

шпоргалка / Himiya
.doc|
№1. Предмет, сод-ие и задачи курса Задача дисциплины – развитие активного мышления на основе системно-структурного подхода, с использованием современного уровня хим. науки с целью подготовки будущих специалистов к творческому освоению профилирующий дисциплины. Химия изучает химическую форму движения, под которой понимают качественное изменение вещ-в, т.е. превращение одних вещ-в в другие. Химия – это наука о вещ-ах и законах их превращений. Курс химии должен дать студентам современное научное представление о хим-ом уровне организации материи и присущей этому уровню хим. форме движения материи, показать значение химии в производстве товаров народного потребления, определении их качества. Изучение химии имеет большое значение в подготовке высококвалифицированных специалистов товароведов, т.к. различные хим. реакции лежат в основе производства и в процессе хранения продуктов питания и непищевых товаров. |
№2. Химия как наука естествознания. Естествознание рассм-т природу как единое целое. Поэтому естествознание – это наука из последовательно вложенных друг в друга осн. частей отдельных естеств.-научн. дисциплин. как химия, физика и т.д. В этом ракурсе химия как наука, имеющая в своем основании физику. она непосредственно представляет основание для биологии. Физика→Химия→Биология→Психология (занимает центральное место)→Физика Учение о системе начинает развитие химия. Его также называют системным подходом или системный анализ. 1830-е годы Берцелиоз выдвинул электрохимическую теорию (молекулы образуются за счет электростатического притяжения атомов). 1840 г. – Жерар – молекула состоит из атомов и их групп, так тесно взаимосвязываются, что их самостоятельное существование без существенного изменения их качества невозможно. Распад молекул приводит к качественно новым веществам. CH4 (CH3+CH3→C2H6) Жерар назвал молекулы унитарной (единой) системой. Существует два вида множества: 1.Суммативное (адитивное) суммирование; 2.Системное (система) множество элементов или частей, в которой все элементы не только тесно взаимосвязаны, но влияют друг на друга и качественно преобразуют друг друга. М. сделать вывод: Что включая в себя один и тот же элемент, система делает его различным в зав-сти от связанных с ним пр-пов. Сохр-яя субординацию ест. наук, м сделать вывод. что одну науку м. выводить из другой. |
(4) Этот способ послужил основой 3-й концептуальной химии. Учение о химическом процессе и предмет изучения не структура молекулы, а организация кинетической системы, в которой структура молекулы представлена как частность. Появились новые способы изучения, возникло производство многоатомарных материалов, синтетический каучук из нефтегазового сырья, этиловый спирт, азотные удобрения из азота воздуха и т.д., то есть появилась технология нефтехимических производств. Процесс развития химии в форме последовательного появления и сосуществования четырех концептуальных систем обусловлен 2-я категориями внешних факторов.
- атом элемента - молекула вещества - система реагирующих веществ - высокоорганизованная каталитическая система Эта иерархия природы – иерархия 4-х концептуальных систем химии: - учение о составе - структурная химия - учение о химическом процессе -эволюционная химия Внутренними противоречиями, являющимися стимулами развития химии, выступают противоречия между эмпирическими и теоретическими знаниями, между теориями разной степени общности, между истиной и заблуждениями, между различными научно-исследовательскими прогами.
|
||||||||||||||||||||||||||||
|
№4. химия и экспертиза потр.тов-в. Химия и экспертиза потребительских товаров. Мы живем в рыночной экономической системе и для правильного функционирования рынка, экономики страны важной составляющей является производство продукции, индификация товара. Сейчас на рынке большое кол-во фальсифицированного товара, которое необходимо выявить. Идентификация-соответствие продукции нормативным документам и требованиям. Область применения идентификации широко: сертификация, формирование ассортмента, контроль качества, экспертиза, оценка качества. Выбор метода идентификации: измерительный-физико-химические метоы, органолептический-чисто товароведный,но понимание что такое запах-пары соответствующего вещества-с точки зрения химии,вкус вещества на языке также связан со строением молекул соотв вещ-ва. С помощью химических знаний создаются комплексные запахи (парфюмерия), производственные запахи(запах свежеиспеченного хлеба, запах груши в лимонаде), химия определяет потребителские св-ва товаров. Существуют измерительные методы для определения действительных значений показателей качества с помощью технических у4стройст,к измерительным относят хроматографический метод,спектральный,фото-электроколориметрический,потенциометрический,рефрактометрический,микроскопический. Таким образом химия и экспертиза потреб товаров тесно связаны др с др |
№8. Природа и образование хим. св. (ММО). Энергия и длина связи. Обр-ие из атомов мол-л, молекулярн. ионов, ионов, кристалл-х, аморфных и др. вещ-в сопр-ся уменьш-ем энергии по ср-ию с невзаимодейств-ми атомами. При этом миним. энергии соотв-ет опред. расположение атомов друг отн-но друга, кот. отвечает существенное перераспр-ние электрон. плотности. Силы, удерж-щие атомы в нов. образованиях, получили обобщенное название «химическая связь». Согласно электрон. теории валентности, хим. связь возникает за счет перераспр-ния электронов валентн. орбиталей, в рез-те чего возн-ет устойчивая электрон. конфигурация благородн. газа(октет) за счет обр-ния ионов(В. Коссель)или обр-ия общ.электрон.пар (Г. Льюис) По своей природе хим. св. предст-т собой взаимод-вие м/у положит-но заряж-ми ядрами и отриц-но заряж-ми электронами, а также электронов друг с другом. Для описания хим. связи широко исп-ся 2 метода:метод молекулярных орбиталей(Малликен, Хунд) и м.валентных связей(Гейтлер, Слетер, Лондон).Согласно ММО, молекула – сов-сть «+» ядер и «-» электронов. Описать молекулу по ММО – значит опр. тип ее орбиталей, их энергию и выяснить хар-р распредел-я электронов по орбиталям.Электрон может нах-ся в связ-щей или разрыхляющей области. В соотв., бывают связ-щиеи и разрыхл-щие МО. Связыв-щая МО хар-ся повышенной электронной плотностью в пространстве меж ядрами, поэтому такая орбиталь энергетич. более выгодна. Наоборот, на разрыхляющей орбитали эл. плотность конц-ся за ядрами, эта МО менее выгодна. МО, образов-ые перекрыванием АО по оси располож-я ядер наз-ся сигма-орбиталями. Сигма-связь хар-ся тем, что их эл. плотность конц-ся меж связ-ми атомами по линии, проходящей через их центры. Отвечают макс-но возможной степени перекрывания АО, поэтому прочны, возможно вращ-е вокруг сигма-связи. Пи-связь – концентр-е эл. плотности не меж атомами, а над и под плоскостью, проходящей через сигма-связь( возникает при перекрывании эл. облаков по обе стороны от линии соед-я атомов. Хим. связь хар-ся энергией и длиной. Мерой прочности связи служит энергия, затрачиваемая на разрушение связи, или выигрыш в энергии при образовании соединения из отдельных атомов (Eсв). Так, на разрыв связи H–H затрачивается 435 кДж∙моль–1, а на атомизацию метана CH4 – 1648 кДж∙моль–1, в этом случае EC–H = 1648 : 4 = 412 кДж. Длина связи (нм) – расстояние между ядрами в том или ином соединении. Обычно длина связи и ее энергия антибатны: чем больше длина связи, тем меньше ее энергия. Хим. связь обычно изображается черточками, соединяющими взаимодействующие атомы; каждая черта эквивалентна обобщенной паре электронов. В соединениях, содержащих более двух атомов, важной характеристикой является валентный угол, образуемый химическими связями в молекуле и отражающий ее геометрию.
|
№6. Хар-ка энерг. сост. ē в атоме. Правила и пор-к заполн. АО. Заселение электронами атомн. орбиталей осущ-ся согласно пр-пу наим. энергии, правилу Клечковского, пр-пу Паули и правилу Гунда. Принцип наименьшей энергии требует, чтобы электроны заселяли АО в порядке увеличения энергии элекронов на этих орбиталях. Это отражает общее правило – максимуму устойчивости системы соответствует минимум ее энергии.
Пр-ло
Клечковского: увеличение Энергии и
соот-но заполн-е орбителй происх-т в
порядке возр-ния суммы квант. чисел
n+l,
а при сумме
n+l
в порядке возр-ния числа n.
Соотв-но подоболочки выстр-ся в след.
ряд: 1s<2s<3s<3p<4s
Принцип
Паули
запрещает в многоэлектронном атоме
находиться электронам с одинаковым
набором квантовых чисел. Это означает,
что два любых электрона в атоме (или
молекуле, или ионе) должны отличаться
друг от друга значением хотя бы одного
квантового числа, то есть на одной
орбитали может быть не более двух
электронов с различными спинами
(спаренных электронов). Каждый подуровень
содержит 2l + 1 орбитали, на которых
размещаются не более 2(2l + 1) электронов.
Отсюда следует, что емкость s-орбиталей
– 2, p-орбиталей – 6, d-орбиталей – 10 и
f-орбиталей – 14 электронов. Если число
электронов при заданном l просуммировать
от 0 до n – 1, то получим формулу
Бора–Бьюри, определяющую общее число
электронов на уровне с заданным n: Эта формула не учитывает межэлектронное взаимодействие и перестает выполняться при n ≥ 3.
Орбитали
с одинаковыми энергиями (вырожденные)
заполняются в соответствии с правилом
Гунда:
наименьшей энергией обладает электронная
конфигурация с максимальным спином.
Это означает, что если на p-орбитали
три электрона, то они располагаются
так:
|
||||||||||||||||||||||||||||
|
№3. Осн. проблема химии и общ. сп-бы ее решения. Осн. проблема химии: 1) получение в-в с заданным комплексом свойств 2) выявление сп-бов управления св-вами вещ-в Важнейшей особенностью основной проблемы химии является то, что она имеет ограниченное и строго определенно число способов решения. Речь идет не о частных способах решения качественного разнообразия веществ, а об общих способах решения качественного разнообразия и качественных превращений веществ. Речь идет о зав-сти св-в вещ-в от 4-х осн. факторов: 1) от элементного и молекулярного состава веществ; 2) от структуры веществ; 3) от кинетических факторов, влияющих на реакционную способность; 4) от высоты химической организации веществ. (1) Этот способ появился в древней натурфилософии и существовал 2 тыс. лет вплоть до работ Р. Бойля. За это время было предложено два различных объяснения разнообразия тем природы. - Демокрит, Лукреций Кар, Эпикур предложили для этого атомистское объяснение, высказав гениальную догадку, что атомы первоматерии различны по массе, объему, конфигурации. - Аристотель, Эмпедокл попытались объяснить видимое разнообразие тел с антиатомистских позиций, считая разнообразие возникшим от различных сочетаний уже известных свойств стихий. Оба эти способа предусматривали некие абсолютные начала, следовательно, 1-й способ решения основной проблемы химии: субстанция – акциденция. Отличительные черты способа: - умозрительность, приобретающая силу абстракции, но лишенная эмпирических (опытных) начал. - логическая дедукция, претендующая на всеобщность объяснения, но не опирающаяся на критерий практики. - выбор объекта-первоматерии или субстанции вместо веществ с их спецификой качественных изменений. Особенности 1-го способа, взятые вместе, настолько выделяют его относительно последующих, что возникает некая идея о границе между «научной химией» и «не вполне научной», существовавшей до работ Бойля. (2)Этот способ появился во 2-й половине XVII века – Бойль. Преобразования в области производства, произошедшие в эпоху Возрождения, вызвали коренные преобразования в области естествознания. Например, низвержение первого бесполезного для практики способа решения проблемы химии. И появление нового способа с принципиально новым экспериментальным подходом к изучению природы. Исследования Бойля привели к выводу о том, что качества и свойства тел не имеют абсолютного характера и зависят от того, из каких материальных объектов эти тела составлены. - Этот вывод отверг представления об элементах-стихиях. - Утвердил представления об элементах, как о простых, далее неразложимых телах, из которых состоят все химические соединения. - На основе признания материальности элементов впервые установил общность ранее разрозненных учений об атомах и элементах. – Идея о том, что наименьшей частицей вещества является состоящая из атомов корпускула (молекула). и способ решения проблемы: состав – свойства. Этот способ положил начало экспериментальной химии, определяя ее как науку о химических элементах и их соединениях. Это первый уровень научных химических знаний. И на нем появилась технология основных неорганических веществ. Возникшее при этом учение о составе – первая концептуальная система химии. (3) 3-й способ был вызван переходом от мануфактурного к фабричному производству. Основой же фабричного производства в 18-м и первой половине 19-го века была легкая (текстильная) промышленность. Переход стимулировал переработку огромной массы веществ растительного и животного происхождения, состав которых крайне однообразен: 5 – 7 химических элементов на все разнообразие + 12 элементов-катализаторов. В этих условиях, а также в результате открытия изомеров и полимеров, появилась идея о том, что свойства и качественное разнообразие веществ обуславливает не только состав, но и еще каким-то фактором, этим фактором явилась структура. Появился новый способ решения проблемы – не только от состава, но и от структуры. Этот 3-й способ стимулировал целый ряд возникших теорий, а теории положили начало ΙΙ концепции. Поднявшись на новый уровень знаний, химия превратилась из науки аналитической в науку синтетическую. Поэтому историки называют этот период «триумфальным маршем синтетического синтеза», так как химия отметила получение синтетических красителей в текстильной промышленности, получение лекарств, искусственного шелка. Т. о. на этом уровне развития химии возникла технология органических веществ и ее появление было ошеломляющим, так как подобные материалы до этого производили в ограниченном количестве при переработке огромного количества сырья, полученного с огромными затратами самого низкопроизводительного с/х труда. Триумф был недолгим. С развитием автомобилестроения возникли новые требования к материалам, нужны стали в больших количествах со строгозаданными свойствами (высокооктановое топливо, моторные масла, смазочные материалы, каучук и т.д.). Для получения этих материалов в больших количествах способ, основанный на структурной химии, оказался недостаточным, т.к.: 1)не обеспечивал экономически приемлемого выхода продукции 2) ориентировался на активные исходные вещества, получаемые из растительного сырья 3) не располагал необходимыми возможностями для управления процессом синтеза, поэтому под влиянием возникших требований, химия была вынуждена перейти к 4-му способу решения проблемы. |
№5. Уровни орг-ции материи. Двойств. природа микрочастиц. Основа окр. действ-сти – материя. Физ. мир хар-ся очень большим диапазоном масштабов. Хар-ый размер иссл. объекта – наиболее естествен. признак уровней структ. организации материи (второстеп. признак: масса, время протекания процесса).3 уровня: 1)микромир(d<10-10м;m=10-10кг;t=10-22c,квант.механика); 2)мегау-нь(d>107м;m=1020кг;t=не выводит, астрофизика); 3)макроуровень(d=10-10-107м;m=10-10-1020c,класс.мех-ка). Между ними нет жестких границ. ФОРМЫ МАТЕРИИ:1)невеществен.(mo=0;Voне равно 0;теория электромагн. поля Максвелла; занимает все пространство - делокализация; бесконечна; основатель Аристотель; характеризует волновые свойства) 2)веществ.(mo не равно 0;Vo=0;законы Ньютона; локализация, т.е. можно определить местоположение; корпоскулярная сущность; основатель Демокрит характеризует корпуск. свойства микрочастиц) Для описания электромагнитного излучения привлекают как волновые, так и корпускулярные представления Луи де Бройль предложил (1924) распространить корпускулярно-волновые представления на все микрочастицы, т.е. движение любой микрочастицы рассматривать как волновой процесс. Математически это выражается соотношением де Бройля, согласно которому частице массой т, движущейся со скоростью v, соответствует волна длиной А: А = h/(mv). |
№9. Ковалентная связь. Хим.связь, образованная путем обобществления пары электронов двумя атомами, наз-ся ковалентной связью. Если ков-ая связь образована одинаковыми атомами, н-р Н – Н, О = О, Cl – Cl, N=N, то обобществленные электроны равномерно распределены м\у ними. Такая связь наз-ся ковалентн неполярной связью. Если же один из атомов сильнее притягивает электроны, то электрон. пара смещается в сторону этого атома. В этом случае возникает полярная ков-ая связь. Критерием способности атома притягивать электрон м. служить электроотриц-ть. Чем выше ЭО у атома, тем более вероятно смещение электронной пары в сторону ядра данного атома. Поэтому разность элекроотриц-ти хар-ет полярность связи. Вследствие смещения электронной пары к одному из ядер повышается плотность отрицательного ядра у данного атома и соотв-но атом получает заряд, называемый эффективным зарядом атома б-. У второго атома повышается плотность положительного заряда б+. Вследствие этого возникает диполь, представляющий собой электрически нейтральную систему с двумя одинаковыми по величине положительным и отрицательным зарядами, находящимися на определенном расстоянии (длина диполя) lд друг от друга. Основной кол-венной хар-кой диполя является дипольный момент μ=ql (Дебай). Дипольн. момент величина векторная и предст-т собой векторную сумму дипольн. моментов всех хим. связей в молекуле и несвязанных эл. пар. Ков. связь харак-ся 3-мя св-вами: Насыщаемость. Причиной этого явл-ся св-во насыщения хим. связи, которая и предопределяет формулу в-ва. При приближении 3-го атома к молекуле водорода электрон будет иметь одинаковое направление спина по отношению к одному из электронов в молекуле Н2. Это будет сопровождаться повышением энергии сис-мы, что приведет к отталкиванию 3-го атома. Таким образом в молекуле использованы обе возможности спина, т.е. происходит насыщение хим. связи. Направленность. В соответствии с основным положением метода ВС для образования хим. связи электронные облака ориентируются в направлении максимального перекрывания, при этом в образовании связи участвуют одноэлектронные облака. Учитывая, что форма эл. облака может быть различной, поэтому и способы перекрывания облаков различаются. В зависимости от этих способов перекрывания различают π и σ-типы связей. σ-связь образуется вдоль линии, соединяющей центры атомов. π-связь образуется по обе стороны о линии, соединяющей центры атомов. Таким образом в молекуле N2 имеется 1 σ-связь и 2 π-связи. Следует отметить, что в первую очередь образуется всегда σ-связь. Она является более прочной. Каждая последующая π-связь будет слабее предыдущей. Поляризация. При взаимодействии разных атомов образующаяся хим. связь всегда более или менее полярна. При взаимодействии эл. облаков подобного рода общая эл. плотность всегда смещается к более ЭО элементу. При этом этот элемент приобретает некоторый отрицательный заряд, а элемент, от которого оттянута общая эл. плотность, приобретает некоторый положительный заряд. Такое перераспределение эл. плотности приводит к образованию полюсов или диполя. Длина. Длина связи хар-ет расстояние между атомами в молекуле . Чем меньше длина связи, тем связь прочнее. В зависимости от способов перекрывания различают π и σ-типы связей. σ-связь образуется вдоль линии, соединяющей центры атомов. π-связь образуется по обе стороны о линии, соединяющей центры атомов. Таким образом, в молекуле N2 имеется 1 σ-связь и 2 π-связи. Следует отметить, что в первую очередь образуется всегда σ-связь. Она является более прочной. Каждая последующая π-связь будет слабее предыдущей. |
||||||||||||||||||||||||||||
|
№10. Ионная связь. Ионная химическая связь – электростатическое взаимодействие отрицательно и положительно заряженных ионов в хим. соединении. Такая связь возникает лишь в случае большой разности ЭО атомов, например м/у катионами металлов первой и второй групп периодической системы и анионами неметаллов 6 и 7 групп. св-ваТ.к. электрическое поле иона имеет сферический хар-р, то для ионной связи не характерна направленность. Ионной связи также не св-но насыщаемость, т.к. ион способен взаимодействовать со многими соседними ионами противоположного знака, число кот-х зависит от зарядов ионов и соотношений геометрических размеров. Ионная хим. связь проявляется в твердых вещ-ах с ионной кристаллической решеткой. Т.к. энергия ионизации больше энергии сродства к электрону, то полн. перехода электронов не происх-т даже в случае пары атомов с больш. разностью ЭО. Поэтому и чисто ионная связь не сущ-ет. Можно лишь говорить о доле ионности связи. |
||||||||||||||||||||||||||||||
|
№11. Межмолек. взаимод-вия: электростат., дон.-акцепт., водородн. св и их влияние на св-ва в-в. Когда в-во находится в виде газа, то мол-лы хаот-ски движутся и находятся больш. частью времени на больш. расстояниях др. от др. Силы взаимод-вия м/у ними малы. В жид. и тв. состояниях расстояния м/у частицами в-ва малы и силы взаимод-вия м/у ними велики. Они и удерж-т частицы жид-ти или тв. в-ва др. около др.Силы, удерж-щие эти частицы, имеют электростатическую природу. Если в-во имеет молекулярн. стр-ру, то м/у мол-лами действ-т межмолек. силы или силы Ван-дер-Ваальса. Межмолекулярн. взаимод-вие отличается от хим-го тем, что проявл-ся на знач-но больших расстояниях (500-600 нм). В основе межмолекулярн взаимод-вия лежит электростат. взаимод-вие диполей. Это взаимод-вие мол-л подраздел-ся на: ориентационное, индукционное и дисперсионное. - Индукционное. Индукц. взаимод-вие возникает м/у неполярн. мол-лами, способными к поляризации (СО2) или м/у полярными и неполярными мол-лами. В рез-те их взаимод-вия возн-т индуцированные диполи. Они возн-т потому, что каждый атом создает в близи себя электр. поле, оказывающее поляризующее д-вие на ближайший атом соседней мол-лы. Мол-ла поляризуется, а образовавшийся диполь в свою очередь поляризует соседние мол-лы. Индуцированный диполь усиливает дипольный момент полярной молекулы. -Ориентационное.Ориентац. взаимод-вие проявл-ся м/у полярн. мол-лами. В рез-те беспорядочн. тепл. движения мол-л при их сближении соседние молекулярн. диполи ориент-ся друг к другу противоположно заряжен. полюсам, поэтому м/у ними возникает взаимное притяжение. Чем более полярны мол-лы, тем больше ориентир. взаимод-вие. С повыш-м темп-ры ориентац. взаимод-вие ослаб-ет. -Дисперсионное.Дисперс. взаимод-вие обусл-но взаимод-вием мол-л др. с др. за счет их мгновенных микродиполей. Они появл-ся за счет движения электронов и колебания ядер в мол-лах. Дисперсионное взаимод-вие увел-ся с увел-ем размеров атомов. При сближении мол-л ориентация диполей перестает быть независимой и их появление и исчезновение в разных молекулах происходит в такт друг другу, и сопровождается их притяжением, при отсутствии синхронности происходит их отталкивание. Донорно-акцепторная связь.Если одна из мол-л имеет электрон. пары, а другая свободн. орбитали, то м/у ними возн-ет донорно-акцепторн. взаимод-вие. Т.е. оно осущ-ся за счет готовой электронной пары. Донорно-акцепт. взаим-вие м/у мол-лами обусл-ет переход в-ва из газообр. состояния в жид. или тверд. Водородная связь. Сущ-т особый вид межмолекул. и внутримол-го взаимод-вия с участием атома водорода – водородн. связь. Ее образ-ие обусл-но тем, что в рез-те сильн. смещения электрон. пары к электроотриц. атому атом водорода, обладающий эффективным положит. зарядом, м. взаимод-вовать с др. электроотриц. атомом (F, O, N, реже Cl, Br, S). Энергия такого электростат. взаимод-вия составляет 20–100 кДж∙моль–1. Водородные связи м. б. внутри- и межмолек-ми. Внутримолекулярная водородн. связь образуется, напр-р, в ацетилацетоне и сопров-ся замыканием цикла Мол-лы карб. кис-т в неполярн. раств-лях димеризуются за счет двух межмолек. водородных связей Искл-но важн. роль водородн. связь играет в биол. макромол-лах, таких неорг. соед-х как H2O, H2F2, NH3. За счет водородн. связей вода хар-ся столь высокими по сравнению с H2Э (Э = S, Se, Te) темп-ми плавления и кипения. Если бы водородн. связи отсутствовали, то вода плавилась бы при –100 °С, а кипела при –80 °С.
