядит так: (рис


 Сж
Сж
Сж
Сж



 2
1 2
1
2
1 2
1


Сг Сг
tg =Г tg = Г
2 – лучше растворяется – значит неподвижная фаза удерживает сильнее вещество
1 – вещество выйдет быстрее, т.к. хуже удерживается неподвижной фазой
Для каждого соединения имеется свой коэффициент, и чем больше различие (больше угол), тем лучше разделить вещества.
Регулировать селективность колонки можно изменяя природу неподвижной фазы
Достоинства метода ГЖХ
1. Высокая разрешающая или разделяющая способность (за 0,5-1,0 час можно получить хроматограмму разделения от 10 до 100 соединений.
2. Высокая чувствительность (можно обнаружить вещества с концентрацией 10-10 %)
3. Малый размер пробы, необходимой для анализа (объем 1-10 мкл, 1 мкл=10-6 л)
4. Малая продолжительность процесса хроматографического разделения
5. Высокая точность метода определения количества веществ (средняя относительная погрешность - 5%, а в приборах более высокого класса до 2 %.
6. Простота аппаратурного оформления, доступность прибора.