|
||||||||||||||||||||||||||||||
|
№12. Агрегатн. сост-е в-ва. Зав-ть физ. св-в от типа хим. связи в мол-лах, м/у мол-ми и ионами. В обычных усл-х атомы, ионы и молекулы не существуют индивидуально. Они всегда составляют только части более высокой организации вещества, практически участвующего в химических превращениях, — так называемого агрегатного состояния.В зависимости от внешних условий вещества могут находиться в разных агрегатных состояниях — в газовом, жидком, твердом. Природа сил притяжения частиц, образующих вещество, во всех состояниях электрическая, т.е. прямо или косвенно связана с участием электронов. Переход из одного агрегатного состояния в другое не сопровождается изменением стехиометрического состава вещества, но обязательно связан с большим или меньшим изменением его структуры. В этом смысле переход из одного состояния в другое относится к явлениям химическим. Конечно, здесь, как и всегда, нужно помнить об относительности и условности разграничения, в том числе и разграничения понятий физическое и химическое явление. Если вещество находится при низкой температуре, частицы его образуют правильную геометрическую структуру, в таком случае энергии связей между частицами больше энергии тепловых колебаний, которые не нарушают образовавшуюся структуру, — вещество существует в твердом состоянии. Твердое состояние По степени распространенности среди твердых тел основным является кристаллическое состояние, характеризующееся определенной ориентацией частиц (атомов, ионов, молекул) относительно друг друга. Это определяет и внешнюю форму вещества в виде кристалла. В идеальных случаях кристалл ограничен плоскими гранями, сходящимися в точечных вершинах и прямолинейных ребрах .Одиночные кристаллы — монокристаллы — существуют в природе, их также получают искусственно. Однако чаще всего встречающиеся кристаллические тела представляют собой поликристаллические образования — сростки большого числа по-разному ориентированных мелких кристаллов неправильной внешней формы. Форму кристаллов изучает кристаллография. В соответствии с геометрической формой кристаллов возможны следующие их системы: кубическая, тетрагональная, орторомбическая, моноклинная, триклинная, гексагональная и ромбоэдрическая. Характерной особенностью кристаллических тел, вытекающей из их строения, является анизотропия. Она проявляется в том, что механические, электрические и другие свойства кристаллов зависят от направления внешнего воздействия сил на кристалл.Частицы в кристаллах совершают тепловые колебания около положения равновесия (узла кристаллической решетки). В соответствии с природой составляющих структурных частиц кристаллические решетки могут быть ионными, атомными (ковалентными или металлическими) и молекулярными. Ионная решетка состоит из ионов противоположного знака, чередующихся в узлах. В атомных решетках атомы связаны ковалентной или метеллической связью. Металлы и их сплавы образуют атомно-металлические кристаллы. Различия в типе химической связи кристаллов определяют существенное отличие физических и химических веще-в с ионной, атомно-ковалентной, атомно-металической и молекулярной решеткой. Ве-ва с атомно-ковалентной решеткой характеризу-ся высокой твердостью, а с атомно-металической-пластичностью. Ве-ва с ионной и в особенности с атомно-ковалентной решеткой обладают высокой температурой плавления. В твердом состоянии при обычных условиях находятся вещества с металлическими или ионными связями; вещества с ковалентными связями могут быть в любом агрегатном состоянии. Твердые тела отличаются от газов и жидкостей наличием собственной формы и собственного объема. Их сжимаемость чрезвычайно мала.жидком состоянии Взаимодействия между частицами вещества в жидком состоянии достаточно сильны, чтобы препятствовать беспорядочному перемещению частиц, но все же недостаточны для прекращения их перемещения относительно друг друга.Подобно твердому телу жидкость обладает определенной структурой. Например, структура жидкой воды напоминает структуру льда: молекулы Н2О соединены друг с другом посредством водородных связей, и для большинства молекул сохраняется тетраэдрическое окружение. Однако в отличие от льда в жидкой воде проявляется лишь ближний порядок — за счет изгиба и растяжения водородных связей относительное расположение тетраэдрических структурных единиц оказывается неупорядоченным. Кроме того, вследствие перемещения молекул часть водородных связей разрывается и состав структурных единиц постоянно меняется. Непрерывное перемещение частиц определяет сильно выраженную самодиффузию жидкости и ее текучесть.По структуре жидкое состояние является промежуточным между твердым состоянием со строго определенной периодической структурой во всем кристалле (наличие дальнего порядка) и газом, в котором отсутствует какая-либо структура и движение частиц беспорядочно. Отсюда для жидкости характерно, с одной стороны, наличие определенного объема, а с другой — отсутствие определенной формы. Первое обстоятельство сближает ее с твердыми телами, второе — с газами. У жидкости вблизи температуры затвердевания упорядоченность внутренней структуры становится более четко выраженной. Напротив, по мере приближения жидкости к температуре кипения усиливается беспорядок во взаимном расположении частиц. Структура и физические свойства жидкости зависят от химической индивидуальности образующих ее частицы, а также от характера и интенсивности сил, действующих между ними. Для воды, как мы видели, большую роль в ассоциации молекул играют водородные связи. У неполярных молекул взаимодействие и взаимное расположение обусловливаются дисперсионными силами. Иначе говоря, общие закономерности образования структурных единиц для жидких тел такие же, как и для твердых тел. Отличие заключается в отсутствии жесткости в структуре и дальнего порядка в расположении частиц.Аморфное состояние Рентгеноструктурные исследования показывают, что аморфное состояние подобно жидкому, т.е. характеризуется неполной упорядоченностью взаимного расположения частиц, у аморфных тел нет определенной температуры плавления — в процессе нагревания они постепенно размягчаются и плавятся. Температурный интервал этих процессов для силикатных стекол, например, составляет 200°.У кристаллических тел при температуре плавления совершается такой переход, при котором дальний порядок исчезает и остается лишь ближний порядок. В аморфных телах характер расположения атомов при нагревании практически не меняется. Изменяется лишь подвижность атомов — увеличиваются их колебания. Сначала очень немногие, а потом большое число атомов приобретают возможность менять соседей. Наконец, число этих перемен в единицу времени становится таким же большим, как и в жидкости.Газовое состояние Характерная особенность газового состояния заключается в том, что молекулы (атомы) газа не удерживаются вместе, а свободно движутся в объеме, значительно превышающем объем самих частиц. Силы межмолекулярного взаимодействия проявляются, когда молекулы подходят друг к другу на достаточно близкое расстояние. Слабое межмолекулярное взаимодействие обусловливает малую плотность газов и их основные характерные свойства — стремление к безграничному расширению и способность оказывать давление на стенки сосуда, препятствующие этому стремлению. Вследствие слабого межмолекулярного взаимодействия при малых давлениях и высоких температурах все типичные газы ведут себя приблизительно одинаково. Но уже при обычных температурах и давлениях начинают проявляться индивидуальности газов. Существенные различия, связанные с природой составляющих газ частиц, особенно сильно проявляются при больших давлениях и низких температурах, т.е. в тех условиях, в которых газы практически используются в технике. Повышение внешнего давления и понижение температуры сближает частицы газа, поэтому межмолекулярное взаимодействие начинает проявляться в большей степени.Плазма Существенные изменения претерпевает вещество при нагревании до 3 температур порядка тысяч и миллионов градусов. В этих условиях оно переходит в ионизированный газ — плазму. В общем случае плазма — это смесь беспрерывно перемещающихся атомов, электронов, положи- тельных ионов и даже атомных ядер. Плазма с температурой порядка 10—100 тыс. градусов называется "холодной", с температурой порядка миллиона градусов — "горячей". В последнем случае нейтральные атомы в плазме существовать не могут, и она состоит из смеси электронов, ионов и атомных ядер. Плазма в целом электронейтральна, но обладает электронной и ионной проводимостью. |
№16. Скорость хим. р-ций и ф-ры. от кот. она зависит. Управление скоростью хим. пр-са. Скорость р-ции- число элемент. актов р-ции, происх-х в единицу времени, в еденицу объема для гомоген. р-ции (происх-т равномернопо всему объему) или на ед-цу поверх-ти раздела фаз для гетерогенных (происх-т только на некот. пограничн. поверх-х. на границах фазы). Ср. скорость изменения р-ции:
В
ходе хим. пр-сов конц-ции меняются
пост-но, поэтому опред-т мгновен. ск-сть
р-ции:
Если ск-сть р-ции оценивается увел-м конц-ции одного из пр-тов р-ции, то произведения С «+», а если конц-ция исх. в-в уменьш-ся, то С «-». Факторы, влияющие на скорость химических реакций. 1. Природа реагирующих вещ-в. Больш. роль играет хар-р хим. связей и строение мол-л реагентов. Р-ции протекают в направлении разрушения менее прочных связей и обр-ния веществ с более прочными связями. 2. Концентрация. С увеличением конц-ции (числа частиц в ед-це объема) чаще происходят столкновения молекул реагирующих веществ – ск-сть р-ции возрастает. 3. Темп-ра. При повышении темп-ры на каждые 100C ск-сть р-ции возр-т в 2-4 раза (Пр-ло Вант-Гоффа). 4. Поверхность соприкосновения реагирующих веществ. Для гетероген. с-м (когда в-ва находятся в разн. агрегатн. состояниях), чем больше поверхность соприкосновения, тем быстрее протекает р-ция. Поверхность тв. вещ-в м. б. увеличена путем их измельчения, а для растворимых в-в - путем их растворения. 5. Катализ. В-ва, которые участвуют в р-циях и увеличивают ее ск-сть, оставаясь к концу р-ции неизменными, называются катализаторами. Мех-зм д-вия катализаторов связан с уменьшением энергии активации р-ции за счет обр-ния промежуточных соед-ний. При гомогенном катализе реагенты и катализатор составляют одну фазу (нах-ся в одном агрегатном состоянии), при гетерогенном катализе - разные фазы (нах-ся в различн. агрегатных состояниях). Резко замедлить протекание нежелательных химических процессов в ряде случаев можно добавляя в реакционную среду ингибиторы (явление "отрицательного катализа"). |
№14. Понятие термодин. с-мы. Ф-ции сост-я. Тепл. эф-т хим. пр-сов и изм-е энтальпии. Термодинамическая с-ма- сов-сть макроскопических тел, кот-е взаимод-вуют и обмен-ся энергией как м/у собой, так и с др. телами (внешн. средой). Основа термодинам. м-да – оперд-ие состояния термодинам. с-мы. Состояние термодинам. с-мы задается термодинам-ми пар-рами- сов-сть физ-х величин, хар-х св-во термодинам. системы. Функции состояния опис-ся с пом. термодинам-х координат(темп., давление, концен-ция, масса). Основ. понятия1)внутр. энергия – это общий запас с-мы, кот. складывается из энергии движения и взаимод-вия мол-л и электронов. Она яв-ся ф-цией состояния с-мы, т.е. не зависит от пути пр-са, она зависит от термодинам. координат, 2)теплота- пр-с передачи энергии, она не явл-ся ф-цией состояния с-мы, 3)Работа- пр-с, связанный с упорядоченным движением,4)Энергия Гиббса-критерий самопроизвольного протекания хим. реакций, 5)энтальпия, 6)энтропия. Тепловой эф-т хим-х процессов и изменение энтальпии. Изм-ние энергии с-мы при протекании в ней хим. р-ции при усл-ии, что с-ма не совершает никакой др. работы, кроме работы расширения, наз-ся тепловым эффектом хим. реакции. При пост. давлении (изобарическ. пр-с) тепловой эф-т р-ции равен изм-нию энтальпии с-мы dH. Если с-ма нах-ся и изохорических усл-х, то тепл. эф-т хим. р-ции равен изм-нию внутр. энергии с-мы. Энтальпия – это одна из термодинам. ф-ций, хар-щая систему, нах-мя при пост. давлении.dH=H2-H1=Qp. Если исходные вещ-ва и прод-ты р-ции нах-ся в стандартном состоянии, то тепл. эф-т р-ции наз-ся стандартн. тепл. эф-том р-ции и обозначается dH0. Если в рез-те реакции теплота выделяется, т.е. энтальпия с-мы понижается, то р-ция наз-ся экзотермической. Р-ция, протекающая с поглощением теплоты, т.е. с повыш-м энтальпии с-мы, наз-ся эндотермический. Тепл. эф-т р-ции зависит от темп-ры, поэтому и в индексе обычно указ-ся темп-ра. Для больш-ва р-ций изм-ние тепл. эф-та с изм-нием темп-ры в пределах темп-тур отн-но не велико. В расчетах значения dH считают пост-ным, равным dH298. |
||||||||||||||||||||||||||||
|
№15. Понятие об энтропии. Напр-сть хим. пр-сов. Мерой неупорядоченности состояния с-мы служит термодинам. ф-ция, получившая название энтропии. Состояние с-мы м. характеризовать микросостояниями составляющих ее частиц, т.е. их мгновенными координатами и скоростями разли. видов движения в разл. направлениях. Число микросостояний с-мы называется термодинам. вероятностью с-мы W. Т.к. число частиц в системе огромно, то термодинамическая вероятность системы выражается огромными числами. Поэтому пользуются логарифмом термодинамической вероятности In W. Величина, равная RInW=S, называется энтропией системы, отнесенной к одному молю вещ-ва. Как и универсальная газовая постоянная R, энтропия имеет единицу измерения Дж/(моль*К). Энтропия вещ-ва в стандартном состоянии называется стандартной энтропией S0. В отличие от других термодинамических функций, можно определить не только изменение, но и абсолютное значение энтропии. Это вытекает из высказанного в 1911 году М.Планком постулата, согласно которому «при абсолютном нуле энтропия идеального кристалла равна нулю». Этот постулат получил название третьего закона термодинамики. По мере повышения темп-ры растет скорость различных видов движения частиц, т.е. число их микросостояний и соответственно термодинамическая вероятность и энтропия вещ-ва. При переходе вещ-ва из твердого вещ-ва в жидкое значительно увеличивается неупорядоченность и соответственно энтропия вещ-ва (∆Sпл). особенно резко растет неупорядоченность и соответственно энтропия вещ-ва при его переходе из жидкого в газообразное состояние (∆Sкип). Энтропия увеличивается при превращении вещ-ва из кристалического в аморфное состояние. Энтропия простых вещ-в явл-ся периодической функцией порядкового номера элемента. Увеличение числа атомов в молекуле и усложнение молекул приводит к увеличению энтропии. Например, S0298(O)=161, S0298(O2)=205, S0298(O3)=238,8 Дж/(моль*К). Следует обратить внимание на то, что известно абсолютные значения энтропии многих вещ-в. изменение энтропии системы в результате протекания химической реакции (∆S)=сумме энтропий продуктов реакции за вычетом энтропий исходных вещ-в с учетом стехиометрических коэффициентов. Т.к. энтропия вещ-ва в газообразном состоянии существенно выше, чем в жидком и твердом состояниях, то изменение энтропии положительно, если в результате процесса возрастает число молей газообразных вещ-в. Вследствие высоких значений энтропии газов, последние называют «носителями энтропии». Энтропия характеризует число микросостояний, явл-ся мерой беспорядка в системе. Ее увеличение говорит о переходе системы из более упорядоченного состояния к менее упорядоченному в результате тех или иных, в том числе химических пр-сов.
№25. Буферные р-ры. Амфотерные электролиты. Растворы, рН которых относительно мала изменяется при добавлении небольших количеств кислоты или основaния, называются буферными. Они обычно содержат слабую кислоту и ее соль, например, СНЗСООН+СНЗСООК или слабое основание и его соль, например, NH4OH + NH4C1. Рассмотрим процессы диссоциации в растворе слабой кислоты и ее соли: СНЗСООН СНЗСОО-+ Н+; СН3COONa СНЗСОО-+ Na +. При добавлении кислоты в раствор ее ионы водорода связываются в слабую кислоту СНЗСОО-+ Н+--> СНЗСООН. При добавлении основания в раствор гидроксид-ион связываетcя в слабый электролит (Н2О) Н++ ОН- Н,О. Образование слабых электролитов при добавлении в буферный раствор кислоты или основания и обусловливает устойчивость рН. рН=pKд+lg(a СНЗСОО-|/a СНЗСООН). Так как соль полностью диссоциирована, то [СНзСОО-] = ссоли. Поскольку доля диссоциированной кислоты мала, то можно принять, что концентрация недиссоциированной кислоты примерно равна исходной концентрации кислоты, т.е. скислоты. => рН=pKд+lg(ссоли. |/ скислоты). Буферные растворы образуются при титровании слабых кислот или слабых оснований. Нередко их готовят специально, если необходимо экспериментально определить рН растворов фотометрическим методом или провести химический эксперимент, связанный с выделением или присоединением ионов водорода, при- постоянном значении рН. Название “буферные” обусловлено тем, что такие растворы не изменяют заметно рН при разбавлении или при добавлении неко- торых.количеств растворов сильных кислот или силь- ных оснований. Постоянство рН буферных растворов имеет значение в жизнедеятельности живых организ- мов или растений: кислотность крови или растительных соков поддерживается постоянной из-за буферного действия содержащихся в них составных частей. Незначительное изменение рН при добавлении сильных кислот или. оснований важно в химической технологии и в анализе: кислотность некоторой реак- ционной смеси остается постоянной при введении буферного раствора независимо от выделения или по- глощения в реакции водородных или гидроксильных ионов. Каждый буферный раствор характеризуется по- этому определенной буферной емкостью. Буферная емкость — это способность буферного раствора не изменять заметно рН при добавлении растворов сильной кислоты или сильного основания. Мерой буферной емкости служит обычно количество сильной кислоты или сильного основания, которое небходимо прибавить к раствору буферной смеси, что- бы рН этого раствора изменилось на единицу. Буферирование играет важную роль в природе и технике. В орга- Низме человека рН меняется очень незначительно вследствие буферных свойств растворов во всех системах. Мало изменяется рН морской воды (рН 8,0). При проведении многих технологических процессов рН среды поддерживают постоянным с помощью буферных систем. Таким образом, в воде происходит ее диссоциация (самоионизация) с образованием ионов водорода и гидроксида. При постоянной температуре произведение активностей ионов водорода и гидроксида является величиной постоянной. Важное значение для многих биологических и технологических процессов имеет водородный показатель среды. Его можно рассчитать, а также определить с помощью индикаторов и приборов. Значение рН можно поддерживать на практически постоянном уровне путем применения буферных смесей. |
||||||||||||||||||||||||||||||
|
№17. хим. равновесие в гомоген. и гетероген. с-мах. Смещение хим. равновесия в нужн. напр-ии. При хим. равновесии ск-сть прямой р-ции равна ск-сти обратной р-ции. Химические реакции, протекающие на границе раздела фаз, наз-ся гетерогенными. Если скорость прямой и обратной гетерогенных реакций становится одинаковым, то наступает химическое равновесие в гетерогенной с-ме. Гомогенной реакцией наз-ся реакция, протекающая в однородной среде(в одной фазе). Константа хим. равновесия зависит от природы реагирующих в-в и от температуры и не зависит от давления и концентрации реагентов. Если в гетерогенной р-ции участвуют твёрдые и газообразные в-ва, то в выражение для константы хим. равновесия входят только парциальные давления газообразных компонентов р-ции. Константа равновесия может быть выражена через молярные доли. Однако она допустима лишь для достаточно разряженных с-м, в которых влияние межмолекулярных сил на активность компонентов очень мало. Константа равновесия может быть выражена через парциальные давления: Кр=pАВ/рАрВ, где pАВ, рА, рВ –парциальные давления реагирующих газов в равновесной с-ме. Парциальное давление равно pi=p0Ni, где p0-общее давление газовой смеси. Кр имеет размерность давления и равна Кр=Kfp0Σn, где Σn –сумма коэффициентов р-ции, в которой коэффициенты продуктов идут со знаком плюс, а начальных продуктов-с минусом. Константа равновесия может быть выражена через молярные концентрации С(моль/л). Такой способ удобен для р-ций , идущих при V=const. КC=CАВ/САСВ (размерность концентрации). КС=КР(RT)-Σn, где Σn- сумма коэффициентов продуктов реакции. Если р-ция идет без изменении числа молей и Σn=0, то все выражения констант равновесия будут совпадать, и они будут безразмерные. Константа равновесия для гетерогенной обратимой р-ции KP=pгаза/жидкости=f(T). Выбор параметров, влияющих на равновесие в гетерогенной с-ме, и условия равновесия определяются правилом фаз Гиббса-Коновалова: число степеней свободы в гетерогенной с-ме равно числу компонентов плюс 2 и минус число фаз. Смещение хим. равновесия. Гетерогенное хим. равновесие подчиняется принципу Ле Шателье. При повышении температуры гетерогенное хим. равновесие смещается в сторону эндотермической реакции. При повышении давления или концентрации исходных вещ-в равновесие смещается в сторону образования продуктов реакции, при повышении кон-ции или давления продуктов реакции равновесие смещается в сторону обратной реакции. При повышении общего давления равновесие сдвигается в направлении уменьшения числа молекул газообразных вещ-в.
№24. Гидролиз солей. Типичные случаи. Применим закон действующих масс к самоионизации воды Н20 + Н20↔ОН+3 + ОН- Или Н2О ↔ Н+ ОН-. Константа ионизации воды К=[Н+][ОН-]/[H2O]=1,8*10-16 может быть вычислена по электроической проводимости. Как видно из выражения константы ионизации, вода в обычных условиях ионизирована крайне мало. Поэтому можно считать что концентрация воды [H2O]-величина постоянная; она равна 55,56 моль/л (1000г/л / 18 моль/л) Уравнение для константы ионизации воды ожно записать следующим образом: К[H2O]=[Н+][ОН-] Следовательно, и произведение К[Н2О] янндля данной температуры яв-ся постоянным К[H2O]=1,8*10-16*55,56=10-14.Обозначим произведение К[H2O]=Кв Величина Кв называется ионным произведением воды и является постоянной не только для чистой воды, но и для разбавленных вод,но и для разбавленных растворов любых веществ. С повышением температуры Кв увеличивается, с понижением -уменьшается. Однако для расчетов, относяшихся к комнатной температуре, можно во всех случаях принимать Кв=10-14 Ионное произведение воды - весьма важная величина, так как позволяет для любого водного раствора найти концентрацию Н+ (ОН+3) при известной концентрации ОН-, и наоборот. Для чистой воды концентрация ионов Н+ (ОНз+) равна концентрации ионов ОН-, т.е. среда нейтральная: [Н+] = [ОН-] =√ Кв = 10-7 моль/л. Если концентрация ионов Н+ выше 10-7 моль/л, а концентрация ОН--ионов ниже, то раствор будет кислым. Наоборот, щелочной раствор соответствует концентрации ионов ОН- выше 10-7 моль/л при концентрации ионов Н+ ниже этого значения. Количественное обозначение реакции среды можно упростить, если принять за основу так называемый водородный показатель рН, определяемый как десятичный логарифм концентрации водородных ионов, взятый с обратным знаком: рН = —lg[H+]. Тогда Нейтральная среда рН = 7 Кислая среда. рН < 7 Щелочная среда.... рН > 7 Практически реакцию среды удобно определять с помощью индикаторов — веществ, меняющих свой цвет в зависимости от относительной концентрации ионов Н+ и ОН-. Значение рН. К одним из экотоксикантов, воздействующих на человека в бытовых условиях, относятся соединения алюминия, поскольку приготовление, хранение и употребление пищевых продуктов часто сопряжено с использованием алюминиевой посуды Катион алюминия Аl3+ в нейтральных растворах образует нерастворимый гидроксид Аl(ОН)3 и на его основе гидроксо- и оксосоединения. Образование таких частиц и нерастворимого АlРО4 лимитирует абсорбцию Аl3+ в пищеварительном тракте. рН среды таких продуктов как молоко кислое, кефир, рассол солений и капусты квашенной, а также фруктовые соки имеют рН ниже 5, т.е. кислую среду. Продуктов же, имеющих щелочную среду, в нашем эксперименте было выявлено гораздо меньше: отвар картофельный и кипяченая вода. Остальные продукты имеют рН среды, близкий к нейтральному показателю 6 – 8. Это свидетельствует о том, что в повседневном питании человека преобладают нейтрально-кислые продукты и блюда. Установлено, что наиболее безопасным для здоровья человека является процесс приготовления пищи в алюминиевой посуде при рН от 6 до 8. В такой емкости можно безбоязненно кипятить молоко, готовить каши и некислый борщ, но не стоит хранить в ней пищу больше 2 дней, держать в кастрюле из алюминия квашеную капусту или соленые огурцы, а тем более варить картофель и фруктово-ягодные соки. Этот металл разрушается при воздействии кислот, а также щелочей – образовавшиеся токсины перейдут в еду.
|
||||||||||||||||||||||||||||||
|
№18. Вода. Н2О – равнобедр треугольник, НОН=104,50, lОН=0,957А0. О2 в молек. Н2О имеет sp3-гибридизацию. Хим. связи ОН образованы за счет перекрывания sp3-гибридных орбит О2 и с-орбиталей Н2. У атома О2 в мол-ле Н2О имеются неподеленные пары, кот. могут быть использованы для обр-ния дополнит. Н2 – и донорно-акцепторных связей. Именно это обст-во, а также большой дипольный момент приводит к сильн. взаимод молекул Н2О и полярными молекулами др. вещ-в. плотн=0,998г/см3. Физич св-ва Н2О аномальны, напр-р, плавление льда сопровождается уменьш-м его V на 9%. В рез-те взаимод молекул Н2О между собой обр-ся ассоциаты. В парах часть молекул Н2О сущ-ют в виде димеров (Н2О)2. В конденсиров. фазах каждая молекула Н2О обр-ет 4 Н2 – связи. напр. к вершинам правильного тетраэдра. Длина Н2-связи НО 2,8А, НО – Н 180. Н/О-НО/Н\Н Хим. св-ва Н2О Вода хороший растворитель 1) В воде растворяются газы, например, аммиак. NH3 + H2O -> NH4OH (опыт фонтана) 2) Вода взаимодействует с оксидами металлов. Негашёная известь взаимодействует с водой с выделением теплоты. CaO + H2O -> Ca(OH)4 3) Вода взаимодействует с оксидами неметаллов. SO3 + H2O -> H2SO4 4) Вода взаимодействует c металлами. 2Na + 2H2O -> 2NaOH (опыт металла с водой) Вода является катализатором химических процессов |
№19. Дисперсные системы. Коллоидные растворы. Дисперсные сист. – сист., в кот одно в-во, нах-ся в диспергированном сост-ии, равномерно распределен в массе др. в-ва. Классиф-ция: 1. По степени дисперсности: 1)грубодисп. сист.(0,1 мкм и …): а) суспензии(крахмал в воде);б)эмульсии (масло в воде); 2)коллоидные р-ры(0,1 мкм – 1 мкм); 3) ионно-дисперсные сист или истинные р-ры(меньше 1мкм); 2. По агрегатному сост-ю дисперсн. фазы и дисперсн. среды(газ-жидк, газ-тв, ж-г, ж-ж, ж-тв, ТВ-г, ТВ-ж, ТВ-тв). Коллоидные р-ры(золи) – микрогетерогенные системы, сост из высокомол. дисперсн. фазы и жидкой дисеперс. среды. В зав.от растворителя — дисперсионной среды, т. е. воды, спирта, бензола или эфира и т. п., различают гидрозоли. алкозоли, бензозоли, этерозоли и т. п. Учитывая, что коллоидные растворы могут при известных условиях терять свою текучесть и затвердевать с образованием гелей, то и .названия их будут гидрогели, алкогеди, бензогели и т. п. Строение мицеллы Agl. Образование коллоидной частицы Agl происходит в результате реакции AgN03+KI = AgI +KN03 Молекулы Agl объединяются в практически нерастворимые частицы, в которых ионы Ag+ и I- образуют кристаллическую решетку. Если AgNO3 и KI взяты в эквивалентных количествах, то частицы-кристаллы растут, достигая значительной величины, превосходящей размеры коллоидных частиц^ и быстро выпадают в осадок. Если же одно из исходных .веществ взято в небольшом избытке, то оно служит стабилизатором,'сообщающим устойчивость коллоидным частицам Agl. Так, при избытке AgNOg в растворе будет находиться большое количество ионов Ag+ и NO3-.Ионы Ag+ будут продолжать достраивать кристаллическую решетку ядра, прочно входя в его структуру и сообщая ему эл. заряд(потенциалоопредел. ионы) Частицы с таким относительно высоким зарядом будут; притягивать оставшиеся в растворе противоположно заряженные (называемые поэтому противоионами) ионы NO3. Начнется ' процесс адсорбции протнвоионов, в результате которого установится динамическое равновесие между адсорбированными и-свободными ионами. Основная часть всех противоионов, адсорбированная на ядре коллоидной частицы, образует вместе с потенциал определяющим и ионами адсорбционный слой. Ядро и адсорбционный слой вместе составляют гранулу. Остаток'противоионов удерживается электростатическими силами притяжения вблизи гранулы, образуя диффузный, слой. Грянула вместе с диффузным слоем образует мицеллу. ((AgI)m*nAg+*(n-x)*NO3-)x-xNO3 Коагуляция – пр. объед-я коллоидных частиц в более крупные агрегаты.При броуновском движ-ии мицеллы свободно сближ-ся .В рез-те при коагуляции образ-ся рыхлые агрегаты различной величины. Крупные агрегаты под действием силы тяжести начинают опускаться на дно сосуда. Происходит пр. седиментации |
№20. Растворы. Кл-ция. Сп-бы выр-ния состава. Р-ры – это гомоген. (однофазные) с-мы перемен. состава, состоящие из двух или более в-в (компонентов). По хар-ру агрегатн. состояния р-ры м. б. газообразными, жидкими и твердыми. Обычно компонент, который в данных условиях находится в том же агрегатном состоянии, что и образующийся раствор, считают растворителем, остальные составляющие раствора – растворенными в-вами. В случае одинакового агрегатного состояния компонентов растворителем считают тот компонент, который преобладает в растворе. В зависимости от размеров частиц растворы делятся на истинные и коллоидные. В истинных растворах (часто называемых просто растворами) растворенное вещество диспергировано до атомного или молекулярного уровня, частицы растворенного вещества не видимы ни визуально, ни под микроскопом, свободно передвигаются в среде растворителя. Истинные растворы – термодинамически устойчивые системы, неограниченно стабильные во времени. По соотношению преобладания числа частиц, переходящих в раствор и удаляющихся из раствора, различают растворы насыщенные, ненасыщенные и пересыщенные. По относительным количествам растворенного вещества и растворителя растворы подразделяют на разбавленные и концентрированы. Раствор, в котором данное вещество при данной температуре больше не растворяется, т.е. раствор, находящийся в равновесии с растворяемым веществом, называют насыщенным, а раствор, в котором еще можно растворить добавочное количество данного вещества, — ненасыщенным. Способы выражения концентрации растворов. Конц-ция р-ров опр-ся кол-вом в-ва, заключенным в опред. объеме (или определенной массе)раствора или растворителя в зависимости от того, что выбрано в качестве меры измерения. Жидкости удобнее измерять по объему, а не по массе, поэтому химики чаще всего используют объемные концентрации и, в первую очередь, молярную концентрацию.Молярная концентрация (молярность)- это число молей растворенного вещества, содержащееся в 1 литре раствора |
||||||||||||||||||||||||||||
|
№21. Р-ры неэлектролитов и их св-ва. Раствор – гомогенная многокомпонентная система. Для веществ с близкими свойствами и неограниченной взаимной растворимостью понятия "растворитель" и "растворенное вещество" относительны. Например, для системы этиловый спирт – пропиловый спирт растворителем будет считаться тот компонент, количество которого существенно больше. Если в системе присутствует вода, то часто растворителем называют именно ее. При ограниченной взаимной растворимости растворителем считают тот компонент, структуру которого сохраняет раствор. Растворимостью вещества называется его максимально возможная, равновесная при данной температуре концентрация. В большинстве таблиц она задается в граммах вещества, содержащегося в 100 г растворителя. Раствор, находящийся в равновесии с растворенным веществом, называется насыщенным. Насыщенный раствор представляет собой гетерогенную систему. Наиболее часто используемые способы выражения концентрации растворов: массовая доля (часто в виде процентной концентрации) – отношение массы вещества к массе раствора: молярная концентрация C – количество вещества (число молей) n в литре раствора: С = n вещества/Vр-ра ; моляльная концентрация Cм – количество вещества (число молей) n , приходящееся на 1 кг растворителя: Cм = n вещества/mрастворителя Простейшая модель раствора может быть представлена двумя наборами шариков – мелких, моделирующих молекулы растворителя, крупных, моделирующих молекулы (частицы) растворенного вещества (рис. 4):
На основе такой модели можно описать некоторые важные свойства разбавленных растворов неэлектролитов. Рассмотрим первую систему. Сосуд с чистой водой перегородили на две половинки полупроницаемой мембраной – маленькие молекулы воды могут проникать сквозь поры в мембране, а большие молекулы сахара не могут. Потом в правую половину сосуда насыпали сахара.
обе скорости перехода воды уменьшаются, но справа (где сахар) уменьшается сильнее – молекулы сахара эффективнее "затыкают" отверстия снаружи (справа). Уровень справа поднимется, и давление увеличит скорость перехода справа:
Явление это называется осмосом (от греч. "осмос" – толчок, давление). В 1887 г. Вант- Рассмотрим вторую систему в рамках той же простейшей модели.
Таким образом, растворенное вещество влияет на температуру замерзания растворителя. Определение молекулярной массы по понижению температуры плавления при известной массе растворенного вещества в 1000 г растворителя называется криоскопией. tзам = K Cм = K(g1000)/MG Cм – моляльная концентрация;g – масса вещества, растворенного в G граммах растворителя;M – молекулярная масса растворенного вещества; K – криоскопическая константа растворителя (для воды 1,853). В рамках той же модели рассмотрим третью систему. Замкнутый термостатируемый сосуд частично заполнен водой, остальную его часть занимают только пары воды. Потом в сосуд бросили поваренную соль.
стрелки, обозначающие межфазные переходы молекул воды, снова неравномерно уменьшатся. Равновесие восстановится за счет понижения давления в системе. Давление пара над раствором всегда ниже, чем над чистым растворителем (закон Рауля – 1882 г): (p0 - p1)/p0 = N N – мольная доля растворенного вещества, соответствует соотношению количеств модельных "шариков-молекул" p0 – давление пара чистого растворителя, p1 – давление пара раствора. По этой причине температура кипения раствора выше, чем растворителя (при том же давлении). Применение – стакан с солью между оконными рамами предохранит стекла от запотевания. Определение молекулярной массы по повышению температуры кипения при известной массе растворенного вещества в 1000 г растворителя называется эбуллиоскопией: tкип = E Cм = E(g1000)/MG Cм – моляльная концентрация;g – масса вещества, растворенного в G граммах растворителя;M – молекулярная масса растворенного вещества; E – эбуллиоскопическая константа растворителя (для воды 0,51) Общий вывод: мы рассмотрели три варианта установления равновесия в системе чистый растворитель – раствор. Во всех случаях никакие переходы частиц растворителя не ускоряются, а только замедляются, но неравномерно. При стремлении систем к равновесию достигаются значительные эффекты (осмотическое давление, изменение температуры замерзания и кипения). Перечисленные выше эффекты можно описать, пользуясь фазовыми диаграммами.
Рис.10 Фазовая диаграмма воды при умеренных давлениях .Точка О – тройная точка равновесия газообразного, жидкого и твердого состояний воды. Точка В – критическая точка воды. Кривая ОД соответствует переохлажденной воде. Методы криоскопии и эбуллиоскопии фактически являются следствием из закона Рауля, что можно отразить на фазовой диаграмме воды (рис.11).
Рис. 11 Изображение зависимости давления пара от температуры . 1 – кривая для чистого жидкого растворителя; 2 – кривая для чистого твердого растворителя; 3, 4,5 – давление пара растворителя над растворами с увеличивающейся концентрацией нелетучего вещества. Если раствор состоит из двух летучих компонентов, его можно разделить на составляющие посредством перегонки. Суть процесса в простейшем случае (закон Рауля соблюдается для обоих компонентов во всем диапазоне концентраций) отображена на рис. 12.
Рис. 12 Зависимость температуры кипения жидкого раствора от состава жидкости N и состава пара У. При нагревании до кипения (точка а1) раствора состава N1 первая порция пара будет иметь состав, соответствующий точке b1, т.е. У1 . Полученный конденсат будет обогащен более летучим компонентом, а остающийся раствор обогатится менее летучим компонентом. При последовательных шагах от точек с индексом 1 до точек с индексом 3 и далее влево мы получим почти чистый малолетучий компонент и сконденсированные фракции пара, обогащенные летучим компонентом. Повторяя процедуру с конденсатом пара, можно в итоге разделить раствор на чистые компоненты. В реальных системах часто наблюдаются отклонения от закона Рауля, и на диаграммах температура кипения - состав возникают максимумы и минимумы. Они соответствуют случаю, когда состав пара и жидкости одинаков (нет промежутка ab); смеси, кипящие при постоянной температуре, называются азеотропными. Наиболее известная азеотропная смесь – этанол-вода, кипит при 78,170С, содержит 96% этанола, в то время как чистый этанол (100%) кипит при 78,30С. Такую смесь нельзя разделить перегонкой, и для получения 100%-ного безводного спирта используют другие методы.
|
№22. Растворы электролитов. Слабые и сильные. Ионные р-ции в р-рах электролитов. Вода явл-ся растворителем для многих в-в. Это обусл-но полярным хар-ром мол-л воды и их сп-стью образовывать хим. связи с др. мол-лами. Учитывая широкое распр-ние водных р-ров электролитов и их большую важность для науки и техники, рассмотрим эти растворы более подробно. Слабые электролиты. Константа диссоциации. В р-рах сл. электролитов пр-с диссоциации протекает обратимо и,=> к нему м.б. применен закон действующих масс. Так, для пр-са диссоциации кислоты НАН++А- константа равновесия Kc, равна Kc= Kд= ([Н+ ][ А- ])/[НА]. Константа равновесия для пр-са диссоциации наз-ся константой диссоциации Кд. Н-р, константа диссоциации уксусной кислоты СН3-СООН равна Кд=([H+][СН3СОО-])/[СН3СООН]. СНЗСООНСНЗСОО- + Н+. Константа диссоциации зависит от природы диссоцирующего вещества и растворителя, также от темп-ры и не зависит от конц-ции р-ра. Кривая зависимости константы диссоциации многих электролитов от темп-ры проходит через максимум. Константа диссоциации указывает на прочность мол-л в данном р-ре. Чем меньше константа диссоциации в данном растворителе, тем слабее диссоциирует электролит и тем, след-но, устойчивее его молекулы. Степень диссоциации уменьшается с увеличением конц-ции слабого электролита. Многоосновные слабые кислоты и основания диссоциируют ступенчато, причем константа диссоциации по каждой послед. ступени всегда на несколько порядков ниже, чем по предыдущей. Сильные электролиты. Многие св-ва р-ров, такие как осмотическое давление, темп-ра кипения и замерзания, давление насыщенного пара, зависят как от конц-ции р-ра, т.е. от числа растворенных в нем частиц, так и от взаимного влияния этих частиц друг на друга. Степень взаимод-вия частиц в р-ре тем выше, чем больше плотность их зарядов и чем меньше ср. расстояние м/у ними. В р-рах слабых электролитов взаимод-вие ионов др. с другом отн-но невелико вследствие их незначительной конц-ции. Сильн. электролиты в р-рах диссоциированы практически полностью. Поэтому в уравнении диссоциации электролита стрелка указывает только на прямой пр-с, например: NaClNa++ С1-. В р-рах сильн. электролитов из-за полной их диссоциации конц-ция ионов велика. Поэтому св-ва таких р-ров существенно зависят от степени взаимод-вия входящих в их состав ионов как друг с другом, так и с полярн. мол-лами раств-ля. Взаимод-вие ионов в р-рах сильн. электролитов приводит к тому, что катионы и анионы испытывают взаимное притяжение, а ионы одного знака заряда будут отталкиваться друг от друга. Поэтому в р-ре каждый произвольно выбранный ион окружен в среднем во времени преимущ-но противоположно заряженными ионами, как, напр-р, в ионных кристаллах. С ростом конц-ции электролита более сильное влияние на св-ва р-ра начинает оказывать процесс образования гидратов. В рез-те пр-са гидратации изменяются размеры растворенных частиц, плотность их заряда, а также вязкость всего раствора в целом. С увеличением конц-ции р-ра электролита уменьшается ср. расстояние м/у противоположно заряженными ионами. При этом растет вероятность образования длительно существующих ионных пар, так называемых ионных двойников, основное отличие которых от мол-л заключается в большей длине связи и наличии взаимодействующих с ионной парой молекул растворителя. Итак, при увеличении конц-ции р-ра электролита более важную роль во взаимод-вии м/у растворенными частицами начинают играть близкодействующие силы хим. связи. Природа и энергия этих сил зависит от специфических св-в взаимодействующих частиц. Поэтому с ростом конц-ции увеличивается различие в св-вах р-ров электролитов одинаковой конц-ции, но разного химического состава.
№26. Окисл-восст. р-ции. ОВР- р-ции, при коорых происходит переход электронов от одних атомов, молекул, ионов к другим, что сопровождается изменением степени окисления. Степенью окисления называется условный заряд, который приобретает элемент в молекуле, исходя из предположения, что общая электронная пара притянута к нему или оттянута от него. Окислителем называется в-во, которое принимает электроны, при этом степень окисления в-ва будет уменьшаться. Окислитель восстанавливается. Восстановителем называется в-во, которое отдает электроны, при этом степень окисления элемента повышается. Восстановитель окисляется. Все окисл. и восстанов. условно делятся на 3 группы: 1) безусловные окислители (простое в-во (F2, O2), в-ва, в состав которых входят Эл-ты в высшей СО. 2) безусловные восстановители (все металлы), в-ва, в состав которых входят элементы в низшей СО. 3) в-ва, проявляющие двойственные св-ва (элементы, находящиеся в промежуточной СО). Типы О-В р-ий: 1)О-В р-ия, при крой изм-ся степени окисления атомов элементов, входящих в состав разных в-в (р-ия выше).2) О-В р-ии, при к-рых степени окисл-я изменяют атомы разных элементов одного и того же в-ва. 3)О-В р-ии самоокисления и самовосстановления. (степень окисл-я одного и того же элемента и понижается и повышается). Эти р-ии носят названия р-ии диспропорционирования. Основной практической задачей ОВР является правильный подбор коэффициентов перед молекулам с тем, чтобы число атомов данного элемента было равно числу атомов в обеих частях ур-я. Для этой цели используют 2 метода: 1) Метод электронного баланса (НЕДОСТАТКИ: при написании полуреакции не учитывается реальная форма ионов, не учитывается кислотность среды); 2) метод полуреакций (недостатков 1-го метода нет). Роль О-В процессов в природе и пром-ти. Примеры использования О-В р-ий в нашей спец-ти: идентификация продуктов (содерж-е различных альдегидов в пище), р-ия серебрянного зеркала (взаимод-е альдегидов с комплексным соединением серебра.
|
№23. Электролит. диссоциация воды. Ионное произведение воды. Водородн. пок-ль. Зн-ие pH 1887 г. Шведский ученый Сванте Аррениус предложил теорию электролитической диссоциации в р-рах взамен прежних представлений, согласно которым в-ва в р-ре распадаются на ионы под действием электрического тока. Электролитическая диссоциация не зависит от разности потенциалов, создающей электрический ток, что подтверждается своеобразным течением химических р-ций в электролите между отдельными ионами вне зависимости от того, из каких в-в эти ионы получены. Полярные молекулы воды могут диссоциировать, проявляя при этом свою амфотерность: Н2О↔Н++ОН- или 2Н2О↔Н3О++ОН-. Вторая ступень диссоциации ещё никем не наблюдалась и ионов О2- в воде не обнаружено. Вода является очень слабым электролитом, степень её диссоциации ~2∙10-9, чистая вода почти не проводит электрический ток. Однако на все ионные равновесия в водных р-рах присутствие ионов Н+ и ОН- оказывает влияние и процесс диссоциации воды необходимо всегда учитывать. При диссоциации абсолютно чистой воды, которую мы принимаем за эталон нейтральности, концентрации ионов Н+ и ОН- равны и условие нейтральности мы можем записать следующим образом: КН2О=[H+][OH-]=1,0∙10-14. Введем новую величину, характеризующую среду и равную – lg[H+]. Обозначим её рН и в дальнейшем будем называть её водородным показателем. рН=7 (для нейтральной среды). Для кислотной среды рН<7. Для щелочной среды рН>7. Равновесие электролитической диссоциации воды влияет на общее равновесие в среде электролита, содержащего те ли другие растворенные в-ва. Значение рН. К одним из экотоксикантов, воздействующих на человека в бытовых условиях, относятся соединения алюминия, поскольку приготовление, хранение и употребление пищевых продуктов часто сопряжено с использованием алюминиевой посуды Катион алюминия Аl3+ в нейтральных растворах образует нерастворимый гидроксид Аl(ОН)3 и на его основе гидроксо- и оксосоединения. Образование таких частиц и нерастворимого АlРО4 лимитирует абсорбцию Аl3+ в пищеварительном тракте. рН среды таких продуктов как молоко кислое, кефир, рассол солений и капусты квашенной, а также фруктовые соки имеют рН ниже 5, т.е. кислую среду. Продуктов же, имеющих щелочную среду, в нашем эксперименте было выявлено гораздо меньше: отвар картофельный и кипяченая вода. Остальные продукты имеют рН среды, близкий к нейтральному показателю 6 – 8. Это свидетельствует о том, что в повседневном питании человека преобладают нейтрально-кислые продукты и блюда. Установлено, что наиболее безопасным для здоровья человека является процесс приготовления пищи в алюминиевой посуде при рН от 6 до 8. В такой емкости можно безбоязненно кипятить молоко, готовить каши и некислый борщ, но не стоит хранить в ней пищу больше 2 дней, держать в кастрюле из алюминия квашеную капусту или соленые огурцы, а тем более варить картофель и фруктово-ягодные соки. Этот металл разрушается при воздействии кислот, а также щелочей – образовавшиеся токсины перейдут в еду.
№27 Гетероген. окисл-восст пр-сы. Мех-зм возн-ния двойн. электр. слоя. Электродн. потенциалы. Ур-ие Нернста. Гальв элементы Рассмотрим след ситуацию в сосуде с водой находится металлическая пластина. Два противоположных процесса: электростатическое напряжение м/у ионами металла и водой (гидротация) и электростатическое притяжение со стороны электронного газа опред прочность кристаллической решетки. В результате растворение металла в воде приобретает характер поверхностного явлении, т.е охватывает лишь область на границе металл-жидкость
двойной электр слой образ разность потенциалов, к-рая наз-ся электронным потенциалом. В р-ре соли металла1).Исходная концентрация ионов металла в р-ре меньше С0 (концентрация) соответствующее равновесному состоянию ионов после в р-р метал. Пластинки. В этом случае будут происходить переход металла в р-р и выделение металла . Тк С<С0 преобладаюшая будет первый процесс, поэтому металл будет заряжен отрицательно, а принимающий слой р-ра продолжительно Me 0-nê→Me +n Me +n +nê→ Me 0 2)C>С0 ионы металла выдел-ся из р-ра на поверхность пластинки. Источника электрона нет поэтому металл выдел-ся на пластинке в виде ионов, сл-но поверхность металла положительна 3) С=Со. Вся система нах-ся в состоянии подвижного равновесия. Разность потенциалов м/у раствором и металлической пластинкой равна нулю. Переход ионов из металла в р-р яв-ся обратимым процессом, происходит в изотермических условиях, сл-но в этих условиях система совершает макс полезную работу. Способность системы совершать макс полезную работу наз-ся свободной энергией A=nFe, F-число Фарадея, n-заряд ионов, е-величина электронного потенциала A=RTLnK-RTLn ame/ amen+ R-газовая универсальная постоянная=8,31Дж/мольК T-температура K-константа равновесия обратимой реакции а-активность ионов металла nFe=R+LnK-RT Ln ame/ amen+ e=2.303 RT/nF LgK-2.303 RT/nF Lg ame/ amen+ , 2.303 RT/nF LgK=const=e0-хар-ет электрохимическую природу электрода.Уравнение Нернста e=e0-2.303 R*T/n*F Lg ame/ amen+ Если с=1г ион/литр тогда а=1, значит Lg1=0, тогда е=е0 –стандартный потенциал, наз-ют такой потенциал, к-рый возникает на метал пластинке, находящейся в контакте с одноименными ионами в р-ре с концентрацией равной 1 г-ион/л. За нулевую точку измерения потенциалов принят условно нормальный потенциал водородного электрода Метал элетрода играет роль проводника электронов переходящих из одних атомов в др в одном и тоже р-ре. Инертные электроды: графитовые, платиновые, паладиновые. Окисл-востан электрод получается в том случае когда неактивный металл напр-р платина нах-ся в р-ре вещ-в, м/у к-рыми может происходить окис-воост реакция. Р-р, содержащий одновременно окисленные и восстановленные формы вещ-ва, образует окислит-востанв систему или редокс-систему. Электроды 1и2 рода. Электрод 1 рода- электрод из металла погруженного в р-р, содержащий ион того же металла. Эти электроны обратимо обмениваются относительно одного из ионов. Электроды 2 рода состоят из металла покрытого слоем трудно растворимой соли ипогруженного в р-р каой-либо растворимой соли с тем же анионом , также электроды обратимы относительно обоих ионов Окислит-восстановит потенциал возникает на границе соприкосновения пластины с р-ром, величина и знак потенциала зависит от соотношения концентрации ионов разных зарядов в р-ре Fe2+ ↔Fe 3++ê K= [Fe 3+][ ê]/ [Fe 2+] Введение в систему дополнит кол-ва электронов приводит к уменьшению концентрации окисленной формы и повышается содержание восстановленной, уменьшение концентрации электронов-наоборот
|
||||||||||||||||||||||||||||
|
№28. Св-ва неМе и их соед-й. ЭО. неМе в основном располагаются в конце малых и больших периодов, а число внешних электронов у их атомов равно номеру группы. Сп-сть присоединять электроны в периоде возрастает по мере приближения к благородному газу, а в группе – по мере уменьшения радиуса атома или снизу вверх. Для завершения внешних электронных уровней атомы неМе присоединяют электроны и явл-ся окисл-лями. Активнее всех присоединяет электрон атом фтора. У остальных элементов неМе, эта сп-сть уменьшается в след.порядке: O,Cl,N,S,C,P,H,Si. У атомов этих уменьшение сп-сти присоединять электроны нах-ся в соответствии с уменьшением знач-ий их относительной электроотриц-ти. Вторым после фтора в этом ряду стоит атом кислорода, а не хлора. Электронная стр-ра неМе – правая верхняя часть периодич.системы. НеМе хар-ся тем, что они имеют электронную конфигурацию, начиная с углерода (р-элементы), заканчиваются эл-тами 8-ой группы, электронная конфигурация заканчивается np6 (инертные газы). Чаще всего у неМе степень окисл-я со знаком минус. В виде простых в-в неМе часто встречаются двухатомные и многоатомные молекулы. Галогены сущ-ют в виде двухатомных мол-л с одинарной ковалентной связью. В таком виде они сущ-ют во всех агрегатных состояниях (твердом, жидк., газообразн.). Физические и хим.св-ва неМе определяются их положением в период.системе. неМе располагаются в 8 группе (благородные газы), 7 группе (галогены), 6 группе (халькогены), 5 гр (азот, фосфор, мышьяк), 4 гр(углерод, кремний, германий), 3 гр (бор) и 1 гр (водород). Физ.св-ва. Все простые в-ва – неМе при обычных усл-ях нах-ся либо в газообразном сост-ии (правая часть период.таблицы и водород) в молекулярной форме или в виде атомов (благородн.газы), либо в твердом виде, лишь бром при обычных усл-ях - жидкость. Твердые НемЕ образуют либо огромные макромол-лы – кристаллы (углерод, кремний, и др.), либо относ-но небольшие макромол-лы (В12, S8 ,P4 ). Темп-ры кип-я и плавл-я у неМе 5 – 8 групп возрастают при увеличении атомного номера эл-та в группе. У кремния и углерода закономерность обратная. Хим.св-ва . энергия ионизации увеличивается, а радиус атома уменьшается с увеличением порядкового номера эл-та в периоде. В одной и той же группе наблюдается обратная зав-ть этих величин отпорядкового номера эл-та, что обусловливает увеличение окислительной способности эл-тов в периоде слева направо, а в группе снизу вверх. Наиболее сильными окислителями явл-ся фтор и кислород, хлор и бром, преимущ-но восстановительные св-ва проявляют водород, бор, углерод, кремний, германий, фосфор, мышьяк и теллур. Промежуточные окислительно-восстановит.св-ва имеют азот, сера, йод. Кислород и галогены м образовыватьионные соед-я, а бор, углерод, водород, азот и фосфор – преимущ-но ковалентные соед-я. Благородные газы имеют мало соед-й. Ксенон образует термодинамически устойчивые фториды XeF2, XeF4, XeF6. Сродством атома к электрону Ее наз-ся энергитический эффект присоединения электрона к нейтральному атому Э с превращением его в отрицательный ион Э- . Сродство атома к электрону выражают в кДж\моль; допускается внесистемная единица эВ\моль. Сродство к электрону численно равно, но противоположно по знаку энергии ионизации отрицательно заряженного электрона Э-. Сродство к электрону зависит от электронной конфигурации атома. Наибольшим сродством к электрону обладают р-элементы 7 группы. Сродства к электрону не проявляют атомы с конфигурацией. Сродства к электрону не проявляют атомы с конфигурацией s2 (Be,Mg,Ca) и s2p6 (Ne,Ar,Kr) или наполовину заполненные р-подслоем (N,P,As). Понятие электроотриц-ти (ЭО) имеет условный хар-р. оно позволяет оценить способность атома данного эл-та оттягивать на себя электронную плотность по сравнению с атомами других эл-тов в соединении. Эта способность зависит от энергии ионизации атома и его сродства к электрону. Согласно одному из определений (Малликен) электротриц-ть атома м\б выражена как полусумма его энергии ионизации и сродства к электрону: х = 1\2 (ЕИ + ЕС ). Имеется около 20 шкал электроотриц-ти , в основу расчета значений которых положены разные св-ва в-в. Значения электроотриц-тей разных шкал отличаются , но относительное расположение расположение эл-тов в ряду электроотриц-тей примерно одинаковое. В шкале электроотр-тей по Полингу электр-ть фтора принята равной 4,0. наименьшими знач-ями электроотр-ти хар-ся s-элементы 1 гpуппы, а наибольшими – р-эл-ты 7 группы. Эл-ту нельзя приписать постоянную эл-ть. Она зависит от многих ф-ров, в частности от валентного состояния эл-та, типа соед-я, в к-рое он входит,и др. тем не менее это понятие важно для качественного объяснения св-в хим.связи и соед-й. |
№29. Кислородн. соед-ия неМе.Биогены. Электронная структура неМе-правая верхняя честь период. с-мы. Неметаллы хар-ся тем, что электронную конфигурацию имеют начиная с С. Неметаллы в основном з-элементы. Заполнение электронами послед атом. орбитали р-типа.Они «любят» достраиватьсвою электронную, поэтому степень окисления «-«.В виде постых вещ-в встречаются в виде двуатомных или многоатомных молекул. Оксиды неметаллов относят к кислотным оксидам, которым соответствуют кислоты Неметаллы могут при определенных условиях между собой реагировать, образуя соединения с ковалентной полярной (H2O, HCl) и неполярной связями (CO2). С кислородом неметаллы образуют кислотные оксиды. В одних оксидах они проявляют максимальную степень окисления, равную номеру группы (например, SO2, N2O5), а других – более низкую (например, SO2, N2O3). Кислотным оксидам соответствуют кислоты, причем из двух кислородных кислот одного неметалла сильнее та, в которой он проявляет более высокую степень окисления. Например, азотная кислота HNO3 сильнее азотистой HNO2, а серная кислотаH2SO4 сильнее сернистой H2SO3. С кислородом неметаллы образуют кислотные оксиды. В одних оксидах они проявляют максимальную степень окисления, равную номеру группы (например, SO2, N2O5), а других – более низкую (например, SO2, N2O3). Кислотным оксидам соответствуют кислоты, причем из двух кислородных кислот одного неметалла сильнее та, в которой он проявляет более высокую степень окисления. Например, азотная кислота HNO3 сильнее азотистой HNO2, а серная кислота H2SO4 сильнее сернистой H2SO3. Характеристики кислородных соединений неметалов: 1.Свойства высших оксидов (т.е. оксидов, в состав которых входит элемент данной группы с высшей степенью окисления) в периодах слева направо постепенно изменяются от основных к кислотным. 2.В группах сверху вниз кислотные свойства высших оксидов постепенно ослабевают. Об этом можно судить по свойствам кислот, соответствующих этим оксидам. 3.Возрастание кислотных свойств высших оксидов соответствующих элементов в периодах слева направо объясняется постепенным возрастанием положительного заряда ионов этих элементов. 4.В главных подгруппах периодической системы химических элементов в направлении сверху вниз кислотные свойства высших оксидов неметаллов уменьшаются. Общие формулы водородных соединений по группам периодической системы химических элементов приведены в таблице
С металлами водород образует (за некоторым исключением) нелетучие соединения, которые являются твердыми веществами немолекулярного строения. Поэтому их температуры плавления сравнительно высоки. С неметаллами водород образует летучие соединения молекулярного строения. В обычных условиях это газы или летучие жидкости. В периодах слева направо кислотные свойства летучих водородных соединений неметаллов в водных растворах усиливается. Это объясняется тем, что ионы кислорода имеют свободные электронные пары, а ионы водорода – свободную орбиталь, то происходит процесс, котроый выглядит следующим образом: H2O + HF H3O + F 1.В периодах слева направо у ионов элементов положительный заряд увеличивается. В связи с этим кислотные свойства летучих водородных соединений элементов в водных растворах усиливаются. 2.В группах сверху вниз отрицательно заряженные анионы все слабее притягивают положительно заряженные ионы водорода Н+. В связи с этим облегчается процесс отщепления ионов водорода Н+ и кислотные свойства водородных соединений увеличиваются. 3.Водородные соединения неметаллов, обладающие в водных растворах кислотными свойствами, реагируют со щелочами. Водородные же соединения неметаллов, обладающие в водных растворах основными свойствами, реагируют с кислотами. 4.Окислительная активность водородных соединений неметаллов в группах сверху вниз сильно увеличивается. Например, окислить фтор из водородного соединения HF химическим путем нельзя, окислить же хлор из водородного соединения HCl можно различными окислителями. Это объясняется тем, что в группах сверху вниз резко возрастают атомные радиусы, в связи с чем отдача электронов облегчается. Электроотрицательность. Понятие электроотрицательности (ЭО) является условным. Оно позволяет оценить способность атома, данного элемента оттягивать на себя электронную плотность по сравнению с атомами других элементов в соединении. Очевидно, что эта способность зависит от энергии ионизации атома и его сродства к электрону. Согласно одному из определений (Малликен) электроотрицательность атома х может быть выражена, как полусумма его энергии ионизации и сродства к электрону: х — 1/2(Еи + Ес). Имеется около 20 шкал электроотрицательности, в основу расчета значений которых положены разные свойства, веществ. Значения электроотрицательностей разных шкал отличаются, но относительное расположение элементов в ряду электроотрицательностей примерно одинаково. |
№30. Общ. св-ва Ме. Ме в период. с-ме. Св-ва Ме от электр. оболочек. Крист. решеткаМе. Мет. св. Ме в природе, сп-бы получения. Из 107 эл-тов 85 явл-ся металлами. К эл-там-металлам относ-ся s-эл-ты 1 и 2 групп, все d- и f-эл-ты, а такжн р-эл-ты главных подгрупп:3 (кроме бора), 4 (Ge,Sn,Pb), 5 (Sb,Bi) и 6(Po). Наиболее типичные эл-ты-металлы расположены в начале периодов (начиная со второго). Плотность Ме не может считаться их характерным св-вом. Легкие Ме меньше и технич.ценность в кач-ве легких конструкционных материалов представляют лишь Al,Mg,Be,Ti. Хар-ным св-вом Ме явл-ся пластичность. При сочетании пластичности с малой вязкостью деформация происходит при незначительных нагрузках. Наиболее прочны те Ме, у к-рых деформация осущ-ся только под действием больших нагрузок. След.св-во – твердость. Наиболее твердыми явл-ся Ме группы Cr, наименее – калий, рубидий, цезий. Твердость Ме связана с их тугоплавкостью. Для Ме хар-на большая теплопроводность: собсвенные электроны, находясь в пост.движении, все время сталкиваются с колебающимися ионами и обменив-ся с ними электронами. Усилившееся при нагревании колебание ионов незамедлит-но передается при псредстве электронов с соседними ионами, причем происходит быстрое выравнивание темп-ры по всей массе. Также МЕ присущ металлич.блеск, способность испускать электроны под возд-ем эл\маг.волн, Ме непрозрачны. Хим.св-ва Ме обусловлены хар-ной способностью атомов метал.эл-тов отдавать свои валентные электроны с образованием элементарных положит-но заряженных ионов. Мерой прочности связи электронов в атомах явл-ся величина энергия ионизации. Наименьшее знач-е энергии иониз-ии у щелочных Ме, поятому они явл-ся самыми энергичными восстановителями. Каждый период начинается с Ме с очень низкой темп-рой плавл-я. По мере увелич-я порядк.номера в кажд.периоде tплавл увеличивается и достигает мах в групп хрома, сам.тугоплавкий – вольфрам (33900С).примерно также изм-ся и темп-ра кип-я Ме. Величины темп-ры кип-я и плавл-я связаны с прочностью кристаллич.решеток и с некоторыми другими хар-ными св-вами Ме (н-р, полиморфизм). Мет.связь ненасыщаема и ненапрвлена, поэтому Ме имеют координационные решетки с максим-но плотной уппаковкой. Для металлических простых в-в наиболее типичны 3 типа кристаллических решеток: 1)кубическая гранецентрированная,2)гексогональная,3)кубическая объемноцентрированная. Для большинства металлов хар-ен полиморфизм. Это связано с тем, что энергии кристаллических решеток различных металлических стр-р близки. В период.системе при переходе от р-эл-тов к s-эл-там уменьшение числа валентных электронов обуславливает закономерный переход от неМе с различной кристаллической решеткой к Ме, имеющим металлические координацион.кристаллы. в соотв-ии с изм-ем стр-ры и типов хим.связи закономерно изм-ся св-ва простых в-в: плотность, темп-ра кип-я и т.д. Ме относ-ся к металлич проводникам. Изм-е св-в связано с порядковым номером. Мет.связь. Ме отличаются от др.в-в высокой электрич.проводимостью и теплопроводностью. В обычном сост-ии они явл-ся кристаллическими в-вами с высокими координационными числами атомов. Высокая электропроводность объясн-ся тем, что что часть электронов может передвигаться по всему объему куска Ме. Атомы не связаны друг с другом двух электронными локализированными связями. Число валентных электронов атомов не дост-но для подобных связей со всеми соседями. Локализации электронов нет, дост-но увеличить электропроводность. Атомы Ме хар-ся также невысокой энергией иониз-ии. В отличие от ковалентных ионных соед-й в Ме небольшре число электронов одновременно связывает большое число атомных ядер, а сами электроны могут перемещаться в Ме. Рас-м хар-р заполнения энергитических зон металлических кристаллов. У эл-тов с одним s-электроном в кристаллах валентная зона построена из s-атомных орбиталей и заполнена лишь наполовину, поэтому при незначительном заблуждении энергитич.состояние каждого из электронов может меняться в пределах всей энергитич.зоны. Это может произойти при приложении к Ме электрич.поля. в случае эл-тов с двумя s-электронами s-зона полностью заполнена. Если же s и p-уровни в изолированных атомах близки, то в кристаллах соотв-ющие зоны перекрываются. В этом случае число валентных электронов недостаточно для заполнения энергитич.уровней перекрывающихся зон. Т.о., мет.кристаллы образ-ся эл-тами, в атомах к-рых число валентных электронов мало по сравн-ю с числом энергитически близких валентных орбиталей. Вследствии этого хим.связь мет.кристаллов сильно делокализована. К наиболее распр-ым в природе металлам относ-ся алюминий, железо, натрий, калий, магний и титан. Распределение металлов в земной коре м\б равномерным (рассеянные Ме) или неравномерным (в виде месторождений). Платиновые Ме , золото, серебро, ртуть нах-ся в земной коре в свободном виде. Остальные эл-ты нах-ся в виде хим.соед-ий с др.эл-тами (в виде минералов). Ме получают из руд, т.е. исходного сырья, в к-ром содержится эк-ки приемлемое кол-во Ме. По мере истощения руд уменьшается эк-ки приемлемое содержание в них Ме и повышается их стоимость. Предвар-но руда обрабатывается для увеличения конц-ии Ме путем отделения пустой породы и разделения остатка на различные фракции. Последующии операции заключаются в получении соединения Ме , из к-рого удобно выделить Ме тем или иным способом. Восстановление проводят хим. И элекр\трохимическими способами. Хим восстановление заключается во взаимод-ии соед-й Ме с углем, водородом или металлами-восстановителями. Многие Ме производят взаимод-ем соед-ий Ме с другими Ме. Таким способом получают кадмий, олово, хром, серебро, титан и др.Ме. кроме магния восстановителями обычно служат цинк и алюминий. Электролизом из р-ров осаждают медь, никель, серебро, хром, кадмий, индий и др.Ме. электролизом из расплавов осаждаются сильные восстановители, такие, как щелочные Ме, магний и алюминий.
|
||||||||||||||||||||||||||||
|
№31. Хим. св-ва Ме. Сравнит. активность Ме. Коррозия Ме. Скорость коррозии. М-ды борьбы. Будучи восстановителями, Ме могут взаимодействовать с окислителями. Все Ме окисляются фтором и могут окисляться хлором. Большинство Ме (кроме платины и золота) могут окисляться бромом и кислородом в кислой среде. В нейтральной среде кислород не может окислять золото, платиновые Ме, ртуть, серебро. Ионы водорода в кислой среде могут окислять многие Ме, кроме платиновых, ртути, золота, серебра, меди, рения, сурьмы и висмута. реальная возможность окисления того или иного Ме определяется не только термодинамикой, но и кинетикой процесса. Взаимодействие многих Ме с хромом, бромом, кислородом, ионами водорода и др. окислителями тормозится пассивными пленками на поверхности Ме. Большой склонностью к пассивации обладают бериллий, алюминий, d-металлы, IY-YIII групп. Многие Ме катализируют различные химические и электрохимические реакции. Пыль и пары некоторых Ме токсичны. Токсичность характеризуется предельнодопустимыми концентрациями вещ-в в рабочей зоне, ПДК (мг/м3). К наиболее токсичным относятся Ме (ПДК-мг/м3): бериллий, ртуть, свинец, кадмий, серебро, никель, родий, таллий, индий. Т.к. Ме и их катионы имеют вакантные молекулярные орбитали, то большинство из них явл-ся комплексообразователями и вхордят в состав комплексных соединений. Способность к комплексообразованию растет с увеличением заряда иона и уменьшением его радиуса, зависит от природы Ме и наличия вакантных орбиталей у его ионов. наиболее выражена склонность к комплексообразованию у ионов переходных Ме, особенно d –элементов VIII, I, II группы. Комплексные соединения, особенно железо, кобальта, меди, марганца, хрома, ванадия, цинка и молибдена, входят в состав биологических систем, включая ферменты, переносчики крови и т.д. Например, в гемоглобин крови входит комплексное соединение железа. Химические свойства Ме нах-ся в периодической зависимости от их порядковых номеров. Коррозия – это разрушение Ме в результате его физ-хим взаимодействия с окружающей средой. Ме окисляются и образуются продукты, состав которых зависит от условий коррозии. Коррозия – самопроизвольный процесс и соответственно протекает с уменьшением энергии Гиббса системы. Хим-ая энергия реакции коррозийного разрушения Ме выделяется в виде теплоты и рассеивается в окружающем пространстве. Коррозия приводит к большим потерям в результате разрушения трубопроводов, цистерн, металлических частей машин, корпусов судов, морских сооружений и т.п. Безвозвратные потери Ме от коррозии составляют до 15% ежегодного их выпуска. Цель борьбы с коррозией – это сохранение ресурсов Ме, мировые запасы которых ограничены. По механизму протекания коррозийного процесса, зависящему от характера внешней среды, с которой взаимодействует Ме, различают химическую и электрохимическую коррозию. Химическая коррозия характерна для сред, не проводящих электрический ток. При хим-ой коррозии происходит прямое гетерогенное взаимодействие Ме с окислителем окружающей среды. По условиям протекания коррозийного процесса различают: а)газовую коррозию – в газах и парах без конденсации влаги на поверхности Ме, обычно при высоких темп-рах. Примером газовой коррозии может служить окисление Ме кислородом воздуха при высоких температурах; б)коррозию в неэлектролитах – агрессивных органических жидкостях, таких как сернистая нефть и др. электрохимическая коррозия характерна для сред, имеющих ионную проводимость. При электрохимической коррозии процесс взаимодействия Ме с окислителем включает анодное растворение Ме и катодное восстановление окислителя. электрохимическая коррозия может протекать: а)в электролитах – в водных растворах солей, кислот, щелочей, в морской воде; б)в атмосфере любого влажного газа; в)в почве. Хотя механизм протекания коррозийного процесса в разных условиях различен, по характеру разрушения поверхности Ме коррозию можно разделить на равномерную и местную. равномерная, или общая, коррозия распределена более или менее равномерно по всей поверхности Ме, в то время как местная коррозия сосредоточена на отдельных участках и проявляется в виде точек, язв или пятен. Местная коррозия, как правило, более опасна, чем равномерная коррозия, т.к. процесс проникает на большую глубину. Особыми видами коррозии явл-ся межкристаллическая коррозия (коррозия по границам зерен), избирательная коррозия (растворение одного из компонентов сплава) и коррозийное растрескивание (коррозия при одновременном воздействии хим-х реагентов и высоких механических напряжений). Данные виды коррозии особенно опасны, т.к. могут привести к быстрому разрушению машины, аппарата или конструкции. Скорость коррозии выражают несколькими способами. Наиболее часто пользуются массовым и глубинным показателями коррозии. первый из них дает потерю массы (в граммах или килограммах) за единицу времени (секунду, час, сутки, год), отнесенную к единице площади (квадратный метр) испытуемого образца. глубинный показатель коррозии выражается уменьшением толщины Ме в единицу времени. скорость электрохимической коррозии можно также выразить величиной тока, приходящегося на единицу площади Ме. Самопроизвольное разрушение Ме (коррозия) приносит большие убытки. Коррозия протекает по различным механизмам и вызывает разные виды разрушений. При разработке методов защиты от коррозии используют указанные способы снижения скорости коррозии, которые изменяются в зависимости от характера коррозии и условий ее протекания. Выбор способа определяется его эффективностью, а также экономической целесообразностью. Все методы борьбы условно делятся на следующие группы: а)легирование Ме (в состав сплава вводят компоненты, вызывающие пассивацию Ме, в качестве таких компонентов применяют хром, никель, вольфрам и др.); б)защитные покрытия (металлические, неметаллические);в)электрохимическая защита (этот метод основан на торможении анодных или катодных реакций коррозийного процесса); г)изменение свойств коррозийной среды (для снижения коррозийной среды уменьшают концентрацию компонентов, опасных в коррозийном отношении); д)рациональное конструирование изделий (должно исключать наличие или сокращать число и размеры особо опасных, с точки зрения коррозии, участков в изделиях или конструкциях, а также предусматривать специальную защиту Ме этих участков от коррозии. №34. Теория хим. строен. орг. соед. Бутлерова. Структ. пр-пы. Углеродн. скелет, радикал и функц. группа. Осн. положения теории А.М. Бутлерова были опубликованы в 1861 г. в статье «О химическом строении веществ» и в ряде других работ. Сущность этих положений сводилась к следующему. 1.Атомы, входящие в состав молекулы органического вещества, не находятся в беспорядочном состоянии, а соединены между собой в определенной последовательности химическими связями.Порядок и последовательность соединения атомов в молекуле А.М. Бутлеров назвал химическим строением. 2.Соединение атомов в молекуле происходит в соответствии с их валентностью. Свободных валентностей у атомов в молекуле нет. 3.Свойства вещества зависят не только от того, какие атомы и сколько их входит в состав молекулы, но и от того, в какой последовательности они соединены между собой в молекуле (т.е. от химического строения).Различие в химическом строении веществ с одной и той же молекулярной формулой является причиной изомерии, и число возможных изомеров можно предсказать. 4.Атомы и группы атомов, входящие в молекулу, оказывают влияние на химическое поведение друг друга. Особенно заметно такое влияние в том случае, если эти атомы или группы атомов связаны друг с другом непосредственно. 5.Зная свойства вещества, можно установить его строение и наоборот: химическое строение органического соединения может многое сказать о его свойствах. 6.Атомы углерода способны соединяться друг с другом с образованием углерод-углеродных цепей различных видов. Эти цепи могут быть открытыми (прямыми или разветвленными) или замкнутыми (циклическими). Цепи могут содержать одинарные, двойные и тройные связи. 7.Строение молекулы можно выразить при помощи структурной формулы, которая для данного органического вещества является единственной. Углеродный скелет (углеродная цепь) – это последовательность всех химически связанных между собой атомов углерода.Функциональная группа – атом или группа атомов, определяющая принадлежность соединения к данному классу и ответственная за его химические свойства.Радикалы свободные - атомы или химические соединения с неспаренным электроном (обозначается жирной точкой), напр-р H, CH3, C(C6H5)3. Парамагнитны, реакционноспособны. Короткоживущие радикалы — промежуточные частицы во многих химических реакциях. Некоторые радикалы свободные стабильны и выделены в индивидуальном состоянии. С участием радикалов свободных осуществляются важные биохимические процессы, напр. ферментативное окисление. |
№32. Ме и их соед-ия в произ-ве тов-в народн. потр-ия. Макро- и микроэлементы. Ксенобиотики. Все простые вещ-ва можно разделить на металлы и неметаллы поскольку их св-ва существенно разные. Рассмотрим хар-ные св-ва металлов: металлическая связь в кристаллах, металлический блеск, хорошая тепло и электропроводимость, ковкость и пластичность, восстановители, оксиды имеют ионный характерный и при растворении в воде образуют осн-ые р-ры. Физ. св-ва: Ме делятся на легкий и тяжелые. Легкие – s-Ме Al,Скандий,Ti, плотность <= 5 г/см3. Тяжелые – d-Me 5-го и 7-го периода. Ме бывают легкоплавкие(s и p-Ме, а также – d-Me 2-ой группы) и тугоплавкие (t плавления > 1500 С, d-Me 4-ой – 7-ой групп). Хим.св-ва: металлы могут взаимодействовать с окислителем, они могут окислятся F или Cl, также Br и O2 в кислой среде кроме Pt и Au. Взаимодействие многих Ме с хромом, Br, O2, ионами H тормозится пассивными пленками на поверхности металлов. Большой склонностью к пассивации обладают бериллий, Al, d-Ме 4-8 групп. Пыль и пары некоторых Ме токсичны, она характеризуется предельно допустимыми концентрациями веществ в рабочей зоне. К наиболее токсичным относятся бериллий, кадмий, талий, никель. Ме явл-ся комплексообразователями и входят в состав комплексных соединений. Комплексообразователи – атомы или ионы имеющие вакантные орбитали. Способность к комплексообраз. растет с увеличением заряда иона и уменьшением его радиуса. Зависит от природы металла и наличия вакантных орбиталей у его ионов. Комплексное соединение Fe, Co, Cu, Mn, Zn входят в состав биологических систем включ. ферменты-переносчики крови. Применение Ме и их соедин. Na используется в металлургии для получения Ме и удаления мышьяка из свинца, а также в кач-ве жидкого теплоносителя в атомной энергетике и химпроме. Рубидий и цезий при освещении легко теряют электроны, поэтому служат материалами фотоэлементов. Щелочи и соли щелочных Ме применяются в машиностроении для обезжиривания деталей, нейтрализации стоков (NaOH, Na2CO3). В энергетике для водоподготовки (NaOH, NaCl). В металлургии (NaCl,KCl,NaNO3) для защиты о коррозии. Смесь (LiCl,LiOH) при сварке и пайке (LiF). Некоторые соли Na и K используются в кач-ве пищевых добавок, н-р, аскорбат натрия E301; от E322 и выше – эмульгаторы, стабилизаторы, н-р, гидроцитрат натрия E322. Из соединений магния наибольшее применение нашел MgO (наполнитель резины в производстве огнеупоров и стройматериалов). MgCl2 в производстве магнезиального цемента. Бериллий применяется в атомной энергетике как замедлитель нейтронов. Бериллиевая бронза (сплав меди и бериллия), содержащая 2.5% бериллия, из которой готовят пружины и другие упругие элементы приборов и устройств. Mg и Ca явл-ся жизненно необходимыми элементами в организме человека. КСЕНОБИОТИКИ, чужеродные для организмов соединения (промышленные загрязнения, пестициды, препараты бытовой химии, лекарственные средства и т. п.). Попадая в окружающую среду в значительных количествах, ксенобиотики могут воздействовать на генетический аппарат организмов, вызывать их гибель, нарушать равновесие природных процессов в биосфере. Изучение превращений ксенобиотиков в организмах, путей их детоксикации и деградации (с помощью микроорганизмов и др.) важно для организации санитарно-гигиенических мероприятий, мер по охране природы. Микро и макроэлементы смотреть физиологию питания.
№35. Кл-ция орган. соед-й по углеродн. скелетуи функц. группам. Гомология и гомолог. ряды. Изомерия. ИЮПАК. Классификация орг. соединений по углеродному скелету:1.Ациклические соединения – соединения с открытой цепью углеродных атомов. Она м.б. прямой или разветвленной.2.Циклические соединения – соединения, в кто-х углеродные атомы образуют циклы.1)карбоциклические и ароматические,2)гетороциклические – циклы содержат гетороатомы. по функциональным группам выделяют соединения со смешанными функциями. Орг. соединения содержат 2 и более различн. функциональных групп. К особенностям органических соединений можно отнести существование гомологических рядов, у кот-х каждый последующий член м.б. произведен от предыдущего добавлением одной определенной для данного ряда группы атомов. Н-р, в гомологическом ряду предельных углеводородов такой группой явл-с СН2. Гомологический ряд хар-ся общей формулой, н-р для предельных углеводородов-СnH2n+2. В тоже время происходит изменение физ-х св-в элементов по мере увеличения. числа групп.Изомерия. Различают структурную и пространственную изомерию. Структурная изомерия обусловлена разным порядком связей углеродных атомов(изомерия скелета) или разным расположением функциональных групп или кратных связей(изомерия положения). Пространственная изомерия обусловлена разными расположениями хим. связей атомов углерода в пространстве и включает геометрическую, оптическую и др. виды изомерии. Пространственные изомеры называют стереоизомерами. К числу геометрических изомеров относятся цис- и транс-изомеры. Оптические изомеры имеют в своем составе один или несколько атомов углерода, связанных с четырьмя разными атомами или группами атомов. Принцип хим. номенклатуры ИЮПАК. В соот-ии с ИЮПАК названия орг. соед. слагаются из словесных обозначений частей, их структуры и знаков, указывающих способ связей этих частей. Основная часть названия соединения состоит из названия самой длинной неразветвленной цепи атомов углерода. Число углеродных атомов в цепи обозначают греческими числительными. С помощью суффиксов и префиксов обозначают классы соединения. Н-р, -ан-предельные соединения, -ин-соединения с тройной связью, -ол-спиртов. С помощью суффикса –ил обозначают радикалы. Функциональным группам, входящим в состав соединений, присвоены соот-ие названия, н-р,СООН-карбокси, ОН_гидрокси. Число одинаковых заменителей обозначают порядковые номера углеродных атомов, у кот-х имеются боковые цепи или функциональные группы |
№33. Органическая химия-химия соед-й углерода. Состав орган. соед-й. элементы органогены. Многообр-ие соед-й углерода. Все вещества химики делят на два больших класса — на орган и неорганические. Органические вещества, в отличие от неорганических, всегда содержат в молекулах один или несколько (часто — очень много) атомов углерода. Существуют, правда, простые неорган вещества, которые также содержат углерод (СО, СО2, угольная кислота и ее соли и др.), но они не обладают свойствами орган соединений и их обычно изучают в курсе общей или неорганической химии. Т.о., основным и обязательным элементом, входящим в состав органических веществ, является углерод. Поэтому такие вещества изучает особая наука — органическая химия, которую А.М. Бутлеров определил как химию соединенийуглерода. Кроме углерода органические вещества почти всегда содержат водород, довольно часто — кислород, азот и галогены, несколько реже — серу, фосфор и некоторые металлы. Такие элементы, как водород, кислород, азот, сера и галогены, называют органогенами, так как они играют важную роль в построении органических соединений. Следовательно, органическая химия изучает соединения углерода со многими элементами. Однако самые простые органические вещества состоят только из углерода и водорода. Такие соединения называют углеводородами. Если в углеводородах один или нескольк атомов водорода заместить на другие атомы или группы атомов, то можно получить разнообразные производные углеводородов. Следовательно, органическую химию можно определить как химию углеводородов и их производных, в состав которых могут входить почти все элементы Периодической системы. Чем вызвано выделение орган. химии в самостоятельную химическую науку? Для этого имеется несколько причин. Первая из них — многочисленность и многообразие органических соединений. Действительно, в настоящее время их известно около 10 млн, число же известных неорганических соединений не превышает 700 тыс. Чем же вызвана эта удивительная многочисленность? Ни один из известных элементов Периодической системы не обладает такими уникальными свойствами, как углерод. Атомы углерода способны соединяться не только с другими элементами, но и друг с другом, образуя часто сложные молекулы, построенные как в виде цепей (линейных, разветвленных), так и различного циклического строения. Эти цепи могут содержать неограниченное число углеродных атомов. При этом атомы углерода связываются друг с другом не только простыми (одинарными) связями, но и двойными, и тройными. Таким образом, различная длина углеродных цепей, их разветвленность, а также разнообразие циклических систем приводят к образованию огромного числа всевозможных органических соединений. 'Вторая причина состоит в сложности и своеобразии органических веществ. Они существенно отличаются от неорганических соединений. Так, при нагревании (даже до сравнительно невысокой температуры) многие органические вещества обычно разлагаются. Их температуры кипения и плавления значительно ниже, чем у неорганических веществ. Кроме того, все органические соединения горючи (за некоторым исключением), тогда как большинство неорганических вещ-в этим св-вом не обладают. Для орг. соед-ний характерно большое разнообразие хим. превращений. Для соврем-го пер-да орг-ой хим хар-но дальн разв строения орг-их соед с привлеч квантовой мех-ки, шир-м исполь-ем физ-их мет-в исслед-ия строения орг-их молекул, разраб нов способов получ, выдел и очистки орг-их в-в. Значение органической химии для современного человека трудно переоценить. Основные продукты питания (белки, жиры, углеводы)— органические вещества, одевает и обувает человека также органическая химия. Вспомните натуральные (хлопок, лен, шерсть, шелк), искусственные (ацетатные и вискозные) и синтетические (капрон, лавсан, анид и др.) волокна. Для изготовления обуви широко применяются различные каучуки, натуральная кожа и кожзаменители. Сейчас трудно найти хотя бы одну отрасль промышленности, которая обходится без органического сырья или продуктов органического синтеза. Так, топливная промышленность использует уголь, газ, нефть, торф, сланцы и древесину. В то же время нефтехимическая промышленность из нефти и газов (природного и попутных) получает автомобильное и авиационное топливо (бензин, керосин и мазут), смазочные масла, мономеры для произв-ва полимеров, разл. растворители, присадки к топливам и др. Лакокрасочная промышленность производит лаки, олифу, органические красители и пигменты, а текстильная и кожевенная — одежду и обувь из натурального и синтетического органического сырья. Нельзя забывать и о том, какой богатый ассортимент лекарств. препаратов дает нам фармац. пром-сть. В больших масштабах проводится синтез полимерных материалов с разнообразными свойствами. Без этих материалов трудно представить строительство, автомобильный, авиационный и железнодорожный транспорт, машиностроение, электрои радиоэлектронную промышленность, сельское хозяйство и медицину, ядерную энергетику и космонавтику. Благодаря орган. синтезу мы получаем разнообразные поверхностно-активные вещества (ПАВ), металлорганические соединения, различные клен и герметики, биол-ски активные вещества, консерванты и антисептики, ср-ва защиты и роста растений. Органическая химия сегодня тесно соприкасается с биологией, медициной, физикой и другими науками. Успехи теоретической и экспериментальной органической химии имеют исключительно важное значение и для понимания биологических процессов, протекающих в живых организмах. Со временем орг. химия стала настолько всеобъемлющей наукой, что ее отдельные разделы превратились в самостоятельные научные направления. Так возникли биохимия, химия высокомолекулярных соединений, химия гетероциклически х соединений, химия углеводов, витаминов, фармацевтическая химия и т. д. сырьеве ист-ки получ орг-их соед Для получения органических соединений используют как природные источники, так и методы синтеза. К первым относятся нефть, природный и попутные газы, каменный уголь, торф, сланцы, древесина, продукты лесного и сельского хозяйства, к вторым — органический и микробиологический синтезы. Если из природных источников органические вещества выделяют в основном в готовом виде, перерабатывая природное сырье, то органический синтез позволяет получать их искусственно — из других органических соединений или даже из отдельных элементов, последовательно создавая необходимую стр-ру мол-лы. Однако эти два способа получения не следует противопоставлять друг другу. Часто бывает так, что органические вещества, выделенные из природного сырья, нуждаются в дальнейшей “достройке”, но уже химическим путем, путем синтеза. Нефть — один из основных видов природного органического сырья. Переработкой нефти получают бензины, керосин и другие разновидности топлива для двигателей, смазочные масла в т.д. Крекинг нефтепродуктов кроме жидких углеводородов дает газообразные непредельные углеводороды (алкены). Природный и попутные газы — ценное сырье для получения различных органических веществ. Природный газ содержит в основном метан (до 95%), а также небольшое количество других предельных углеводородов (этан, пропан, бутан). Каменный уголь — источник очень многих органических продуктов. При его переработке можно получить сотни различных органических веществ. Уголь — один из перспективных видов хим. сырья, так как его мировые запасы намного превышают запасы нефти и газа. Битуминозные и горючие сланцы дают химической промышленности такие ценные пр-ты, как сланцевое масло, из кот. получают бензин и другие виды топлива, а также битум и смолы, содержащие разл. фенолы. Торф является источником предельных углеводородов, органических кислот (в основном щавелевой), фенола. Целлюлоза — основная часть древесины. Из целлюлозы получают бумагу, искусственное волокно, спирты, эфиры, эфирные масла и другие продукты. Сельскохозяйственное сырье — источник получения многих органических продуктов: крахмала, глюкозы, спирта, витаминов, скипидара, канифоли и др. Промышленные отходы производства также могут использоваться для получения многих органических продуктов. При этом первостепенное значение имеет утилизация отходов производства. Это важная экологическая и экономическая проблема. В данном случае необходимо решать две задачи: выделение индивидуальных органических веществ из сложной смеси и их дальнейшее практическое использование. Органический синтез позволяет получать огромное число разнообразных органических соединений, которые нельзя получить из природных источников. Методы синтеза используют при производстве органических красителей и лекарственных препаратов, полимерных соединений, поверхностно-активных веществ и других продуктов. Органический синтез дал огромное многообразие органических веществ, при этом органические продукты получают с высокой степенью чистоты. Орган. синтез рождается вначале в лаборат. условиях, а затем переносится в промышленные. Микробиологический синтез. В последние годы ценным источником органических веществ становится микробиологический синтез. В его основе лежит способность некоторых микроорганизмов в процессе своей жизнедеятельности синтезировать довольно сложные органические вещества, которые не удавалось получить химическим путем. Микробиологический синтез применяют для получения белков, ферментов, антибиотиков и других веществ. |
||||||||||||||||||||||||||||
|
№36. Углеводороды. Углеводороды — наиболее простые органические соединения, молекулы которых построены только из двух элементов — углерода и водорода.По характеру связей между углеродными атомами алифатические углеводороды могут быть предельными (насыщенными) и непредельными (ненасыщенными).1Алканы, или парафины, — алифатические предельные углеводороды, в молекулах которых атомы углерода связаны между собой простой (одинарной) о-связью. Оставшиеся валентности углеродного атома, не затраченные на связь с другими атомами углерода, полностью насыщены водородом. Поэтому предельные (насыщенные) углеводороды содержат в молекуле максимальное число водородных атомов. Номенклатура. Первые четыре представителя предельных углеводородов имеют эмпирические названия: метан, этан, пропан, бутан. Начиная с пентана названия углеводородов образуют от греческих числительных* с добавлением суффикса -а»: пентан, гексан, гептан, октан, нонан, декан, ундекан и т.д.Общее (родовое) название предельных углеводородов — алканы. Алкан называют в таком порядке: вначале указывают (цифрой) место расположения радикалов, затем их перечисляют в алфавитном порядке (метил, этил), а в конце добавляют название главной цепи. Изомерия. Для алканов характерен самый простой вид изомерии — структурная изомерия.Первые четыре члена гомологического ряда метана — газообразные вещества, начиная с C5Hi2 — жидкости, а алканы с числом углеродных атомов 16 и выше при обычной температуре—твердые вещества.Увеличение молекулярной массы приводит к повышению температуры кипения и плавления и увеличению плотности. Алканы — соединения неполярные и трудно поляризуемые. Эти свойства, как мы увидим далее, определяют их химическое поведение.В алканах все углеродные атомы связаны между собой прочными а-связями. Оставшиеся валентности атомов углерода полностью насыщены атомами водорода. Поэтому алканы являются соединениями насыщенными и не вступают в реакции присоединения.Алканы — малоактивные соединения. Они не измняются при действии на них концентрированных кислот, щелочей, металлов (в том числе и щелочных) и окислителей при обычных условиях. Таким образом, для предельных углеводородов различают два основных типа химических реакций:1)реакции замещения водорода с разрывом связи С-Н;2)реакции расщепления молекулы алкана с разрывом связей С-С и С-Н.В реакциях первого типа легче происходит замещение атома водорода, связанного с третичным угаеродным атомом, труднее — со вторичным и совсем трудно — с первичным. Это можно объяснить, если сопоставить энергии связей С-Н у первичного (около 420 кДж/молъ), вторичного (390 кДж/моль) и третичного (370 кДж/моль) атома углерода. Алкены, или олефины — алифатические непредельные углеводороды, в молекулах которых между углеродными атомами имеется одна двойная связь.Алкены содержат в своей молекуле меньшее количество водородных атомов, чем соответствующие им алканы ( с тем же числом углеродных атомов), поэтому такие углеводороды называют непредельными или ненасыщенными.Номенклатура. По систематической номенклатуре родовое название этиленовых углеводородов — алкены. Названия отдельных гомологов по этой номенклатуре производят заменой суффикса -ан в соответствующих алканах на суффикс -ен (этан — этен, пропан — пропен, бутан — бутен и т.д.). Изомерия. Для алкенов характерны два вида структурной изомерии. Кроме изомерии, связанной со строением углеродной цепи (изомерия углеродного скелета), появляется изомерия, которая зависит от положения двойной связи в этой цепи (изомерия положения). Помимо структурной изомерии в ряду этиленовых углеводородов возможен еще один вид изомерии — цис-, транс-изомерия. Алкены легче воды и плохо растворимы в ней (однако лучше, чем соответствующие алканы), но хорошо — в органических растворителях. Пары алканов имеют характерный резкий запах.Алкены малополярны, но легко поляризуются. Этилен и пропилен горят слегка коптящим пламенем.Алкены в отличие от алканов обладают высокой химической активностью. Их свойства определяются двойной углерод-утл сродной связью, состоящей из сигма- и пи-связей. Пи-Связь как наименее прочная и более доступная при действии реагента разрывается, а освободившиеся валентности атомов углерода затрачиваются на присоединение атомов, из которых состоит молекула реагента.Для алкенов наиболее типичны реакции присоединения.Двойная связь, имеющая высокую электронную плотность, обычно выступает в качестве донора электронов. Поэтому реакции присоединения, протекая по гетеролитическому типу, являются реакциями эяектрофильного присоединения. Алкены вступают также в реакции полимеризации и окисления. Диеновые углеводороды(алкадиены, или диолефины) — непредельные органические соединения, содержащие в молекуле две двойные связи,Молекулы диеновых углеводородов содержат на два атома водорода меньше, чем молекулы алкенов. Поэтому общая формула диеновых углеводородов СnН2n-2. Номенклатура, По систематической номенклатуре диеновые углеводороды называют, как и этиленовые, но заменяют суффикс -ен на -диен (две двойные связи). Изомерия. Изомерия диенов зависит от различного положения двойных связей в углеродной цепи и от строения углеродного скелета. Диены, содержащие в молекуле несопряженные (изолированные) двойные связи, проявляют свойства обычных алкенов. В то же время десны с сопряженными двойными связями обладают высокой реакционной способностью и отличаются рядом особенностей. Однако для lex и других диеновых углеводородов характерны прежде всего реакции присоединения. Алкины {ацетилены) — алифатические непредельные углеводоро-i, в молекулах которых между атомами углерода имеется одна трой-связь.Алкины образуют гомологический ряд с общей формулой С„Н2п_2.Номенклатура. По названию первого представителя гомологичес->го ряда алкинов — ацетилена — эти углеводороды называют ацети-ювыми.По систематической номенклатуре алкины называют, заменяя суф-)икс -ан (в алканах) на суффикс -ин. Изомерия. Изомерия алкиновых углеводородов определяется строки углеродной цепи и положением в ней тройной связи. от двух до четырех углеродных атомов— газы, начиная с С5Н8 — жидкости, а высшие алкины (с Ci6H30 и выше) — твердые вещества. Положение тройной связи существенно влияет на температуру кипения Химические свойства алкинов определяются тройной связью и осо-жностью ее строения. Алкины способны вступать в реакции присое-1ения, замещения, полимеризации и окисления.
|
№37. Р-ция полимеризации и сополимеризации олфеинов. Каучуки. В следствии разрыва пи-связи, молекулы алкена могут соединяться др. с др., образуя макромолекулы с высокой молекулярной массой.СН2=СН2 – (-СН2-СН2-)n. Это реакция полимеризации – р. соед-я нескольких молекул мономера. при кот. не происходит выдел-я побочных низкомол-х продуктов. Диеновые у-в легко вступают в реакции полим-ции с образованием каучукоподобных высокомолек-х продуктов. Н2С=СН-СН=СН2 + Н2С=СН-СН=СН2 +… - -СН2-СН=СН-СН2-СН2-СН=СН-СН2-…Каучуки и резина относятся к эластомерам. Каучуки делят на нату рольный (природный) и синтетические.Натуральный каучук (НК) представляет собой высокоэластичную массу, получаемую из млечного сока (латекса*) некоторых тропичес-i ких деревьев (гевеи бразильской и др.) и растений. Натуральный каучук— природный непредельный полимер (C5H8)n со средней молекулярной массой от 15 тыс. до 500 тыс.НК — полимер изопрена.Наиболее важным отличием натурального каучука является его высокая эластичность — способность к большим деформациям под действием внешней нагрузки и восстановление формы после снятия нагрузки. Натуральный каучук растворяется во многих углеводородах, образуя вязкие растворы. Подобно диенам, он вступает во многие реакции присоединения.Каучук — пластический материал. Изделия из него обладают рядом недостатков: при повышении температуры он становится липким, теряет форму, а при низкой температуре — эластичность. Поэтому каучук нельзя использовать в природном виде.Для придания каучукам прочностных свойств, эластичности и термостойкости их подвергают обработке серой—вулканизируют.В результате каучук превращается в резину. Высокая потребность промышленности в каучуке привела к тому, что большая часть его производится синтетическим путем. Некоторые виды синтетического каучука не уступают натуральному, а по некоторым свойствам даже превосходят его.Синтетические каучуки (СК) — синтетические аналоги натурального каучука, получаемые полимеризацией мономеров (каучукогенов) — дивинила, изопрена, хлоропрена и др. Часто используют метод со-полимеризации двух мономеров и более.СК:1.Бутадиеновые каучики(-СН2-СН=СН-СН2)n, 2.Бутадиен – стирольные каучики (-СН2-СН=СН-СН2-)n-(-СН2СН(С6Н5))m,3.Бутадиен-нитрильные каучуки (-СН2-СН=СН-СН2-)n-(-СН2СН(CN)-)m,4.Изопреновые к.(-СН2-С(СН3)=СН-СН2-)n, 5. Хлоропреновые к.(-СН2-С(Cl)=СН-СН2-)n, 6. Полисульфидные к.(конденсация дигалогенопроизводных с полисульфидами щелочных металлов).Высокомолекулярные соединения (ВМС) — это химические вещества, которые состоят из большого числа повторяющихся группировок (мономерных звеньев), соединенных между собой химическими связями.Высокомолекулярные соединения часто называют просто полимерами (от греч. poly — много, meros — часть).Число элементарных звеньев в макромолекуле (п) является одной из главных характеристик полимера и называется степенью полимеризации (Р) полимера. Иногда макромолекулы могут быть построены не из одинаковых, а из разных элементарных звеньев. Такие полимеры называют сополимерами в отличие от гомополимеров, которые содержат в макромоле-кулярной цепи одинаковые элементарные звенья.
№40. Гетероциклические соединения. Гетероциклические соединения – орг. в-ва, содержащие цикл, в состав которого, кроме атомов углерода, входят атомы других элементов (гетероатомы), наиболее часто N, О, S, реже — Р, В, Si и др. Многообразие типов ГС чрезвычайно велико, т. к. они м. отличаться др. от др. числом атомов в цикле, природой, числом и расположением гетероатомов, наличием или отсутствием заместителей либо конденсированных циклов, насыщенным, ненасыщенным или ароматическим хар-ром гетероцикл. кольца. Неароматические ГС по хим. св-вам близки к своим аналогам с открытой цепью; некоторые различия обусл-ны эфф-тами напряжения в цикле и пространств. эфф-тами, связанными с циклической структурой. Так, окись этилена (I) и тетрагидрофуран подобны алифатическим эфирам простым, а этиленимин (III) и пиролидин (IV) — алифатическим вторичным аминам:
Ароматичность проявляется у ГС (гл. образом 5- и 6-членных), содержащих, подобно другим ароматическим соединениям, замкнутую систему 4n+2 p -электронов. Химия таких ГС, сохраняя известное сходство с химией ароматических соединений бензольного ряда, опр-ся в основном специф. хар-ром каждого гетероциклического ядра. К важнейшим ароматическ. ГС относятся фуран (V), тиофен (VI), пиррол (VII), пиразол (VIII), имидазол (IX), оксазол (X), тиазол (XI) и пиридин (XII). Большое значение имеют также ГС, конденсированные с бензольными ядрами, — бензофуран (кумарон; XIII), бензпиррол (индол; XIV), бензтиофен (тионафтен; XV), бензтиазол (XVI), бензпиридины — хинолин (XVII) и изохинолин (XVIII), дибензпиридин (акридин; XIX):
Ароматический хар-р фурана, тиофена, пиррола и их бензпроизводных опр-ся участием неподелённой электронной пары гетероатома в обр-нии замкнутой системы шести p-электронов. В кислой среде гетероатом присоединяет протон и система перестаёт быть ароматической. Поэтому такие ГС, как фуран, пиррол и индол, не выдерживают действия сильных кислот (тиофен устойчив к кислотам вследствие меньшего сродства серы к протону):
В 6-членных гетероциклах неподелённая электронная пара гетероатома не участвует в образовании ароматической системы связей. Поэтому пиридин — гораздо более сильное основание, чем пиррол, и с кислотами образует устойчивые соли:
Некоторые важные ГС м. б. получены из каменноугольной смолы, напр-р пиридин и его гомологи, хинолин, изохинолин, индол, акридин, карбазол и др.; гидролизом растительных отходов (шелуха подсолнечника, солома и т. п.) получают фурфурол. Однако наибольшее значение имеют синтетические м-ды, которые весьма разнообразны и специфичны; они рассмотрены в статьях, посвященных отдельным представителям ГС. При синтезе чаще всего исходят из соединений с открытой цепью. Для некоторых 5-членных гетероциклов известны взаимные превращения. Так, фуран, пиррол и тиофен переходят друг в друга при действии соответственно H2O, NH3 или H2S при 450° над Al2O3. Юрьева реакция, превращение фурана в его аналоги — пиррол (X = NH), тиофен (X = S), селенофен (X = Se):
Осущ-ся пропусканием паров фурана соотв-но с аммиаком, сероводородом или селеноводородом над окисью алюминия (Al2O3) при 400—450 °С. Выход 30—40%. В Ю. р. вступают также гидрированные аналоги и гомологи фурана. Например, тетрагидрофуран почти колич-но превращается в тетрагидротиофен. Открыта в 1935 советским химиком Ю. К. Юрьевым (1896—1965). Применяется в орган. синтезе для получения некоторых замещенных пирролов. Роль ГС в пр-сах жизнед-сти растительных и животных организмов искл-но велика. К ГС отн-ся такие в-ва, как хлорофилл растений и гемин крови, компоненты нуклеиновых кислот, коферменты, некоторые незаменимые аминокислоты (напр-р, пролин и триптофан), почти все алкалоиды, пенициллин и некоторые другие антибиотики, ряд витаминов, например кобаламин (витамин B12), никотиновая кислота и её амид (витамин PP), растительные пигменты (антоцианины) и т. д. К ГС принадлежат многие широко применяемые в медицине синт. лекарств. в-ва, такие, как антипирин, амидопирин, анальгин, акрихин, аминазин, норсульфазол и другие. Г. с. широко применяют в различных отраслях промышленности (растворители, красители, ускорители вулканизации каучука и т. д.). |
№38. Циклоалканы. циклоалкены, циклоалкадиены. Циклоалканы(СnH2n)состоят из циклов, построенных из метиленовых групп, кот. связаны между собой сигма-связями.т.е.эти соединения изомерны этиленовым углеводородам. циклоалканы,содержащие пяти-и шестичленные циклы(циклопентан),составляют основную массу некоторых сортов нефти(бакинской)входят в состав эфирных масел и др.природных соедин.Получение: циклоалканы выделяют из нефти, некоторых природных в-в или получают синтетическим путем. Действием цинковой пыли в спиртовом растворе на 1,3-дихлорпропан получают циклопропан CH2C CH2 H2C +Zn= H2C / +ZnCI CH2CI CH2 1,3-дихлор пропан циклопропан Циклоалканы в химическом отношении во многом напоминают алканы. они сравнительно малоактивны и склонны к реакциям замещения. но низшие алканы вступают в реакцию присоединения, которые связаны с разрывом кольца.Циклоалкены(CnH2n-2) -непредельные алициклические соединения могут содержать в цикле одну или две 2-й связи.циклоалкодиены(CnH2n-4) такую формулу имеют циклы с одной тройной связью.Получение:циклопентен можно синтезировать действием спиртовой щелочи на бромциклопентан. Хим.св-во:подобно этиленовым углеводородам, циклоалкены вступают в реакции присоединения.они ведут себя как диеновые системы.например:при действии брома на циклопентадиенидет1,4-присоединение с образованием 1,4-дибромпроизводного,эта реакция названа необратимым катализом,потому,что из циклогексана и бензола нельзя каталитически получить обратно циклогексен. Терпены, углеводороды, молекулы которых построены из изопреновых звеньев C5H8, то есть имеют состав (C5H8) n, где n=2, 3, 4...; относятся к обширному классу природных соединений — изопреноидов. По числу изопреновых звеньев в молекуле Т. подразделяются на монотерпены C10H16 (обычно называемые просто терпенами), сесквитерпены (полуторные терпены) C15H24, дитерпены C20H32, тритерпены C30H48 и т. д. Т. обнаружены практически во всех тканях растений (содержатся в эфирных маслах, скипидаре, смолах, бальзамах), найдены в продуктах жизнедеятельности некоторых бактерий и грибов, в секреторных выделениях насекомых. Обычно Т. сопутствуют их кислородсодержащие производные (спирты, эфиры, альдегиды, кетоны, кислоты и др.), часто называют терпеноидами. По строению молекулы различают Т. ациклические (с открытой цепью углеродных атомов), например мирцен, и циклические, содержащие одно или несколько неароматических колец, например лимонен, камфен, пинены.Монотерпены — бесцветные с характерным запахом жидкости (tkип150—190°С), сесквитерпены — бесцветные вязкие жидкости или легкоплавкие кристаллические вещества (tkип 230—300 °С) с более слабым, но более стойким, чем у монотерпенов, запахом. Например, запах имбиря определяется присутствием сесквитерпенового углеводорода цингибирена, запах липы — сесквитерпеновым спиртом фарнезолом. Активное начало цитварного семени — сесквитерпеноид сантонин. К производным дитерпенов относятся, например, смоляные кислоты (абиетиновая, левопимаровая и др. кислоты), содержащиеся в канифоли, и спирт фитол, сложный эфир которого — составная часть хлорофилла. Тритерпен сквален найден в печени акулы. Установлено, что тритерпеновую структуру имеют также стерины и гормоны; так, показано образование стероидного гормона холестерина из сквалена. Некоторые тетратерпеноиды (каротин и ксантофиллы) являются красящими веществами растений (см. Каротиноиды). Политерпенами можно считать гуттаперчу и каучук натуральный.Т. практически не растворимы в воде, хорошо растворимы в неполярных органических растворителях; легко окисляются, полимеризуются, гидрируются, галогенируются, изомеризуются. Ациклические Т. легко (например, под действием разбавленных минеральных кислот) превращаются в циклические. Обратный переход осуществляется в более жёстких условиях: например, мирцен получают в промышленности пиролизом -пинена при 500 °С. Из природного сырья Т. и терпеноиды обычно выделяют ректификацией, вымораживанием (например, ментол из мятного масла) и др. методами. Многие терпеноиды получают в промышленности из более доступных Т. или из химического сырья. Так, содержащиеся в скипидаре (в количестве до 95%) Т. используют для производства камфоры (выделяемой также из масла камфорного лавра), терпинеола, терпингидрата, гераниола, карвона; цитраль, выделяемый из некоторых эфирных масел, получают также окислением линалоола и в значительных количествах синтезируют из изопрена, ацетона и ацетилена. Т. и терпеноиды в индивидуальном состоянии или в виде эфирных масел и смол широко используют в качестве компонентов парфюмерных композиций и пищевых эссенций, в медицине (ментол, гераниол, терпнигидрат и др.). Из Т. получают также смазочные масла, инсектициды, например полихлорпинен и полихлоркамфен, флотационное масло, иммерсионные жидкости. Каротиноиды, жёлтые, оранжевые или красные пигменты (циклические или ациклические изопреноиды), синтезируемые бактериями, грибами и высшими растениями. Животные обычно не образуют К., но используют их для синтеза витамина А. К К. относятся широко распространённые в растениях каротин и ксантофиллы; ликопин (С40Н5б) — в плодах томатов, шиповника, паслена; зеаксантин (С40Н56О2) — в семенах кукурузы; виолаксантин и флавоксантин — в плодах тыквы; криптоксантин (C40H56O) — в плодах дынного дерева; физалин (C72H116O4) — в цветках и плодах физалиса; фукоксантин (С40Н56О6) — в бурых водорослях; кроцетин (C20H24O4) — в рыльцах шафрана; тараксантин (C40H56O4) — в цветках львиного зева, белокопытника и др. Относительное содержание различных К. меняется в процессе развития растений и под влиянием условий среды. В клетке концентрация К. наиболее высока в пластидах. К. способствуют оплодотворению растений, стимулируя прорастание пыльцы и рост пыльцевых трубок. К. участвуют в поглощении света растениями и восприятии его животными; играют большую роль в процессах фотосинтеза, а также в переносе кислорода в растениях. Число и положение двойных связей в молекулах К. определяют их окраску (известно свыше 150 К.-пигментов). При большем числе двойных связей К. поглощают в длинноволновой части спектра; цвет их ярко-оранжевый или красный. Каротин (от лат. carota — морковь), C40H56, оранжево-жёлтый пигмент из группы каротиноидов; предшественник витамина А. Синтезируется растениями; особенно много его в листьях при переходе растений к цветению. Богаты К. корни моркови (отсюда название — из них он впервые был выделен; в некоторых сортах 31 мг% на сырое вещество), плоды шиповника (от 2 до 16 мг%), смородины, рябины, облепихи, актинидии и др. В растениях встречается главным образом Р-каротин, который в качестве предшественника витамина А в 2 раза активнее изомерных ему - и -каротина. Биосинтез витамина А происходит только в животном организме. Особенно богата этим витамином печень китов и некоторых рыб (в 100 кг китовой печени содержится около 100 г витамина А — дневная доза для 50 тыс. человек). К. улучшает плодовитость животных, рост и развитие молодняка, предохраняет животных от ксерофтальмии. Роль К. в растительном организме недостаточно ясна. По-видимому, К. играет существенную роль в процессах фотосинтеза, дыхания и роста растений. К. легко образует перекиси, в которых молекула кислорода присоединяется по месту двойной связи и затем может участвовать в окислении различных соединений. Ликопин - вещество из группы каротиноидов, в которую входит и витамин А. Ликопин придает плодам томата красную окраску. В ходе тщательного и всестороннего исследования этого вещества выяснилось, что ликопин обладает выраженными антиоксидантными свойствами. Другими словами, он способен предотвращать повреждение наших клеток так называемыми свободными радикалами. Эти молекулярные соединения образуются в организме постоянно. Снижение защитных свойств организма приводит к нежелательным последствиям. Активно разрушаются клеточные мембраны, повреждается молекула ДНК. Результатом этого бывают злокачественные опухоли, сердечно-сосудистые заболевания и другие патологии. Таким образом ликопин усиливает действие комплекса других антиоксидантов, и является уникальным природным средством для профилактики сердечно-сосудистых и онкологических заболеваний. |
||||||||||||||||||||||||||||
|
№41 Галогенопроизводные. Фреоны. Галогенопроизводными называют орг.соед-ния, в мол-лах кот-х один или неск-ко атомов водорода замещены на галогены. В зав-сти от числа водородных атомов в углеводороде, замещенных на галогены, различают моно- и полигалогенопроизводные(ди-).Хар-р радикала, связанного с галогеном, определяют предельность или непредельность галогенопроизводных. Моиогалогенопроизводные предельных углеводородов (моногалогеналкилы) Строение Моногалогенопроизводные предельных углеводородов (моногалогеналкилы) образуют гомологический ряд, который выражают общ. формулой СлН2и+1Х (X — галоген). Атом галогена м. б. связан с первичным, вторичным или третичным углеродным атомом. Поэтому моногалогеналкилы делят на первичные (I), вторичные (II) и третичные (III):
Строение галогеналкилов м. представить структурной (I) или электронной (II) формулами. Например, для хлористого метила они имеют вид:
Электрон. ф-ла пок-ет, что атом галогена в галогенопроизводных имеет три пары неподеленных электронов. Номенклатура. По систем. номен-ре название галогенопроизводных составляют из названия углеводорода и галогена с добавлением цифры, обозначающей его место в цепи. При этом цепь нумеруют так, чтобы атом углерода, связанный с галогеном, получил наименьший номер. Если имеются и другие заместители, то их перечисляют в алфавитном порядке.Для названия простых моногалогенопроизводных используют номенклатуру радикалов, с которыми связан атом углерода. Например:
Изомерия. Структурная изомерия моногалогенопроизводных зависит от строения углеродной цепи и положения в ней атома галогена:
Химические свойства Хим. связь С-Х в галогеналкилах отл-ся повышенной полярностью. Это связано со значит. электроотрицательностью атома галогена, связанного с углеродным радикалом. Поэтому в мол-ле галогеналкилов электронная плотность смещается в сторону атома галогена, проявляющего индуктивный эффект:
Этим объясняется выс. реакционная сп-сть галогеналкилов в хим. р-циях. Галогеналкилы вступают в реакции нуклеофильного замещения, реакции отщепления. 1. Реакции нуклеофильного замещения (Sn). Поляризованность связи С-Х способствует замещению галогена X на нукдеофильные реагенты. 2 Реакции отщипления.отщепление 2-ух атомов от одного и того же вещ-ва без замещения их на другие атомы. 3. Реакции восстановления. При восстановлении галогеналкилов в присутствии катализаторов образуются алканы 4. Реакции образования магнийорганических соединений. Эти соединения получают взаимодействием магния с галогеналкилами в среде абсолютного диэтилового эфира. Дн- и полигалогенопроизводные предельных углеводородов (полигалогеналкилы)Строение. Ди- и полигалогенопроизводные алканов могут иметь одинаковые или разные атомы галогенов у одного и того же (I) или у разных (II) углеродных атомов:
Номенклатура. Названия полигалогенопроизводных алканов по систематической номенклатуре принципиально не отличаются от названий моногалогенопроизводных. Например:
По рациональной номенклатуре названия полигалогенопроизводных производят от названий радикалов (метилен СН2, этилен -СН2-СН2-, этилиден Н3С-СН ) и соответствующих галогенов:
Изомерия. Хлористый этилиден и хлористый этилен — изомеры. Галогенопроизводные, у которых атомы галогена находятся при одном углеродном атоме, называют геминальными. Если же они расположены при разных атомах углерода — вицинальными. Физические свойства. Дигалогенопроизводные—тяжелые масла или кристаллические вещества, нерастворимые в воде. Физические свойства некоторых полигалогенопроизводных. Полностью фторированные алканы (перфторуглероды) выдерживают высокие температуры (до 500°С), стойки к действию кислот, щелочей и окислителей, не обладают токсичными свойствами. Температуры кипения перфторуглеродов незначительно отличаются от температуры кипения алканов с тем же числом углеродных атомов, но отличаются от температуры кипения других галогенопроизводных. Химические свойства. В химическом отношении ди- и полигалогенопроизводные очень схожи смоноалкилгалогенами. Например, в реакциях нуклеофильного замещения вицинальные дигалогенопроизводные образуют двухатомные спирты:
В промышленных масштабах вырабатывают галогенопроизводные парафинов. Из метана получают метилхлорид, метиленхлорид, хлороформ, четырёххлористый углерод и др. продукты. Метиленхлорид и четырёххлористый углерод являются хорошими растворителями. Хлороформ используют для синтеза тетрахлорэтилена, хлорфторпроизводных, ценного мономера тетрафторэтилена и прочих. Хлорированием этана производят гексахлорэтан и др. хлорпроизводные. Продукт хлорирования твёрдых парафинов хлорпарафин-40 (около 40% Cl) используется в качестве пластификатора, хлорпарафин-70 (около 70% Cl) - для пропитки бумаги и тканей повышенной огнестойкости.. Хлористый метил H3C-CI, применяют в органическом синтезе в качестве метилирующего средства, растворителя в производстве бутилкаучука. Хлористый изоприл (CH3)2CHCI применя-ся в синтезе кумола(растворитель).Фреоны (хладоны) — насыщенные газообразные или жидкие фторуглероды или полифторуглероды (часто содержат также атомы хлора, реже—брома). Растворимы в органических растворителях, не растворяются в воде. Негорючи, взрывобезопасны, химически инертны, малотоксичны. Применяют в качестве хладагентов в холодильных установках, газов-наполнителей в аэрозольных устройствах, растворителей, компонентов огнетушащих составов (особенно при тушении пожаров различных электрических устройств — АТС, ЭВМ и др.). Надежными противопожарными средствами являются 1,2-дибром-1,2-тетрафторэтан СВгF2-СВгF2 и бромтрифторметан СВг3-F2. Проблема сохранения озонового слоя земли. Токсические свойства галогенопроизводных зависят от характера галогена и от строения углеводородной цепи, с которой он связан.Хлорпроизводные углеводородов представляют большую опасность для окружающей среды. Их применение во многих областях промышленности (нефтеперерабатывающей, авиационной, строительной, электронной, в сельском и коммунальном хозяйстве) делает неизбежным их попадание в атмосферу.В последние годы при широком использовании аэрозольных и противопожарных устройств в атмосферу попадает огромное количество различных фреонов. В земной атмосфере они могут вступать во взаимодействие с озоном, который защищает Землю от интенсивного ультрафиолетового излучения. Пестициды относятся к различным классам органических и неорганических соединений. Большинство представляет собой органические вещества, получаемые синтетическим путём. Среди них важное место принадлежит хлорорганические и фосфорорганические П.Хлорорганические Писцициды обладают общим токсическим действием на организм; они обычно поражают внутренние органы (печень, почки) и нервную систему. Признаки отравления мало специфичны: общая слабость, головокружение, тошнота, раздражение слизистых оболочек глаз и дыхательных путей Галогенопроизводнве непредельных углеводородов. Строение.в молекулах непредельных галогенопроизводных двойная связь может находится в различном положении по отношению к атому галогена X: H2C=CH-X H2C=CH-CH2-X H2C=CH-(CH2)n-CH2-X Номенклатура.название непредельных галогенопроизводных по истематической номенклатуре составляют таким образом, чтобы цифрой было указано не только положение атома галогена в углеродной цепи, но и положение двойной связи.Нумерацию цепи начинают с двойной связи.По рациональной номенклатуре используют названия непредельных радикалов и галогенов:H2C=CH-CI(хлорэтен, хлористый винил)CHCI=CHCI(1,2-дихлорэтен)Изомерия. Непредельные галогенопроизводные могут существовать виде изомеров, которые отличаются строением углеродной цепи и положением в ней двойной связи и атома галогена:H3C-CH=CH-CHBr-CH3(4-бромпентен-2) Физические св-ва Непредельные галогенопроизводные могут быть в газообразном или жидком состоянии. Химические св-ва. Непредельные галогенопроизводные в химическом отношении активнее обычных галогеналкилов.Двойная вязь входящая в состав галогенпроизводных, позволяет им вступать в реакции присоединения, полимиризации и окисления. 1Реакции электрофильного присоединения. А Гидрирование. При гидрировании непредельных галогенпроизводных образуются галогеналколы H2C=CHCI+H2cтрелкаH3C-CH2CI хлорэтан хлорэтан б Галогенирование. Из моногалогенпроизводных полигалогенпризводные: H2C=CHCI+CI2стрелкаH2CCI-CHCI хлорэтан 1,1,2-трихлорэтан в гидрогалогенирование ,2Реакции нуклеофильного замещения(Sn), 3Реакции полимеризации. Непреде-е галогенопроиз-ые образуют высокомолукуляр-е ве-ва.
№49. Карбоновые к-ты аром. ряда. Ароматические карбоновые кислоты — органические вещества, молекулы которых содержат карбоксильную группу, связанную непосредственно с бензольным ядром или атомом углерода боковой цепи'.С6Н5-СООН(бензойная кислота),С6Н5-СН2-СООН(фенилуксусная к-та). В зависимости от числа карбоксильных групп ароматические кар-боновые кислоты делят на одно-, двух- и многоосновные.Одноосновные ароматические кислоты — кристаллические вещества, плохо растворимые в воде (однако лучше в горячей, например бензойная кислота). Температуры плавленияикипения этих кислот выше, чем у кислот алифатического ряда с тем же числом углеродных атомов. Многоосновные ароматические кислоты не растворимые в воде кристаллические вещества.Химические свойства ароматических кислот определяются карбоксильной группой и связанным с ней бензольным ядром. Для ароматических кислот характерны все основные химические превращения, которые были рассмотрены для кислот алифатического ряда. . Бензойная кислота (бензенкарбоно-вая) C6Hs-COOH — белое кристаллическое вещество с т. пл. 122,3°С. Растворяется в горячей воде. Применяют в качестве антисептического и консервирующего средства. Натрийбензоат может использоваться как ингибитор коррозии. Бензойную кислоту применяют в производстве красителей, лекарственных препаратов, душистых веществ, бензоил-хлорида. Фталевая кислота (о-фталевая, или 1,2-бензендикарбоновая) одна из наиболее важных двухосновных ароматических карбоновых! кислот. Получают окислением нафталина кислородом воздуха в приутствии V205. Применяют в произв-ве красителей, эфиров. Гидроксикислоты — производные углеводородов, в молекуле которых содержатся две функциональные группы — карбоксильная и спиртовая. Н-р, НОСН2-СООН(гидроксиуксусная к-та). Молочная кислота, или а-гидроксипропионовая , СН3-СНОН-СООН Молочную кислоту применяют в текстильной и кожевенной промышленности, а также в медицине (обладает бактерицидным действием). Молочная кислота крайне гигроскопична. Винная кислота, или дигидроксиянтарная, НООС-СНОН-СНОН-СООН встречается в виде двух оптических изомеров, рацемата и мезовинной кислоты. D-Винная (виннокаменная) кислота не ядовита и используется в пищевой промышленности в качестве вкусового средства (при изготовлении лимонада), химических разрыхлителей теста. Применяют ее в аналитической химии (для определения ионов калия) и в медицине.Лимонная кислота (З-гидроксипентантриоловая)Хорошо растворяется в воде, спирте; содержится в лимонах (до 6-7%), листьях махорки (до 10%), смородине. Получают в промышленности биохимическим путем (лимоннокислым брожением глюкозы под действием специальных грибков), а также из махорки.Не ядовита. Применяют в пищевой и текстильной промышленности, в медицине (для приговлення кровяной плазмы при переливании крови).Лимонная кислота участвует в важном метаболическом цикле — цикле Кребса (цикл лимонной кислоты, или цикл трикарбоновых кислот) |
№43. Фенолы. Фенолы – гидроксисоединения, в молекулах которых ОН-группы связаны непосредственно с бензольным ядром.
В зависимости от числа ОН-групп различают одноатомные фенолы (например, вышеприведенные фенол и крезолы) и многоатомные. Среди многоатомных фенолов наиболее распространены двухатомные:
Как видно из приведенных примеров, фенолам свойственна структурная изомерия (изомерия положения гидроксигруппы). Физические свойства. Многие фенолы при комнатной температуре кристаллические вещества (мета-крезол - жидкость). Они плохо растворимы в воде, хорошо р-ряются в водных р-рах щелочей. Получение. 1. Гидролиз галоаенбензолов. При нагревании хлообензола с гидроксидом натрия и дальнейшей обработке продукта к-той образуется фенол: С6Н5-Сl + 2NaOH -> С6Н5-ОН + NaCI 2. Кумопьный способ. При каталитическом окислении иэопропилбензола (кумола) кислородом воздуха образуются фенол и ацетон:
Химические свойства. 1. Кислотные св-еа фенола проявляются в р-циях с щелочными металлами и щелочами: C6H5OH + Na => C6H5ONa + 1/2Н2Т, С6Н5ОН + NaOH => C6H5ONa + H2O. Фенол - слабая кислота Он выделяется из растворов фенолятов под действием углекислого газов: C6H5ONa + СО2 + Н2О=> C6H5OH + NaHCO3. Кислотные свойства фенолов ослабляются при введении в кольцо заместителей I рода и усиливаются при введении заместителя II рода. 2 Образование сложных эфиров. В отличие от спиртов, фенолы не образуют сложных эфиров при действии на них карбоновых кислот; для этого используются хлорангидриды кислот: С6Н5ОН + СНз-СО-СL => C6H5-O-CO-СНз + HCI. 3 Реакции электрофильного замещения в феноле протекают значительно легче, чем в ароматических углеводородах а) Галогвнирование. При действии бромной воды образуется осадок 2.4,6-трибромфенола:
Это качественная реакция на фенол. б) Нитрование. Под действием 20%-ной азотной кислоты фенол легко превращается в смесь орто- и лара-нитрофенолов Если нитровать фенол концентрированной азотной кислотой, то образуется 2,4,6-тринитрофенол - сильная кислота (пикриновая): в) Реакция поликонденсации с формальдегидом с образованием фенолформальдвгидных смол:
Применение Фенол применяется при производстве фенолформальдегидных смол, в фармацевтической промышленности и как антисептик (карболовая кислота). Гидрохинон (1,4-диоксибензол) – проявитель в фотографии. Гидрохинон является сильным восстановителем. Как и фенол, он обладает слабым дезинфицирующим действием. Гидрохинон не придает воде запаха, привкус появляется при концентрации несколько граммов в 1 дм3.Гидрохинон при содержании 100 мг/дм3 стерилизует воду. В организме гидрохинон окисляется в п-бензохинон, который превращает гемоглобин в метгемоглобин. Поликонденсация - реакция образования полимера из мономеров с одновременным образованием побочных низкомолекулярных продуктов реакции (воды, спирта и др.). Конденсационные смолы — к ним относят полимеры, получаемые реакцией поликонденсации. Фенолоформальдегидные смолы. Эти высокомолекулярные соединения образуются в результате взаимодействия фенола (CбН5ОН) с формальдегидом (СН2 = О) в присутствии кислот (НС1 и др.) или щелочей (NaOH, NH4OH) в качестве катализаторов. Образование фенолоформальдегидных смол происходит согласно схеме:
Процесс сопровождается выделением воды. Фенолоформальдегидные смолы обладают замечательным свойством: при нагревании они вначале размягчаются, а при дальнейшем нагревании (особенно в присутствии соответствующих катализаторов) затвердевают. Из этих смол готовят ценные пластические массы — фенопласты: смолы смешивают с различными наполнителями (древесной мукой, измельченной бумагой, асбестом, графитом и т. п.), с пластификаторами, красителями, и из полученной массы изготовляют методом горячего прессования различные изделия. В последние годы фенолоформальдегидные смолы нашли новые области применения, например, производство строительных деталей из отходов древесины, изготовление оболочковых форм в литейном деле.
№46 Непред. одноосн. к-ты. Орг. стекло. 1-основные непредельные кислоты содерж. радикал с кратной связью, соедин. с карбоксильной группой. Для 1-основных карбоновых кислот с 1 связью формула: CnH2n-1COOH. Для алкодиеновых и алкеновых кислот CnH2n-3COOH. H2C=CH=COOH – акриловая (пропеновая). CH3-CH=CH2-CH=CH3-COOH – сорбеновая. По систематической номенклатуре названия 1-основных непред. кислот образуются из названия соответ. непред. углеводородов с добавлением «-овая». Изомерия – связана со строением углеводородов в цепи и положением в ней 2-ной связи. CH2=CH-CH2-COOH (бутентриовая), CH3-CH=CH-COOH (бутендваовая). Получение: 1. введение карбоксильной группы содерж. кратную связь CH2=CH-CH2Cl+KCN -> CH2=CH-CH2-CN -> CH2=CH-CH2-COOH. 2. введение кратной связи.
№47. Важн. предст-ли: муравьиная. уксусная…
Карбоновые
кислоты,
класс органических соединений,
содержащих карбоксильную группу
(карбоксил)
В зависимости от природы радикала, связанного с группой — COOH, К. к. могут принадлежать к алифатическому (жирному), алициклическому, ароматическому или гетероциклическому ряду. К. к. широко распространены в природе в свободном состоянии и в виде производных (главным образом сложных эфиров). Так, в летучем масле герани содержится пеларгоновая кислота, в лимонах — лимонная. В состав животных и растительных жиров и масел входят глицериды высших нормальных К. к. жирного ряда, из которых преобладают пальмитиновая кислота, стеариновая кислота и олеиновая кислота. К. к., их производные, а также многочисленные соединения, содержащие наряду с карбоксильной иные функциональные группы (например, аминокислоты, оксикислоты и др.), имеют большое биологическое значение и находят разнообразное практическое применение. Муравьиную и уксусную кислоты, например, применяют при крашении и печатании тканей; уксусную кислоту и уксусный ангидрид — в производстве ацетилцеллюлозы. Аминокислоты входят в состав белков. В медицине используют салициловую кислоту, n-аминосалициловую кислоту (ПАСК) и др. Карбоновые кислоты являются слабыми кислотами, поэтому их соли подвергаются обратимому гидролизу. В зависимости от числа карбоксильных групп в молекуле, карбоновые кислоты подразделяются на одноосновные, двухосновные и т. д. Карбоновые кислоты, как и неорганические кислоты, со спиртами образуют сложные эфиры (см. § 173), в виде которых часто встречаются в природных продуктах. Уксусная кислота СН3—СООН (безводная)—жидкость с острым раздражающим запахом (темп. кип. 118,1 СС); при +16,7 °С застывает в кристаллическую массу, по виду напоминающую лед (100%-ная, или «ледяная» уксусная кислота). Смешивается с водой в любых соотношениях. Широко применяется как приправа I к пище и консервирующее средство. В продажу поступает пищевая уксусная кислота в виде 80% (уксусная эссенция) и 9%' (уксус) водного раствора. Давно известен натуральный или винный уксус — продукт, содержащий уксусную кислоту и получающийся при скисании виноградного вина (в результате микробиологического окисления содержащегося в вине этилового спирта). Уксусная кислота используется также во многих синтезах и как растворитель. Теперь и пищевую и техническую уксусную кислоту Получают преимущественно синтезом из ацетилена — присоединением к нему воды по реакции Кучерова (стр. 486) и окислением образующегося уксусного альдегида. Высшие жирные кислоты. К ним относятся предельные и непредельные карбоновые кислоты с открытой цепью атомов углерода, содержащие 16, 18 и более С-атомов; такого рода кислоты входят в состав природных жиров (см. § 173). Важнейшими являются предельные кислоты пальмитиновая С15Н31СООН, или CH3(CH2)14COOH, и стеариновая C17H35COOH, или CH3(CH2)16COOH, а также непредельная С17Н33СООН, или СН3(СН2)7СН=СH(СН2)7СООН — олеиновая кислота. Высшие предельные кислоты — воскообразные вещества, непредельные — жидкости (напоминающие растительное масло). Натриевые и калиевые соли высших жирных кислот называются мылами (например, C17H35COONa — стеарат натрия, C15H31COOK — пальмитат калия и т. д.). Натриевые мыла — твердые, калиевые — жидкие.
Бензойная
кислота простейшая одноосновная кислота ароматического ряда. Бесцветные кристаллы (пластинки) (темп, плавл. 121,5°С). Антисептик. Применяется для консервирования пищевых продуктов, а также во многих органических синтезах. Щавелевая кислота НООС—СООН — простейшая двухосновная карбоновая кислота. Кристаллическое вещество (безводная — темп, плавл. 189 °С; дигидрат С2Н2О4.2Н2О—темп, плавл. 101,5°С); растворяется в воде; ядовита. В виде кислой калиевой соли содержится во многих растениях. Применяется при крашении тканей. Терефталевая кислота НООС—С6Н4—СООН. Двухосновная карбоновая кислота ароматического ряда. Ее структурная формула:
Из терефталевой кислоты и этиленгликоля получают синтетическое волокно лавсан. Конденсационные смолы — к ним относят полимеры, получаемые реакцией поликонденсации. Полиэфирные смолы. Примером таких смол может служить продукт поликонденсации двухосновной ароматической терефтале-вой кислоты с двухатомным спиртом этиленгликолем:
Полиэтилентерефталат — полимер, в молекулах которого многократно повторяется группировка сложного эфира. В СССР эту смолу выпускают под названием лавсан (за рубежом — терилен, дакрон). Из нее готовят волокно, напоминающее шерсть, но значительно более прочное, дающее несминаемые ткани. Лавсан обладает высокой термо-, влаго- и светостойкостью, устойчив к действию щелочей, кислот и окислителей. Молочная кислота может служить примером соединений со смешанными функциями — проявляет свойства кислоты и спирта (спиртокислота). Она образуется при молочнокислом брожении сахаристых веществ, вызываемом особыми бактериями. Содержится в кислом молоке, рассоле квашеной капусты, силосе.
Салициловая кислота НО—С6Н4—СООН — аналог молочной кислоты в ароматическом ряду. Имеет строение;
Относится к соединениям со смешанными функциями — прояв ляет свойства кислоты и фенола (фенолокислота). Антисептик. Используется (особенно ее соли и эфиры) как лекарственное вещество, а также во многих синтезах.
|
№44. Оксосоединения. альдегиды и кетоны-производные у/в, в молекуле кот-ых содерж-ся 1 и более карбонильных групп (=С=О). Карбонильную группу иногда называют оксогруппу (оксосоединения). Альдегиды-соед-ия, в молекулах кот-ых (=С=О) связана с одним радикалом и атомом водорода, исключение муравьиный альдегид Н-СНО. –СНО альдегидная группа. Кетоны соед-ия, в молекулах кот-ых карбонильная группа связана с 2-мя радикалами. В зав-ти от хар-ра радикала, связанного с карбонильной группой альдегиды и ктоны м.б. предельными и непред-ми. Строение: Осн-ые хим-ие св-ва альдегидов икетонов определяет карбонильная группа двойная связь кот-ой как и в алканах, двойная связь из сигма и пи-связей. Двойная связь карбонильной группы сильно пояризована. Наличие частиц «+» обуславливаются хим-ие св-ва, разнообразие хим-их реакций. По систем-ой номенк-ре название альдегидов сост-ся из названия соотв-щего алканов с добавлением суф-са –аль, а нумерация углеродной среды начин-ся с альдегидной группы углерода (нах-ся в начале цепи). Н-р: Н-СНО-предельный у/в- метаналь. По рацион-ой номенк-ре все альдегиды расм-ся как производные уксусного альдегида(СН3-СН2-СНО-метилуксусный альдегид). Кетоны по систематич-ой номенк-ре называют слова, образ-ые с помощью суф-са –он. Название алкана добав-ся суф. –он, с указанием положения карбонильной гр в углеродной цепи, нумерация начинают с того конца цепи к кот-му ближе расположен карбонил. Н-р: СН3-СО-СН3 – пропанон(ацетон). По рац-ой ном-ре кетоны называют согласно названию радикалов, связ-ых с карбонильной группой добавляя слово кетон. Н-р: НС3-СН2-СН2-СН2-СО-СН3-1)систем назв-гексанон 2; 2) рац. назв метилбутилкетон. Изомерия: для альдегидов обусловлена изменением в стр-ре углеродной цепи С-С-С-С-СНО(пентаналь)-валерьянальдегид; С>С-С-СНО- (изовалерьян альдегид)-3 метилбутаналь. Для кетонов опред-ся не только стр-рой углеродной цепи, но и положением в ней карбонильной группы. НС3-СН2-СН2-СО-СН2-СН3 –изомер гексанон 3; СН3>СН-СО-СН2-СН3-рац-ое-этилизопропил кетон, сист-4 метил пентанон 3.Хим-ие св-ва:1) окисление спиртов: R-CH2OH—R-CH2-CHO; R-CH-R`--R-CO-R`;2) дегидрирование спиртовR-CH2OH—R-CH2-CHO,R-CHO-R`--R-CO-R`; 3) оксосинтез(присоединение воды и оксида углерода(2))R-СH=СН2+СО+Н2О—R-CH2-CH2-CHO, R-СH=СН2+СО+Н2О—R-CH-CH3-CHO ;4) разложение карбонильных кислот(пары карб-ых кислот при пропускании над оксидами нек-ых Ме при темп-ре 450 образ-ся соли, кот-ые подвер-гся пиролизу(тенрмическое разложение) и обр-ся альдегиды и кетоны). Физ-ие св-ва: Хим. св-ва: И альдегиды и кетоны явл-ся хим. активными, эта реакционная способность обусловлена карбонильной группой. Для альдегидов и кетонов хар-ны реакции присоедин. (идет реакция сильного нуклиафильного замещ.), замещ. (с образ-ем оксидов), окисл., полмериз. (при нагревании все альдегиды циклизируются), конденс., восстанов.
№45. Карбоновые к-ты
Карбоновые
кислоты,
класс органических соединений,
содержащих карбоксильную группу
(карбоксил)
В зависимости от природы радикала, связанного с группой — COOH, К. к. могут принадлежать к алифатическому (жирному), алициклическому, ароматическому или гетероциклическому ряду. По числу карбоксильных групп в молекуле различают одно-, двух- и многоосновные (соответственно моно-, ди- и поликарбоновые) кислоты. Кроме того, К. к. могут быть насыщенными (предельными) и ненасыщенными (непредельными), содержащими в молекулах двойные или тройные связи. Большинство К. к. имеет тривиальные названия, многие из которых связаны с их нахождением в природе, например муравьиная, яблочная, валериановая, лимонная кислоты. По Женевской номенклатуре наименования К. к. производят от названий углеводородов с тем же числом атомов углерода, прибавляя окончание «овая» и слово «кислота», например метановая кислота (муравьиная), этановая кислота (уксусная) и т.д. Нередко К. к. рассматривают как производные углеводородов; например, кислоту строения HC º С — COOH называют ацетиленкарбоновой кислотой. Кислотные свойства обусловлены способностью К. к. к диссоциации в водном растворе: RCOOH Û RCOO- + H+. Как правило, К. к. слабее минеральных. Константы диссоциации одноосновных насыщенных кислот жирного ряда при 25°С изменяются от 1,7×10-4 (муравьиная кислота) до 1,3.10-5 (высшие гомологи). Сила К. к. существенно зависит также от электрофильности радикала, связанного с карбоксилом. Введение электроотрицательных заместителей (например, NO2, CN, Cl) в положение, соседнее с карбоксильной группой, резко повышает кислотность, например циануксусная кислота CNCH2COOH примерно в 200 раз сильнее уксусной кислоты CH3COOH. По мере удаления от карбоксила влияние заместителей ослабевает. Дикарбоновые кислоты сильнее монокарбоновых, причём влияние одного карбоксила на другой тем больше, чем они ближе расположены друг к другу. Так, в ряду кислот щавелевая кислота HOOC—COOH сильнее малоновой кислоты HOOCCH2COOH, которая, в свою очередь, сильнее янтарной HOOC (CH2)2COOH, и т.д. Кислотность непредельных кислот выше, чем предельных; влияние двойной связи тем сильнее, чем она ближе расположена к карбоксилу. Так, акриловая кислота CH2=CH—СООН в 4 раза сильнее пропионовой CH3—CH2—COOH. Ароматические кислоты сильнее предельных алифатических (например, константа диссоциации бензойной кислоты 6,5.10-5). К. к. — жидкие (например, низшие жирные кислоты) или твёрдые (например, высшие жирные и ароматические кислоты) вещества (см. табл.). Низшие члены насыщенных К. к. жирного ряда хорошо растворимы в воде, средние члены (C4 — C10), а также ароматические кислоты — ограниченно, высшие жирные кислоты в воде не растворимы; как и ароматические кислоты, они хорошо растворяются в спирте, эфире, бензоле. Наиболее важные химические свойства К. к. — способность превращаться в производные. При взаимодействии с основаниями К. к. дают соли: RCOOH + NaOH ® RCOONa + H2O. При действии на К. к. спиртов в присутствии минеральных кислот легко образуются эфиры сложные: RCOOH + R'OH ® RCOOR' + H2O; при действии галогенангидридов минеральных кислот (например, PCl3, POCl3, SOCl2)— галогенангидриды К. к. RCOX (X — атом галогена). При нагревании кислот с водоотнимающими средствами получаются ангидриды К. к. (RCO)2O. Галогенангидриды и ангидриды К. к. применяют как ацилирующие агенты. Отщепление воды от аммониевых солей К. к. (1) и реакция галогенангидридов с аммиаком (2) приводят к амидам кислот: 1) RCOONH4 ® RCONH2 + H2O 2) RCOCI + 2NH3 ® RCONH2 + NH4CI. Методы получения К. к. весьма многочисленны. Окислением первичных спиртов и альдегидов получают К. к. с тем же числом атомов углерода. Окисление кетонов сопровождается разрывом связи С—С; из циклических кетонов образуются дикарбоновые кислоты, например адипиновая кислота из циклогексанона:
Насыщенные углеводороды могут быть подвергнуты деструктивному окислению с образованием смеси продуктов, в том числе и карбоновых кислот. Этим методом из 1 m парафина обычно получают около 350 кг К. к. Окисление боковой цепи жирно-ароматических углеводородов либо многоядерных ароматических углеводородов приводит к ароматическим К. к.; например, фталевая кислота получается окислением о-ксилола или нафталина:
Ненасыщенные углеводороды окисляются по месту двойной связи:
Важный метод синтеза К. к. — гидролиз их нитрилов, легко получаемых взаимодействием галогенопроизводных углеводородов с цианистым натрием: RCI + NaCN ® RCN ® RCOOH. В настоящее время промышленное значение приобрёл метод синтеза К. к. карбонилированием, т. е. введением группы CO в органические соединения:
Некоторые К. к. получают из природных продуктов. Так, щелочным гидролизом (омылением) жиров получают соли высших жирных кислот (мыла) и глицерин. Лимонную кислоту получают из ботвы хлопчатника и из стеблей махорки (после выделения из них никотина). Многие К. к. получают сбраживанием углеводов в присутствии бактерий определённого вида (маслянокислое, молочнокислое, лимоннокислое и др. виды брожения). К. к. широко распространены в природе в свободном состоянии и в виде производных (главным образом сложных эфиров). Так, в летучем масле герани содержится пеларгоновая кислота, в лимонах — лимонная. В состав животных и растительных жиров и масел входят глицериды высших нормальных К. к. жирного ряда, из которых преобладают пальмитиновая кислота, стеариновая кислота и олеиновая кислота. К. к., их производные, а также многочисленные соединения, содержащие наряду с карбоксильной иные функциональные группы (например, аминокислоты, оксикислоты и др.), имеют большое биологическое значение и находят разнообразное практическое применение. Муравьиную и уксусную кислоты, например, применяют при крашении и печатании тканей; уксусную кислоту и уксусный ангидрид — в производстве ацетилцеллюлозы. Аминокислоты входят в состав белков. В медицине используют салициловую кислоту, n-аминосалициловую кислоту (ПАСК) и др. Понятие ацильного радикала. Комплекс представляет собой димер, состоящий из двух идентичных полипептидных мономеров 1 и 2. Каждый из мономеров включает все ферменты, катализирующие биосинтез жирных кислот; он не является, однако, функциональной единицей (в состав последней входят фрагменты обоих мономеров, при этом половина одного мономера взаимодействует с "комплементарной" половиной другого). Синтазный комплекс одновременно синтезирует две молекулы жирных кислот . Имеются два типа синтазных комплексов, катализирующих биосинтез жирных кислот ; оба находятся в растворимой части клетки. У бактерий, растений и низших форм животных, таких, как эвглена, все индивидуальные ферменты синтазной системы находятся в виде автономных полипептидов; ацильные радикалы связаны с одним из них, получившим название ацилпереносящий белок ( АПБ ). Высшие жирные К. к. широко применяют как сырьё для производства мыла, лаков и красок, поверхностно-активных веществ, как эмульгаторы в производстве каучуков, как пластификаторы в производстве резин и др. Адипиновая кислота — один из исходных продуктов в производстве полиамидного волокна (найлона), терефталевая — в производстве полиэфирного волокна (лавсана, терилена), полимерный нитрил акриловой кислоты (орлон) применяют как синтетическое волокно, близкое по свойствам к натуральной шерсти. Полимеры и сополимеры эфиров метакриловой кислоты используют как органическое стекло. Мыла, соли высших жирных кислот (см. Карбоновые кислоты). В производстве и быту М. (или товарными М.) называют технические смеси водорастворимых солей этих кислот, часто с добавками некоторых др. веществ, обладающие моющим действием. Основу смесей обычно составляют натриевые (реже калиевые и аммониевые) соли насыщенных и ненасыщенных жирных кислот с числом атомов углерода в молекуле от 12 до 18 (стеариновой, пальмитиновой, миристиновой, лауриновой и олеиновой). К М. часто относят также соли нафтеновых и смоляных кислот, а иногда и др. соединения, обладающие в растворах моющей способностью. Не растворяющиеся в воде соли жирных кислот и щёлочноземельных, а также поливалентных металлов называются «металлическими» М. Водорастворимые М. — типичные мицеллообразующие поверхностно-активные вещества. При концентрации выше определённого критического значения в мыльном растворе наряду с отдельными молекулами (ионами) растворённого вещества находятся мицеллы — коллоидные частицы, образованные скоплением молекул в крупные ассоциаты (см. также Полуколлоидные системы). Наличие мицелл и высокая поверхностная (адсорбционная) активность М. обусловливают характерные свойства мыльных растворов: способность отмывать загрязнения, пениться, смачивать гидрофобные поверхности, эмульгировать масла и др.
№50. Углеводы. Углеводы, или сахара – ОС, сод-ие в молекуле одновременно альдегидную или кетонную группу и несколько спиртовых групп. Т.е. углеводы явл-ся многоатомными альдегидоспиртами или кетоноспиртами. У-в – довольно обширная группа прир.орг.в-в, кот. играют большую роль в жизни чел, жив,растений. Они широко распространены в природе, особенно в раст. мире, и сост. осн. массу орг. в-ва. У-в входят в состав нуклеотидов. У-в – один из основных видов продуктов питания. Все у-в делят на две группы: 1.Простые у-в(моносахариды, монозы). В зав. от числа углеродных атомов в молекуле моносахаридов различают тетрозы, пентозы, гексозы. Если моносахариды сод-т альдегидную группу, то они относ-ся к классу альдоз, если кетонную – к классу кетоз. 2.Сложные ( полисахариды).Подвергаются гидролизу с образованием простых у-в. Они дел-ся: 1) низкомол-ые, сахароподобные у-в(олигосахариды), растворимы в воде и сладкие на вкус; 2) высокомол-ые, несахароподопные у-в, несладкие на вкус и нерастворимы в воде. В зав. от состава: 1) гомополисахариды(из остатков одн. моносахарида); 2) гетерополисахариды. Монохы СnH2nOn: гексозы С6Н12О6, пентозыС5Н10О5. Из гексоз наиб. значения имеют глюкоза(альдегидная группа), и фруктоза(кетонная гр.). Они имеют одну и ту же общую формулу, но различное строение, они изомеры. Две формы моноз – цепная и циклическая явл-ся таутомерными и способны самопроизвольно переходить одна в др. Моносахариды – тв. кристаллические в-ва, обычно сладкого вкуса. Гигроскопичны, хорошо растворяются в воде, оптически активны. Моносахариды проявляют св-ва спиртов. Моносахариды проявляют св-ва спиртов, карбонильных соед-ий и циклических полуацеталей. В откр. форме монозы уч-ют в осн. в реакциях, хар-ых для альденидов и кетонов, а в циклической – для многоатомных спиртов. Пентозы (С5Н10О5) входят в состав древесины, древесный сахар
|
||||||||||||||||||||||||||||
|
№52. Амины. Амины- органические соединении, которые можно рассматривать как производные аммиака, в котором атомы водорода (один, два или три) замещены на углеводородные радикалы. Амины делятся на первичные — R-NH2;, вторичные - R-NH-R', третичные - R-N(R')-R". Четвертичные соли [R4N]+xr - органич аналоги аммониевых солей В зависимости от природы радикала амины могут быть алифатическими (предельными и непредельными), алициклическими, ароматическими, гетероциклическими. Общая формула предельных алифатических аминов Сn2n+3N Номенклатура. Названия аминов производят чаще всего по принципам рациональной номенклатуры,от названий соответствующих углеводородов, добавляя к ним приставку амино- или окончание -амин: СН3-NН2 - метиламин, C6H5-NH2 - фениламин, (CH3)2NH - диметиламин, C6H5-NH-CH3 - метилфениламин, (СНз)зМ - триметиламин. Изомерия. 1. Изомерия углеродного скелета (начиная с бутиламина). 2 Изомерия положения аминогруппы (начиная с пропиламина). Физические свойства. Метиламин, диметиламин и триметиламин - газы, средние члены алифатического ряда - жидкости, высшие - твердые вещества. Низшие амины имеют резкий запах. Получение. 1 Нагревание алкилгалогенидов с NНз под давлением дает смесь солей первичных, вторичных и третичных аминов и четвертичной соли СН3Сl + NH3 => CH3NH2 + HCI => (CH3NH3)CI СН3Сl + CH3NH2 =>(CH3)2NH + HCI => [(СН3)2NН2]Сl, CH3CI + (CH3)2NH=> (CH3)3N + HCI -> [(СН3)3NН]Сl. CH3CI + (CH3)3N -+ [(CH3)4N]CI Соли аминов дегидрогалогенируются при действии щелочей: [CH3NH3)CI + NaOH =>CH3NH2 + NaCI + H2O 2. Ароматические амины получают восстановлением нитросоединений: C6H5NO2 + 6{Н] => C6H5NH2 + 2H2O. Для восстановления можно использовать цинк в кислой среде или алюминий в щелочной среде. основными свойствами, причем алифатические амины являются более сильными основаниями, чек аммиак, а ароматические - более слабыми. Этс объясняется тем, что радикалы СНз-, C2H5- увели чивают электронную плотность на атоме азота, а фенильный радикал C6H5- уменьшает ее. Щелочная реакция растворов аминов объясняется образованием гидроксильных ионов при взаимодействии аминов с водой: R-NH2 + H2O s* [R-NH3]* + ОН" Амины в чистом виде или в растворах взаимодействуют с кислотами, образуя соли: CH3NH2 + H2SO4=>[CH3NH3]HSO4, C6H5NH2 + HCI => [C6H5NH3]CI. Соли аминов - твердые вещества, хорошо растворимые в воде и плохо растворимые в неполярных органических растворителях. При действии на соли аминов щелочей происходит их разложение с выделением свободных аминов. [CH3NH3]CI + NaOH -> CH3NH2 + NaCI + H2O 2. Горение. Амины сгорают в кислороде, образуя азот, углекислый газ и воду: 4C2H5NH2 +15O2 = 8СО2 + 2N2 + 14H2O. 3. Реакции с азотистой кислотой, а) Первичные алифатические амины при действии азотистой кислоты превращаются в спирты. R-NH2 + NaNO2 + HCI = R-OH + N2 + NaCI + H2O. б) Первичные ароматические амины при действии HNO2 превращаются в соли диазония: C6H5NH2 + NaN02 + 2HCI => [C6H5-NЕN]*Cl + NaCI + 2H2O в) Вторичные амины (алифатические и ароматические) дают нитрозосоединения: R2NH + NaNO2 + HCI => R2N-N=O + NaCI + H2О. Анилин C6H5NH2 - важнейший из ароматических аминов. Он представляет собой бесцветную маслянистую жидкость, малорастворимую в воде. Для качественного обнаружения анилина используют его реакцию с бромной водой, в результате которой выпадает белый осадок 2,4,6-триброманилина. Полиамидные смолы. Полимеры этого типа являются синтетическими аналогами белков. В их цепях имеются такие же, как в белках, многократно повторяющиеся амидные —СО—NH— группы. В цепях молекул белков они разделены звеном из одного 1 С-атома, в синтетических полиамидах — цепочкой из четырех и более С-атомов. Волокна, полученные из синтетических смол, —■ капрон, энант и анид — по некоторым свойствам значительно превосходят натуральный шелк. В текстильной промышленности из них вырабатывают красивые прочные ткани и трикотаж. В технике используют изготовленные из капрона или анида веревки, канаты, отличающиеся высокой прочностью; эти полимеры применяют также в качестве основы автомобильных шин, для изготовления сетей, различных технических тканей. Капрон является поликонденсатом аминокапроновой кислоты, содержащей цепь из шести атомов углерода:
Энант — поликонденсат аминоэнантовой кислоты, содержащей цепь из семи атомов углерода. Анид (найлон или перлон) получается поликонденсацией двухосновной адипиновой кислоты НООС—(СН2)4—СООН и гексаме-тилендиамина NH2—(СН2)б—NH2. Строение цепи анида можно выразить формулой:
|
№54. Простые и сложные белки. Белки – сложные высокомолекулярные орг.соединения, молекулы кот-х построены из остатков α-аминокислот. По хим. составу белки делятся на простые – при гидролизе распадаются на аминокислоты, и сложные – образуются при гидролизе аминокислоты и вещ-в не белковой природы(углеводы, нукл. кислоты);это соед. белкового вещ-ва с небелковыми. Белки – очень сложные азотосодержащие орг. соединения. Их малек. масса составляет величину от 5тыс до нескольких миллионов. В состав их молекул входят: углерод, кислород, азот, водород, сера. Кроме того белки могут содержать небольшое кол-во фосфора, галогенов, металлов. Остатки α-аминокислот связанных м/у собой пептидными связями-CO-NH-. Соединения, построенные из небольшого числа молекул α-аминокислот, наз-я пептидными.1,2,3.4-структ. белка. Определенны набор аминокислотных остатков, их число и последовательность соединения в полипептидной цепи определяет основу строения белков-их первичную структуру. Сведения о составе, т.е. наборе аминокислотных остатков и последовательность их соединения друг с другом в полипептидной цепи можно получить, изучая продукты гидролиза белков. Гидролиз проводят, действуя на белки ферментами. При этом белки вначале превращаются в пептоны, а затем – в полипептиды, кот-е расщепляются на пептидные цепи и на α-аминокислоты. Полипептидные цепи имеют в большинстве случаев спералевидную форму. Устойчивость отдельных витков цилиндрической спирали обеспечивает множественными водородными связями, кот-е возникают м/у атомом карбонильной группы и водородом аминогруппы. Такое пространственное расположение полипептидной цепи наз-ют вторичной. Спирали сворачиваются в клубки подобно гибкой спирали электрической плитки. В рез-те происходит пространственная упаковка частицы-глобулы. Такое образование относят к третичной структуре. Пространственное объединение нескольких полипептидных макромолекул м/у собой с образованием единой, большой субмолекулы наз-ся четвертичная структура. Значение белков. Белки играют важную биологическую роль, являясь основным веществом, из которого построены клетки животных и растительных организмов. Белки участвуют в важнейших процессах жизнедеятельности организма – строительных, каталитических, энергетических, обменных, защитных. Белки входят в состав ферментов, гормонов, нуклеотидов. Они явл-ся необходимой составной частью пищевого рациона человека и животных. Хим св-ва. Белки обладают амфотерными св-ми. В щелочной среде они проявляют кислотные св-вс, а в кислой – основные. Многие белки растворяются в воде, в кислотах и щелочах. Реакция осаждения. Если белки нагреть до 100 С или подействовать на них кислотами, щелочами или растворами некоторых солей, то в их молекулах происходит необратимые изменении, кот-ые наз-ся денатурацией. Процесс денатурации приводит к разрушению водородных и некоторых ковалентных связей. Биуретовая реакция связана с присутствием в молекуле белка пептидных связей. При обработке белка концентрированным раствором щелочи и насыщенным раствором сульфата меди появляется фиолетовое окраш-ние. Ксантопротеиновая р-ция. действии на белки конц-ой азотной к-ты появляется желтая окраска. Она связана с образованием пр-тов нитрования ароматических ядер, содержащихся в молекуле белка. Сульфгидрильная реакция. При нагревании белков с рас-ом плюмбита натрия выпадает черный осадок сульфида свинца. №56. Гравиметрический анализ Гравиметрический метод основан на определении массы исследуемого вещ-ва, анализируемого образца путем измерения точного взвешивания.+)метод простой в выполнении, высокая точность, хорошая воспроизводимость,-)трудоемкий, продолжительный, самый старый. По способу отделения определяемого компонента выделяют 4 метода.1)метод осаждения. Определяемый компонент рас-ра вступает в хим. реакцию с прибавленным реагентом, образуя малорастворимый продукт – осадок, кот-й отделяют, промывают, высушивают, прокаливают, взвешивают на аналитических весах.2)метод отгонки. Определяемый компонент выделяют из анализируемой пробы в виде газообразного вещ-ва и измеряют массу газа или массу осадкаМетод прост, универсален, используется при анализе многих тов-в в лаб-ях. Он явл-ся обязательным для определения содержания воды.3)метод выделения. Определяемый компонент выделяют из рас-ра.4)инструментальный метод основан на измерении массы анализируемого вещ-ва при его непрерывном нагревании заданным температурным интервалом. Основные операции весового взвешивания. 1)отбор средней пробы вещ-ва и подготовка его к анализу,2)взятие навески,3)растворение,4)осождение с пробой на полноту осождения, 5)фильтрование, 6)промывание осадка с пробой,7)высушивание и прокаливание осадка, 8)взвешивание, 9)вычисление рез-ов анализа. Правило отбора средних проб зависит от особенностей анализ-го материала и от цели определения. Методика отбора средней пробы предусмотрена Гостами и в ТУ.Средняя проба д.б. составлена из большого числа мелких порций взятых в разных местах анализируемого мат-ла. Навеса-кол-во вещ-ва необходимого для выполнения анализа. Чем больше навеска, тем точнее рез-т анализа. Навеска д.б. меленькой.
|
№55. Кл-ция физ. и физ-хим. м-дов анализа. Аналит. химией называют науку о методах кач-ного и колич-ного исследования составов веществ или их смесей. Кач-ный анализ – метод обнаружения катионов и ионов. Колич-ный анализ заключ. в определении колич-ва содержания отдельных составных частей сложного вещества, результаты выраж-ся в %. Вначале определ. кач-ный анализ, затем колич-ный. Классификация методов: 1. В зависимости от массы вещества требуемого для провед. анализа. – макрометоды (m>0,1г), полумикрометоды (0,1÷0,01г), микрометоды (m=10-2 – 10-3), ультрамикрометоды (m=10-3 - 10-6), субмикрометоды (m=10-6 - 10-9) 2. отличаются свойствами опред. компонента – хим., физ., хим-физ., биологич (микрохим., микробиологич). Хим. методы основаны на хим. превращениях, кот-ые протек. в реакциях и приводят к образ. осадков окруж. соедин. и газообразующих веществ. Хим. процессы используемые в целях хим.анализа наз-ют аналит.реакциями. Вещества, вызыв. хим. превращ. наз-ся реактивами (реагентами). Многие хим.методы считаются заслуженной классической хорошей проверкой, но они не всегда удовлетворяют современным требованиям особенно при проверке чистоты вещества. Н-р, в расщепленных материалах содерж. B и Cd и других опасных примесей не должно превышать миллионных долей %. При приготовлении полупроводников если на 10 мил. Атомов Ge приходится не более 1 атома примесей (мышьяк, сурьяна). Недостатки: большинство хим.методов не обладают чувствительностью для кач. или колич. обнаруж. о исследовании материалов. Для контроля в экологических процессах нужны быстрые методы анализа, позволяющие управлять ходом процесса, поэтому появляются экспресс-методы. Физ. Методы основаны на существенном определении зависимости между физ.свойствами веществ и хим.состоянием. В физ.методах имеют важное знач. оптические методы (спектральный, люминесцентный, рефрактометрический методы анализа). При спектральном анализе о присутствии того или иного элемента судят по наличию в спектре линий хар-ых для этого спектра. Спектральный анализ отличается высокой чувствительностью и быстротой выполнения. Впервые использовали в 1800 г.. Фактический метод люминесцентный (флуористентный) используют свечение исследуемого объекта, возникающий под действием УФ лучей, источниками которых служит кварцевая лампа. Некоторые вещества обладают люминесцентными св-вами, другие вещества могут люминесцировать после обработки спец.реактивами. Люминесцентный метод анализа харак-ся очень высокой чувствительностью (до 10-10 – 10-13 г люминесцирующих примесей). Рефрактометрический метод основан на зависимости между показателями преломлений концентрированного растворенного вещества и его молекулярного строения. По показанию преломления можно установить хим.природу вещества, концентрацию, степень чистоты анализируемого материала. К физ.методам анализа относятся также радиометрический, масс-спектрометрический, рентгеностр. Радиометрический основан на изучении радиоактивного излучения того или иного элемента. Рентгеностр. анализ используют для изучения строения веществ (λ<10м). Масс-спектр. анализ позволяет определить отдельную массу молекул и радикалов с помощью масс-спектрометров. Физ.-хим. методы основаны на наблюдении физ. явлений происходящих при хим. реакциях: колорометрический (сравнивает интенсивность окраски исследуемого и стандартного раствора), электровес (измеряет массу Ме выделяющегося на катод в результате электролиза), кондуктометрический(электропроводность в котором протек. реакция), потенциометрический (потенциал электрода каждого элемента находящегося в исслед. растворе), полярографический (учитывает изменения силы тока с ростом напряжения в анализируемом растворе в приборе полярографе), хромотографический (позволяет разделять смеси веществ на отдельные компоненты). Существуют методы фазовых разделений веществ или ионов: осаждения, соосаждения, экстракции, зонной плавки, методы основанные на удалении компонентов в виде газов. Содержание той или иной составной части анализируемого вещества в результате целого ряда операционных измерений выполнение их может быть связано с ошибками (при отборе и обработке средней пробы). Ошибки анализа подразделяются на системные и случайные. Систематические обусловлены причинами связанными с применением методов анализа, поэтому их можно предусмотреть или избежать или внести в вычисления поправку. Важнейшие виды систематический ошибок: 1. все ошибки обусловлены недостаточным избранием метода анализа, которые снижают точность определения, устранить их достаточно сложно 2. ошибки зависят от квалификации работников и от тщательности выполнения отдельных аналитических операций. |
||||||||||||||||||||||||||||
|
№57. Титриметрический анализ. Сущность метода заключ. в измерении V р-ра того или иного реагента, израсходованного на реакцию с анализируемым компонентом. Для этих целей используют титрованные р-ры, концентрация кот-ых известны. Титром наз-ся масса вещ-ва содержащегося в 1 мл (1 см3) титрованного р-ра (в г/мл и г/см3). Определения проводят способом титрования, т.е. постепенного приливания титр-ого р-ра к р-ру анализируемого в-ва, V кот-го точно измерен. Титрование прекр-ся при достижении т. экв-ти, т.е. достижение экв-ви реагента титруемого р-ра и анализируемого компонента. Существует несколько разновидностей титрометрич. анализа: кислотно-основное, осадительное, комплексонометрич., окисл-восстан. титрования. В основе кисл-овнов титр-ия лежит реакция нейтрализации. М-д позволяет концентрацию к-ты или катионов, гидролизирующихся с образ-ем ионов H, титрованием р-ром щелочи или определить концентрацию оснований, в т.ч. анионов, гидролизирующихся с образованием гидроксид-ионов титрованием р-рами кислот. Т. экв-ти устанавливается при помощи кислотно-основн индикаторов изменяющих окраску в опред. интервале PH (н-р, можно определить карбонатную жесткость воды. c1v1=c2v2, где v1,v2-объемы анализируемого и титр-ого р-ов, с2-молярная концентрация экв-ов вещ-ва в титр-ом р-ре, с1-определяемая молярная концентрация экв-ов ионов в анализируемом р-ре). При осадительном титр-нии анализируемый р-р титруется реагентом, образующим с компонентом титров-ого р-ра малораствор. соединение. Т. экв-ти опред-ся с помощтю индикатора, образующ. с реагентом окрашенное соедин-ие. При коплексонометрич. титр-нии определяемый компонент в р-ре титруется р-ом комплексона, чаще всего этилендиаминотетроуксуснуй к-ты (ЕДТА, комплексона 2) или ее двунатриевой соли (комплексона 3 или трилона Б). Комплексоны явл-ся лигандами (лиганды это простые и сложные анионы, где простые – F,Cl,Br,I, сложные – CN, NO3) и образуют со многими катионами комплексы. Индикаторами т. экв-ти обычно служат лиганды, образующие с анализируемым ионом окрашенное комплексное соединение. Метод комплексоно-метрич. титрования используется для опред. общей жесткости воды. О-В титр-ние заключ. в титр-нии р-ра восстановителя титро-ным р-ом окислителя или наоборот. В качестве титр-ных р-ов окислителей нашли применение р-ры перманганата калия (пермангонатометрия), дихромата калия (дихроматометрия) и йода (йодометрия). Из титр-ных р-ов восстановителей следует отметить р-ры гидрозина (гидрозинометрия). При пермангонатометрическом титр-нии в кислой среде Mn (VII) малиновая окраска переходит в Mn (II) бесцветный р-р. В пермангонатмет. титр-нии можно определить содержание нейтритов в р-ре, при дихроматомет. титр-нии индикатором служит дифиниламин, окраш-щий р-р в синий цвет при избытке дихромат-ионов. I-метрич титр-ние используется для анализа р-ов окислителей.
№60. Спектр. м-ды анализа. Методы, основанные на изучении спектров излучения, получили название эмиссионных спектральных методов анализа. В методе эмиссионной спектроскопии проба вещества нагревается до очень высоких температур (2000-15000˚С). Вещество, испаряясь, диссоциирует на атомы или ионы, которые дают излучение. Проходя через спектрограф, излучение разлагается на компоненты в виде спектра цветных линий. Сравнение этого спектра со справочными данными о спектрах элементов позволяет определить вид элемента, а по интенсивности спектральных линий – количество веществ. Метод дает возможности определять микро- и ультрамикроколичества вещества, анализировать несколько элементов, причем за короткое время. Разновидностью эмиссионного анализа явл-ся эмиссионная пламенная фотометрия, в которой исследуемый раствор вводят в бесцветное пламя горелки. По изменению цвета пламени судят о виде вещ-ва, а по интенсивности окрашивания пламени – о концентрации вещ-ва. Анализ выполняют с помощью прибора – пламенного фотометра. Метод в основном используется для анализа щелочных, щелочноземельных Ме и магния. Методы, основанные на изучении спектров поглощения лучей анализируемыми вещ-вами, получили название абсорбционно-спектральных. При прохождении света через раствор свет или его компоненты поглощаются или отражаются. По величине поглощения или отражения лучей судят о природе и концентрации вещ-ва.
|
№58. Кл-ция физ-хим. м-дов.Электро-хим. м-ды. Физико-химические и физические (инструментальные) методы анализа включают оптические, хроматографические, электрохимические и некоторые другие (например, радиометрические, термические, масс-спектрометрические, пикнометрические, ультразвуковые и т. д.).К достоинствам инструментальных методов анализа относятся: низкие предел обнаружения (1—КГ9 мкг) и предельная концентрация (до -КГ1 г/мл) определяемого вещества; селективность (можно определять составные компоненты смеси без их разделения и выделения); быстрота проведения анализов, возможность их автоматизации и компьютеризации; объективность результатов. К недостаткам следует отнести сравнительно большую ошибку определения (порядка -5%; в ряде случаев — до 20%, в то время как при химическом анализе ошибка определения обычно составляет -0,1—0,5%), а также сложность применяемой аппаратуры и ее высокую стоимость. ческих методов анализа, включающую полярографические, гю-тендиометрические, кулоно-, кондуктометрические и другие методы анализа. Известно много электрохимических методов и их модификаций, для которых предложены разные принципы классификаций: 1)измеряемый электрический или электрохимический параметр: Полярография основана на получении и использовании зависимости между силой постоянного тока и напряжением, подаваемым на электроды. В качестве индикаторного электрода используется ртутный капающий электрод. Потенциометрия основана на измерении потенциала индикаторного электрода в зависимости от концентрации определяемого иона. Кондуктометрия основана на изучении зависимости между электропроводностью раствора и концентрацией в нем ионов. 2) процессы, протекающие на них: а) методы, основанные на протекании электродной реакции потенциометрические,вольтамперометрические, кулонометрические и др.); б)методы, не связанные с протеканием электродной реакции (кондуктометрические и др.). 3) по способу выполнения анализа, выделяют: а) прямые методы (прямая потенциометрия, ионометрия, кулонометрия, вольтамперометрия и др.); б) косвенные методы (титриметрические с электрохимическими способами индикации конечной точки титрования — потенцио-, амперо-, кул о неметрическим и др.); в) инверсионные методы (инверсионная вольтамперометрия и др.). Потенциометрический метод анализа относится к электрохимическим. Это метод определения концентрации ионов в раст-ре, основанный на измерении потенциала электрода, погруже кого в исследуемый раствор. Поскольку потенциал отдельного электрода измерить невозможно, то в действительности измеряют разность потенциалов между электродами или э. д. с. гальвг ческого элемента. Следовательно, для выполнения потенциометрического анализа необходимо составить соответствующую гальваническую ячейку (пару). Она состоит из двух электродов, помещенных в анализируемый раствор. Потенциал одного из этих электродов — индикаторного — зависит от концентрации определяемого иона. Потенциал другого — электрода сравнения постоянен не зависит от состава анализируемого раствора. В аналитической практике используют две разновидности потенциометрического анализа. Первая — прямая потенциометрия измерение потенциала индикаторного электрода и нахожде] концентрации определяемого иона по его величине. Вторая — потенциометрическое титрование — измерение потенциала индикаторного электрода (точнее э.д.с. гальванической пары) в процессе титрования анализируемого раствора. Зависимость потенциала индикаторного электрода от состава раствора используют для нахождения объема в конечной точке титрования. При проведении кислотно-основных реакций в качестве индикаторного используют стеклянный электрод Объем титранта в конечной точке титрования (к.т.т.) в потенциометрическом титровании находят разными способами. Наиболее простым является построение кривой титрования в координатах потенциал —объем титранта (интегральная кривая) и графическом нахождении объема титранта в к.т.т. Другой способ состоит в построении кривой титрования в координатах. Для получения этой зависимости (дифференциальной кривой) на оси ординат откладывают отношение Д£/ДК(где ДК — объем очередной порции титранта; — изменение потенциала, вызванное прибавлением этой порвд титранта). Кривая титрования, построенная таким приемом, имеет острый максимум в конечной точке титрования. В настоящее время выпускаются установки для автоматическо-1 го потенциометрического титрования. Кондуктометрический анализ основан на исп-ии зав-ти меж электропроводностью р-ров электролитов и их концентрацией. Об электропроводностью р-ра – проводиков 2 рода – судят на осн-ии измер-я их эл. сопротивл-я в электрохим. ячейке. кот. представляет собой стекл. сосуд с 2-мя впаянными в него электродами, меж кот. нах-ся испытуемый р-р электролита. Через ячейку проп-ют переменный эл. ток. Эл. сопротивление R=ρ*l/S=l/k*S, где ρ-уд. эл. сопр-е, k=1/ ρ – уд. электропров-ю. Эквивалентная электропроводность λ – это эл. проводимость слоя р-ра электролита толщ. 1см, нах-ся меж одинак эл-дами с такой площадью , чтобы объем р-ра сод-ал 1 г-экв раствор. в-ва. λ=1000k/c и молярная эл-сть μ=100k/c. Полярография.параметры – потенциал полуволны Е1/2 и величина диффузного тока iD=Кс
№61. Хромотография. Хроматография явл-ся одним из наиболее широко используемых методов определения и разделения сложных смесей в-в, а также концентрация исходных в-в. Хр-я основана на сформулированном М.С.Цветом в 1903г. Она основана на общ.принципе – распределение компонентов смеси (р-ра) м\у 2-мя фазами, одна из к-рых неподвижна и имеет развитую поверхность, а другая представляет поток, фильтрующийся ч\з неподвижный слой. В основе хр-ии лежит сорбция 9процесс поглощения газов, паров или растворенных в-в жидкими или твердыми поглотителями). Различают след-ие виды сорбции: 1)абсорбция – поглощение всего объемом жидкого поглотителя газов, паров растворенных в-в.2)адсорбция – поглощение поверхностью поглотителя пог8лощающихся в-в происходит на границе раздела соприкасающихся фаз. 3)каппилярная конденсация – образование жидкой фазы в порах и каппилярах твердого поглотителя.
|
№59. Оптичексие м-ды Оптические методы анализа –это методы основанные на измерении оптических свойств веществ и излучений, взаимодействия электромагнитного излучения с атомами или молекулами анализируемого вещества, вызывающего излучение, поглощение или отражение лучей. Классификация оптические методы анализа основываются на измерении оптических свойств вещества(испускание, поглощение, отражение, преломление, рассеивание, поляризация) Классификация по признакам:1по изучаемым объектам(атомный или молекулярный спектральный анализ),2 по характеру взаимодействия электромагнитных излучений с веществом:,1).атомно-абсорбционный-измерение поглощение монохромного излучения, атомами определенного вещества в газовой фазе после атомизации вещества,2)эмиссионный спектральный анализ-измерение интенсивности света излучаемого веществом при энергитическом возбуждении,3)пламенная фотометрия -используется газовое пламя в качестве источника энергитического возбуждения источника,4)молекулярно адсорбционный- измерение светопоглощения молекул или ионами излучаемого вещества,5)люминесцентный -измерение интенсивности излучения люминесценции-(испускание излучение веществом под воздействием различных видов возбуждения),6)спектральный анализ с использованием эффекта комбинационного рассеивания света(раман-эффект)-на измерение интенсивности излучения при влении комбинационного рассеивания света.,7) нефелометрический- на измерении рассеивание света частиц света дисперсной системы (среды), 8)турбидиметрический-на измерении ослабление интенсивности излучения при его прохождении через дисперсную среду,9)рефрактометрический анализ-основан на измерении показателей света преломления веществ,10)интерферометрический-на изучении явления интерференции света,11)полярометричекий- на измерении велечены оптического вращения, угла вращения плоской поляророметрии света оптически активными веществами(орган-и активн-е вещества D-глюкоза или L- глюкоза)3.по области используемого электромагнитного спектра:1)спектроскопия(спектрофотометр) в УВИ области спектра(от 200-400 нм, видимый 400-600 нм),2)инфракрасная спектроскопия (ИК)рассматриваемого интервала длина волны 0,76-100 мкм (1мкм),3)рентгеноскопия.4 по природе энергитических переходов:1)электрон спектор (в УВ и облости) возника-е при изменении электрич-х состояний частиц,2)колебатель-й спектор( охватывает ИК область и комбинацию рассеивания света)- измеряет измерение колебательных состояний частиц, 3)вращательный -охватывает ИКи микроволн-ую область электромагнитного излучения при изменении вращательного состояния молекул.Сущность колориметрического. Методы основаны на уравнивании интенсивностей световых потоков, прошедших через стандартный и исследуемый растворы, с помощью уравнительных диаграмм. Приемниками обоих световых потоков в фотометрии является глаз исследователя, в фотоэлектроколориметрии - фотоэлемент.Закон Бугера-Ламберта-Бера. Поглощение света имеет избирательный характер. Величина оптической плотности раствора имеет различное значение при разных длинах волн. Поэтому для фотометрического анализа важно знать спектр поглощения определяемого вещества, т.е. зависимость поглощения от длины волны. Если отложить на оси абцисс длину волны, на оси ординат -молярный коэффциент поглощеня ( или оптическую плотность раствора), то получим кривую, показывающую распределение поглощающей способности вещества по спектру. Т. е. спектр поглащения данного вещества. Спектр поглощения дает возможность выбрать оптимальную длину волны, которой соответствует максимум светопоглощения данным вещ-вом. Сущность нефелометрического. совокупность методов измерения интенсивности рассеянного в данной среде видимого или ультрафиолетового света с целью определения концентрации, размера и формы диспергированных частиц в дисперсных системах. Сущность рефрактометрического. В основе данного метода лежит способность сред различно преломлять проходящие через них лучи света. Преломление света оценивается величиной показателя преломления n, она зависит от природы вещества, его концентрации, длины волны падающего света, температуры. Преломление света является следствием взаимодействия его с веществом( поляризация атомов и молекул вещества) 3-и типа поляризации:1) электронная поляризация,2) атомная поляризация,3)ориентационная поляризация. Количественный рефрактометрический анализ основан на зависимости показателя преломления от состава системы. Эта зависимость устанавливается экспериментальным путем измерения показателя преломления для ряда эталонных систем известного состава. Строится калибровочный график в координатах показатель преломления-состав системы. С помощью данного графика можно измерить показатель преломления системы неизвестного состава и по графику определить его состав. Приборы–рефрактометры и интерферометры. Сущность поляриметрического. Оптически активные вещества, имеющие асимметричную молекулярную или кристаллическую структуру, поворачивают плоскость поляризации линейно поляризованного света на угол a . Угол вращения плоскости поляризации луча зависит от природы оптически активного вещества, концентрации (для растворов), длины волны света, температуры, природы растворителя. Принципиальные схемы используемых приборов. Фотометр выполнен виде одного блока. На металлическом основании закреплены узлы фотометра, которые закрываются кожухом. Кюветное отделение закрывается съемной крышкой. В фотометр входят фотометрический блок, блок питания, микропроцессорная система. На боковой стенке фотометра расположена розетка для подключения к фотометру термопечатающего устройства типа УТП-2. Рефрактометр -на корпус выведены маховик и заглушка направляющие типа «ласточкин хвост» для установки рефрактометрического блока, в верхней части корпуса размещен окуляр, корпус закрыт крышкой. На которой смонтированы светофильтр и зеркало. рефрактометрический блок состоит из 2-х частей: верхней и нижней. Нижняя неподвижная часть является измерительной, а верхняя-осветительной призмой.(линза склеенная, сетка, светофильтр, шкала, стекло защитное)Нефелометр-прибор для количественного химического анализа мутных сред, основанный на измерении интенсивности света рассеянного взвешенными частицами среды. в которых на окуляр попадает рассеянный свет, направленный под углом 90° к пучку падающего света. Кроме того, в фотоэлектрических колориметрах (например, ФЭК-Н-57, ФЭК-56-2) предусмотрены приспособления для использования их как нефелометров.( набором светофильтров; применение различных светофильтров с узкими спектральными диапазонами пропускаемого света позволяет определять по отдельности концентрации разных компонентов одного и того же раствора, производятся в монохроматическом свете того участка спектра, который наиболее сильно поглощается данным веществом в растворе, нижние границы определяемых концентраций в зависимости от рода вещества составляют от 10-3 до 10-8 моль/л.) поляриметр Он состоит из одной призмы Николя, выполняющей функцию поляризатора света , и призмы – анализатора . Призму-анализатор можно поворачивать относительно плоскости поляризации света. Вращая анализатор относительно плоскости поляризации, находят такое его положение, при котором интенсивность света в окуляре наибольшая. Угол поворота анализатора и является углом вращения плоскости поляризации. Он имеет знак минус, если вращение происходит против часовой стрелки, знак плюс – при вращении по часовой стрелке. |
||||||||||||||||||||||||||||
|
№7. Целостные св-ва атомов. Атомом называют мельчайшую химически неделимую электронейтральную частицу вещества. Молекулы состоят из атомов. В настоящее время известно 110 видов атомов. В центре атома находится положительно заряженое ядро, обладающее значительной (в масштабах атома) массой и маленькими размерами. Различные атомы отличаются друг от друга значением заряда ядра. Атомы, обладающие одинаковым значением заряда ядра могут иметь различные значения относительной атомной массы, но проявляют одинаковые химические свойства. Об атомах одного вида говорят, что они являются атомами одного химического элемента. Химический элемент- это вид атомов с определенным зарядом ядра. Следовательно, заряд ядра является важнейшей характеристикой атома и определяет его химические свойства. Заряд ядра атома каждого химического элемента, выраженный в единицах заряда электрона (в единицах СИ заряд электрона -1,6x10-19 Кл), равен его порядковому номеру в таблице ПСХЭ Менделеева. Так как электронное строение элементов изменяется периодически, то соответственно периодически изменяются и свойства элементов, определяемые их электронным строением, такие как энергия ионизации, размеры атомов, окислительно-восстановительные и другие свойства. В данном параграфе рассмотрена лишь периодичность энергии ионизации, сродства к электрону, электроотрицательности и размеров атомов. Периодичность других свойств будет рассмотрена позднее. Энергия ионизации. Энергия, необходимая для удаления одного моля электронов от одного моля атомов какого либо элемента, называется первой энергией ионизации /j. В результате ионизации атомы превращаются в положительно заряженные ионы. Энергию ионизации выражают либо в килоджоулях на моль (кДж/моль), либо в электронвольтах (эВ). Энергия ионизации характеризует восстановительную способность элемента. Первая энергия ионизации определяется электронным строением элементов и ее изменение имеет периодический характер. Энергия ионизации возрастает по периоду. Наименьшие значения энергии ионизации имеют щелочные элементы, находящиеся в начале периода, наибольшими значениями энергии ионизации характеризуются благородные газы, находящиеся в конце периода. Пики на кривой зависимости энергии ионизации от порядкового номера элемента наблюдаются у элементов с законченными s-подоболочками (Be, Mg) и d-подоболочкой (Zn, Cd, Hg), и p-подоболочкой, в АО которой находится по одному электрону (N, P, As). Минимумы на кривой наблюдаются у элементов, имеющих на внешней подоболочке по одному электрону (щелочные металлы В, Al, Ga, In). В одной и той же группе энергия ионизации несколько уменьшается с увеличением порядкового номера элемента, что обусловлено увеличением размеров атомов и расстояния внешних подоболочек от ядра. Кроме первой энергии ионизации, элементы с многоэлектронными атомами могут характеризоваться второй I2, третьей I3 и более высокой энергией ионизации, которые равны соответственно энергии отрыва молей электронов от молей ионов Э+, Э2+ и т. д. Энергии ионизации возрастают с увеличением их номеров, т.е. h< h< h- Особенно резкое увеличение энергии ионизации наблюдается при отрыве электронов из заполненной подоболочки. Сродство к электрону. Энергия, которая выделяется при присоединении моля электронов к молю атомов, называется сродством к электрону. Э+е=Э-
Электроотрицательность. Для характеристики способности атомов в соединениях притягивать к себе электроны введено понятие электроотрицательности (ЭО). Учитывая, что эта способность атомов зависит от типа соединений, валентного состояния элемента, эта характеристика имеет условный характер. Однако ее использование полезно для объяснения типа химических связей и свойств соединений. Имеется несколько шкал ЭО. Согласно Р. Малликену (США) ЭО равна полусумме энергии ионизации и энергии сродства к электрону. Сложность использования подхода Малликена заключается в том, что нет надежных методов количественного определения энергии сродства к электрону. Поэтому Л. Полинг (США) предложил термохимический метод расчета ЭО на основе определения разности энергии диссоциации соединения А — В и образующих его молекул А — А и В — В. Он ввел относительную шкалу ЭО, приняв ЭО фтора, равной четырем. ЭО элементов возрастает по периоду и несколько убывает в группах с возрастанием номера периода у элементов I, II, V, VI и VII главных подгрупп, III, IV и V — побочных подгрупп, имеет сложную зависимость у элементов III главной подгруппы (минимум ЭО у А1), возрастает с увеличением номера периода у элементов VII — VIII побочных подгрупп. Наименьшие значения ЭО имеют s-элементы I подгруппы, наибольшие значения — f-элементы VII и VI групп. Атомные радиусы. Атомы не имеют строго определенных границ. Поэтому абсолютное значение радиуса атома определить невозможно. Можно условно принять за радиус атома теоретически рассчитанное значение расстояния от ядра до наиболее удаленного от него максимума электронной плотности (орбитальный радиус атома) или половину расстояния между центрами двух смежных атомов в кристаллах (эффективные радиусы атомов). Наблюдается периодичность изменения атомных радиусов, особенно у s- и p-элементов. У d- и f-элементов кривая изменения радиусов атомов по периоду имеет более плавный характер. В одной и той же группе с увеличением номера периода атомные радиусы, как правило, возрастают в связи с увеличением числа электронных оболочек. Однако увеличение заряда ядра при этом оказывает противоположный эффект, поэтому увеличение атомных радиусов с увеличением номера периода относительно невелико, а в некоторых случаях, например у р-элементов III группы значение орбитального радиуса у А1 больше, чем у Са.
№42. Спирты. Спиртами, или алкоголятами, называют производные углеводородов, в молекулах которых один или несколько атомов водорода замещены на соответствующее число гидроксильных групп –ОН. Спирты бывают предельными и непредельными. Это зависит от характера радикала, связанного с гидроксильной группой: Н3С-СН2-СН2-ОН Н2С=СН-СН2-ОН пропиловый спирт аллиловый спирт (предельный) (непредельный) от числа гидроксильных групп, входящих в состав молекулы спирта, зависит его атомность. Спирты бывают одноатомные (I), двухатомные (II), трехатомные (III) и многоатомные (VI): СН3-СН2-ОН НОСН2-СН2ОН этиловый спирт этиленгликоль I II НОСН2-СН(ОН)-СН2ОН НОСН2-(СНОН)4-СН2ОН глицерин гексит III IV ОДНОАТОМНЫЕ ПРЕДЕЛЬНЫЕ СПИРТЫ (АЛКАНОЛЫ) Строение. Изучение спиртов лучше начать с рассмотрения предельных одноатомных спиртов, имеющих общую формулу СnН2n+1ОН или R-OH. В зависимости от характера углеродного атома (первичный, вторичный или третичный), с которым связана гидроксильная группа, спирты могут быть первичными (I), вторичными (II) или третичными (III):
СН3-СН2-СН2-ОН пропиловый спирт изопропиловый спирт I II
трет-бугшовый спирт III Одновалентная спиртовая группа –СН2ОН называется первичной,двухвалентная >СН-ОН — вторичной и трехвалентная ->С-ОН -третичной. Строение молекулы спирта можно представить структурной (I) или электронной (II) формулами. Для метилового спирта (метанола) они имеют вид: Смотри стр.144 Артеменко
Атом кислорода в гидроксильной группе, обладая значительной электроотрицательностью, оттягивает электронную плотность связи О-Н в свою сторону. Поэтому такая связь становится частично поляризованной: на атоме кислорода появляется частичный отрицательный, а на атоме водорода — частичный положительный заряды: Смотри стр.145 Артеменко
Эта поляризация может меняться в зависимости от свойств радикалов, связанных с гидроксилом. Так, обычные алкильные радикалы, обладая донорными свойствами, понижают поляризацию связи О-Н. В результате подвижность атома водорода гидроксильнои группы в молекуле спирта несколько меньше, чем в воде. Однако в ряду одноатомных предельных спиртов более «кислым» является метиловый спирт. Еще более слабо выражена кислотность у вторичных и особенно у третичных спиртов (за счет влияния трех радикалов): Смотри стр.145 Артеменко
Т.о, по снижению поляризации связи О-Н спирты можно расположить в такой ряд: СН3ОН ) первичные ) вторичные ) третичные. Однако если ввести в спиртовый радикал атомы или группы, обладающие электроноакцепторными свойствами, то кислотные свойства спиртов усиливаются. Номенклатура и изомерия Номенклатура. Названия простых спиртов обычно производят от названий радикалов, с которыми связана гидроксильная группа: Смотри стр.145 Артеменко
По систематической номенклатуре одноатомные предельные спирты называют по названию соответствующего алкана с добавлением суф фикса -ол. Главную цепь нумеруют с того конца, к которому ближе расположена гидроксильная группа: Смотри стр.146 Артеменко
Иногда спирты рассматривают как производные простейшего спирта — метилового СН3ОН, который иногда называют карбинолом: Смотри стр.146 Артеменко
Изомерия. Изомерия спиртов зависит от строения углеродной цепи и положения в ней гидроксильной группы. Если у первых двух членов гомологического ряда одноатомных предельных спиртов — метилового и этилового — изомеры отсутствуют, то для следующего гомолога с формулой С3Н7ОН существуют два изомера: Смотри стр.146 Артеменко
Спирт с молекулярной формулой С4Н9ОН имеет четыре изомера, которые отличаются друг от друга не только различным расположением гидроксильной группы в углеродной цепи, но и строением: Смотри стр.146 Артеменко
Получение. Предельные одноатомные спирты получают следующими способами. 1. Гидратацией (присоединением воды к алкенам). Реакцию проводят в присутствии катализаторов. Если использовать в качестве катализатора серную кислоту (сернокислотная гидратация), реакция идет в две стадии Смотри стр.147 Артеменко
При получении этилового спирта этот метод вытеснил сернокислотную гидратацию. Гидратация алкенов имеет важное промышленное значение. Она позволяет получать спирты из доступного и дешевого сырья — газов крекинга. Так, из 1 т этилена можно получить 1,4 т этилового спирта. Впервые в нашей стране этот спирт начали получать гидратацией этилена с 1952г. 2.Гидролизом моногалогенопроизводных. Реакцию проводят, нагревая галогеналкилы с водой или водным раствором щелочей Смотри стр.147 Артеменко
3.Восстановлением альдегидов, кетонов и сложных эфиров. В присутствии катализаторов (Ni, Pt, Pd, Co) альдегиды и сложные эфиры при восстановлении переходят в первичные спирты, в кетоны — во вторичные: Смотри стр.147 Артеменко
4. Взаимодействием магнийорганических соединений с карбонил-содержащими веществами. 5. Гидролизом (омылением) сложных эфиров. 6. Спиртовым брожением (расщеплением) моносахаридов под влиянием ферментов. Так можно получать этиловый спирт. 7. Метиловый спирт (метанол) получают из синтез-газа. Процесс идет при 220-3 00°С и сравнительно невысоком давлении с использованием катализатора из оксидов меди и цинка Смотри стр.148 Артеменко
Физические свойства. Предельные одноатомные спирты от С1, до С12 — жидкости. Высшие спирты от С13 до С20 — мазеобразные, а от С21 и выше — твердые вещества. Спирты C1—C3 смешиваются с водой во всех соотношениях и имеют характерный (алкогольный) запах. Спирты С4 и С5 обладают сладковатым удушливым, а с С6 по С11 — неприятным запахом. Твердые спирты лишены запаха и практически нерастворимы в воде. Все спирты легче воды (их плотность ниже единицы). Температура кипения спиртов нормального строения повышается с увеличением молекулярной массы. Спирты нормального строения кипят при более высокой температуре, чем спирты с изостроением. При одном и том же числе углеродных атомов в молекулах температуры кипения спиртов располагаются в ряд: температура кипения первичных спиртов > вторичных спиртов > третичных спиртов. Температуры кипения спиртов выше, чем галогеналкилов и углеводородов с тем же числом углеродных атомов. Это свойство, характерное для всех гидроксилсодержащих веществ, объясняется тем, что молекулы спирта, как и воды, являются ассоциированными жидкостями за счет возникновения между молекулами спирта водородных связей Смотри стр.149 Артеменко
При растворении спиртов в воде возникает водородная связь между молекулами спирта и воды Смотри стр.149 Артеменко
При этом происходит выделение теплоты и уменьшение общего объема (при смешивании 52 объемов спирта с 48 объемами воды получают не 100 объемов водно-спиртового раствора, а 96,3). В ИК-спектрах валентные колебания неассоциированных (свободных) гидроксильных групп лежат в области 3670-3580 см-1. Участие этих групп в образовании межмолекулярных водородных связей проявляется интенсивными полосами в области меньших частот (3550-3450 см-1). Внутримолекулярная водородная связь проявляется в виде широкой полосы в области 3200-2500 см-1. В УФ-области спектра спирты поглощают при 150-200 нм. Спирты имеют нелинейное строение с углом С-О-Н, равным 110°25'. ДВУХАТОМНЫЕ СПИРТЫ (АЛКАНДИОЛЫ, ИЛИ ГЛИКОЛИ) Двухатомные спирты (гликоли) содержат в молекуле две гидро-ксилъные группы при разных углеродных атомах. Общая формула таких спиртов СnН2n(ОН)2. Первым представителем двухатомных спиртов является этилен-гликоль (этандиол) НОСН2-СН2ОН. Номенклатура. По систематической номенклатуре названия двухатомных спиртов производят от названия предельных углеводородов с добавлением суффикса -диол и указанием положения гидроксильных групп в углеродной цепи Смотри стр.161 Артеменко
По рациональной номенклатуре названия двухатомных спиртов можно составить из названий соответствующих алкеновых углеводородов с добавлением слова «гликоль»: Смотри стр.161 Артеменко
Изомерия двухатомных спиртов зависит от строения углеродной цепи и расположения в ней гидроксильных групп. В зависимости от их положения различают а-гликоли (гидроксильные группы находятся у рядом стоящих углеродных атомов), (3-гликоли (гидроксильные группы расположены в 1,3-положении), у-гликоли (гидроксильные группы в 1,4-положении) и т.д.: Смотри стр.161 Артеменко
Получение. 1. Гидролиз дигалогенопроизводных алканов (с атомами галогена при разных углеродных атомах) Смотри стр.161 Артеменко
2. Окисление этиленовых углеводородов (реакция Вагнера) Смотри стр.161 Артеменко
3. Гидратация а-оксидов Смотри стр.161-162 Артеменко
Физические свойства. Низшие гликоли — сиропообразные, сладкие (от греч. «гликос» — сладкий) на вкус, растворимые в воде вещества. Гликоли кипят при более высокой температуре и имеют большую плотность, чем соответствующие им (с тем же числом углеродных атомов) одноатомные спирты. Это объясняется присутствием в молекуле второй гидроксильной группы, что ведет к образованию дополнительных водородных связей. Химические свойства. В химических реакциях двухатомные спирты могут реагировать одной или двумя гидроксильными группами. 1 . Образование гликолятов. В отличие от одноатомных спиртов двухатомные спирты легко вступают во взаимодействие не только со щелочными металлами, но и с оксидами и гидроксидами металлов. Образующиеся вещества называют гликолятами Смотри стр.162 Артеменко
2. Реакции дегидратации. Эти реакции, как известно, могут быть внутримолекулярными (1) и межмолекулярными (2): Смотри стр.162 Артеменко
В случае межмолекулярной дегидратации (2) процесс может идти и дальше — с образованием полимера Смотри стр.163 Артеменко
Диоксан — прекрасный растворитель для многих органических веществ. Смешивается с водой. Требует осторожного обращения — ядовит. Особенно токсично производное диоксана — диоксин
Чрезвычайно опасно его полихлорпроизводное — 2,3,7,8-тетра-хлорбензопарадиоксин. 3. Образование простых и сложных эфиров. Взаимодействуя со спиртами или кислотами (органическими или неорганическими), гли-коли образуют простые (1) или сложные (2) эфиры: Смотри стр.163 Артеменко
4. Замена гидроксильных групп на галоген. При взаимодействии двухатомных спиртов с галогеноводородами происходит замена гидро-ксильной группы на галоген Смотри стр.163 Артеменко
5. Окисление. При окислении этиленгликоль образует ряд продуктов: Смотри стр.163 Артеменко
Щавелевая кислота часто является предпоследним продуктом окисления большинства органических соединений. 6. Пинаколиновая перегруппировка. Двухатомный третичный спирт — пинакон (2,3-диметилбутандиол-2,3) в присутствии серной кислоты способен перегруппировываться в кетон — пинаколин Смотри стр.164 Артеменко
№7. Целостн. св-ва атомов-m, размер, уст-сть, их изменение по периодам и группам Атомом называют мельчайшую химически неделимую электронейтральную частицу вещества. Молекулы состоят из атомов. В настоящее время известно 110 видов атомов. В центре атома находится положительно заряженое ядро, обладающее значительной (в масштабах атома) массой и маленькими размерами. Различные атомы отличаются друг от друга значением заряда ядра. Атомы, обладающие одинаковым значением заряда ядра могут иметь различные значения относительной атомной массы, но проявляют одинаковые химические свойства. Об атомах одного вида говорят, что они являются атомами одного химического элемента. Химический элемент- это вид атомов с определенным зарядом ядра. Следовательно, заряд ядра является важнейшей характеристикой атома и определяет его химические свойства. Заряд ядра атома каждого химического элемента, выраженный в единицах заряда электрона (в единицах СИ заряд электрона -1,6x10-19 Кл), равен его порядковому номеру в таблице ПСХЭ Менделеева. Так как электронное строение элементов изменяется периодически, то соответственно периодически изменяются и свойства элементов, определяемые их электронным строением, такие как энергия ионизации, размеры атомов, окислительно-восстановительные и другие свойства. В данном параграфе рассмотрена лишь периодичность энергии ионизации, сродства к электрону, электроотрицательности и размеров атомов. Периодичность других свойств будет рассмотрена позднее. Энергия ионизации. Энергия, необходимая для удаления одного моля электронов от одного моля атомов какого либо элемента, называется первой энергией ионизации /j. В результате ионизации атомы превращаются в положительно заряженные ионы. Энергию ионизации выражают либо в килоджоулях на моль (кДж/моль), либо в электронвольтах (эВ). Энергия ионизации характеризует восстановительную способность элемента. Первая энергия ионизации определяется электронным строением элементов и ее изменение имеет периодический характер. Энергия ионизации возрастает по периоду. Наименьшие значения энергии ионизации имеют щелочные элементы, находящиеся в начале периода, наибольшими значениями энергии ионизации характеризуются благородные газы, находящиеся в конце периода. Пики на кривой зависимости энергии ионизации от порядкового номера элемента наблюдаются у элементов с законченными s-подоболочками (Be, Mg) и d-подоболочкой (Zn, Cd, Hg), и p-подоболочкой, в АО которой находится по одному электрону (N, P, As). Минимумы на кривой наблюдаются у элементов, имеющих на внешней подоболочке по одному электрону (щелочные металлы В, Al, Ga, In). В одной и той же группе энергия ионизации несколько уменьшается с увеличением порядкового номера элемента, что обусловлено увеличением размеров атомов и расстояния внешних подоболочек от ядра. Кроме первой энергии ионизации, элементы с многоэлектронными атомами могут характеризоваться второй I2, третьей I3 и более высокой энергией ионизации, которые равны соответственно энергии отрыва молей электронов от молей ионов Э+, Э2+ и т. д. Энергии ионизации возрастают с увеличением их номеров, т.е. h< h< h- Особенно резкое увеличение энергии ионизации наблюдается при отрыве электронов из заполненной подоболочки. Сродство к электрону. Энергия, которая выделяется при присоединении моля электронов к молю атомов, называется сродством к электрону. Э+е=Э-
Электроотрицательность. Для характеристики способности атомов в соединениях притягивать к себе электроны введено понятие электроотрицательности (ЭО). Учитывая, что эта способность атомов зависит от типа соединений, валентного состояния элемента, эта характеристика имеет условный характер. Однако ее использование полезно для объяснения типа химических связей и свойств соединений. Имеется несколько шкал ЭО. Согласно Р. Малликену (США) ЭО равна полусумме энергии ионизации и энергии сродства к электрону. Сложность использования подхода Малликена заключается в том, что нет надежных методов количественного определения энергии сродства к электрону. Поэтому Л. Полинг (США) предложил термохимический метод расчета ЭО на основе определения разности энергии диссоциации соединения А — В и образующих его молекул А — А и В — В. Он ввел относительную шкалу ЭО, приняв ЭО фтора, равной четырем. ЭО элементов возрастает по периоду и несколько убывает в группах с возрастанием номера периода у элементов I, II, V, VI и VII главных подгрупп, III, IV и V — побочных подгрупп, имеет сложную зависимость у элементов III главной подгруппы (минимум ЭО у А1), возрастает с увеличением номера периода у элементов VII — VIII побочных подгрупп. Наименьшие значения ЭО имеют s-элементы I подгруппы, наибольшие значения — f-элементы VII и VI групп. Атомные радиусы. Атомы не имеют строго определенных границ. Поэтому абсолютное значение радиуса атома определить невозможно. Можно условно принять за радиус атома теоретически рассчитанное значение расстояния от ядра до наиболее удаленного от него максимума электронной плотности (орбитальный радиус атома) или половину расстояния между центрами двух смежных атомов в кристаллах (эффективные радиусы атомов). Наблюдается периодичность изменения атомных радиусов, особенно у s- и p-элементов. У d- и f-элементов кривая изменения радиусов атомов по периоду имеет более плавный характер. В одной и той же группе с увеличением номера периода атомные радиусы, как правило, возрастают в связи с увеличением числа электронных оболочек. Однако увеличение заряда ядра при этом оказывает противоположный эффект, поэтому увеличение атомных радиусов с увеличением номера периода относительно невелико, а в некоторых случаях, например у р-элементов III группы значение орбитального радиуса у А1 больше, чем у Са. |
№13. Комплексные соед-ия. Основополагающие представления о комплексных соединениях ввел в науку швейцарский ученый Альфред Вернер (1892). В развитии химии комплесных соединений большую роль сыграли труды Л.А. Чугаева и его многочисленных учеников — И.И. Черняева, А.А. Гринберга, В.В. Лебединского и др. По Вернеру, в большинстве комплексных соединений различают внутреннюю и внешнюю сферы. Например, в комплексных соединениях K2[BeF4], [Zn(NH3)4]Cl2 внутреннюю сферу составляют группировки атомов (комплексы) [BeF4]2- и [Zn(NH3)4]2+, а внешнюю сферу — соответственно ионы К+ и С1-. Центральный атом (ион) внутренней сферы является комплексообразовате-лем, а координированные вокруг него молекулы (ионы) — лигандами. В формулах комплексных соединений внутреннюю сферу (комплекс) часто заключают в квадратные скобки. Общепризнанного определения понятия "комплексное соединение нет. Это обусловлено разнообразием комплексных соединений и их характерных свойств. В лабораторной практике химики чаще всего используют соединения в твердом и растворенном состоянии. Для этих условий можно дать следующее определение комплексных соединений: комплексными называют соединения, в узлах кристаллов которых находятся комплексы, способные к самостоятельному существованию в растворе. Следует отметить, что такое определение, конечно, далеко не исчерпывает существа проблемы и применимо лишь в известны* пределах. Классификация комплексов. По характеру электрического заряда различают катионные, анионные и нейтральные комплексы. В приближении ионной модели заряд комплекса представляет собой алгебраическую сумму зарядов образующих его частиц. Катионный комплекс можно рассматривать как образованный в результате координации вокруг положительного иона нейтральных молекул (Н2О, NH3 и др.). Молекулы Н2О и NH3 в номенклатуре комплексных соединений называют -аква- и -аммин соответственно:
Соединения, содержащие амминокомплексы, называются аммиакатами, содержащие аквакомплексы — гидратами. В роли комплексообразователя в анионном комплексе выступает атом с положительной степенью окисления (положительный ион), а лигандами являются атомы с отрицательной степенью окисления (анионы). Отрицательный заряд комплекса отражают добавлением к латинскому названию комплексообразователя суффикса -am, например:
Нейтральные комплексы образуются при координации вокруг атома молекул, а также при одновременной координации вокруг положительного иона-комплексообразователя отрицательных ионов и молекул. Например:
Электронейтральные комплексы, следовательно, являются комплексными соединениями без внешней сферы. Роль комплексообразователя может играть любой элемент периодической системы. В соответствии со своей химической природой неметаллические элементы обычно дают анионные комплексы, в которых роль лигандов играют атомы наиболее электроотрицательных элементов, например K[PF6], K3[PO4], K3[PS4]. Что же касается типичных металлических элементов (щелочных и щелочно-земельных металлов), то способность к образованию комплексных соединений с неорганическими лигандами у них выражена слабо. Имеющиеся немногочисленные комплексные ионы являются катионными, например [Sr(OH2)6]Cl2, [Ca(NH3)8]Cl2. Амфотерные элементы, которые занимают промежуточ-ное положение между типичными металлическими и неметаллическими элементами, образуют как катионные, так и анионные комплексы, например [А1(ОН2)6]С13 и К[А1(ОН)4]. Классификация лигандов. Лиганды могут занимать в координационной сфере одно или несколько мест, т.е. соединяться с центральным атомом посредством одного или нескольких атомов. По этому признаку I различают монодентатные, дидентатные, тридентатные, ..., полиден-татные лиганды (от лат. dentalus — имеющий зубы). Примерами моно-дентантных лигандов являются ионы С1-, F-, ОН-, молекулы NH3, Н20, СО и др. К бидентантным относится, например, молекула этилендиа-мина H2N—СН2—СН2—NH2 (сокращенное обозначение en). Комплексы с полидентантными лигандами называются хелатными (или клешневидными, от греч. chelate — клешня). Ниже приведены нехелатный и хелатный комплексы меди (II):
В качестве бидентантных лигандов часто выступают также ионы СО2-3, SO2-4 и им подобные:
Многие лиганды могут выступать также в качестве мостиковых атомов (групп атомов) в многоядерных (полимерных) комплексах. Например, центральные атомы октаэдрических комплексов (рис. 61) могут быть соединены посредством одного, двух или трех мостиковых атомов (групп атомов).
Подобная картина наблюдается, например, в соединениях кобальта (III). Структурные формулы некоторых из них приведены ниже:
Комплексные соединения широкого распространены в природе, играют важную роль в биологических процессах. Достаточно упомянуть халатные комплексы — гемоглобин крови (комплексообразователь Fe2+) и хлорофилл зеленых растений (комплексообразователь Mg2+). Комплексные соединения находят самое разнообразное практическое применение. Так, образование хелатных комплексов используется при умягчении жесткой воды и растворении камней в почках; важнейшую роль играют комплексные соединения в аналитической практике, производстве металлов и т.д.
№48. Жиры и масла. жиры – смеси сложных эфиров, образованных трехатомным спиртом – глицерином и высшими к-тами. В образовании сложных эфиров, входящих в состав жира, могут принимать участие разные высшие карбоновые к-ты, но из спиртов только один – глицерин. Поэтому эти эфиры наз-ют глицеридами (ацилглицеринами). Общ.ф-ла жиров имеет вид Н 2С –О – СО – R\
НС – О – СО – R\\
Н2 C – О – СО – R\\\ ,ё Где R\ , R\\ ,R\\\ - радикалы высших карбоновых к-т, входящих в состав жира. В состав жиров в основном входят триглицериды – производные глицерина, остаток к-рого связан с тремя остатками высших к-т (ацилами), но в жирах могут присутствовать и ди- и даже моноглицериды
Формулы
Называя глицериды, вначале нумеруют углеродные атомы в остатке глицерина, а затем указывают положение и название заместителей с добавлением слова «глицерин»:
Формула
Основные источники жиров и масел – животные и растительные организмы. Из них жиры и масла выделяют вытапливанием (нагреванием животных тканей), отжимом (прессованием нагретых семян) и экстрагированием (растворением жиров в хим.растворителях с последующим извлечением). Жиры имеют маслянистую консистенцию (твердую или жидкую). Жиры не растворяются в воде, но растворимы во многих органических растворителях. Химич.св-ва.1)гидролиз (омыление жиров). Под действием воды при высокой темп-ре и давлении или в присутствии минеральных к-т, а также щелочей или ферментов (липазы) происходит процесс расщепления жиров, в рез-те к-рого образуются глицерин и высшие карбоновые к-ты. Если омыление жира проводят водным р-ром щелочи, то вместо свободных к-т получают их соли – мыла.
Реакция
2)окисление жиров. Растительные масла, содержащие глицериды непредельных к-т (с двумя или более двойными связями), легко окисляются кислородом воздуха, образую тонкую прозрачную пленку – линоксин. Такие масла стали называть высыхающими и широко использовать в лакокрасочной пром-ти. 3)гидрогенизация жиров (гидрирование). Жидкие масла можно перевести в твердые – жиры. Процесс насыщения водородом двойных связей проводят в присутствии катализатора – мелкораздробленного никеля.
Реакция
Впервые этот метод был разработан Фокиным. Полученные твердые жиры (саломас, или комбижир) используют для произв-ва пищевого продукта – маргарина и для техничесих целей. Маргарин получают в основном отверждением растительных масел – их гидрогенизацией. Если в процессе гидрогенизации в мол-ле жира сохранилось достаточное кол-во непредельных триглицеридов, то образуется «мягкий» маргарин. Он содержит гораздо больше непредельных триглицеридов, чем сливочное масло. Маргарины различаются содержанием жира. Для приготовления маргарина с содерж-ем жира от 40 до 82% в жировую основу (гидрированный жир – саломас) добавляют водно-молочную смесь. Эти компоненты смешивают и эмульгируют, а затем кристаллизуют. В рез-те получают затвердевшую, однородную и пластичную массу, к-рая поступает потом на фасовку. Маргарин можно получать и другим способом. К высокоплавкому твердому жиру постепенно добавляют жидкое масло в такой пропорции, чтобы полученная смесь плавилась при той же темп-ре, что и натуральное сливочное масло.
№53. Аминокислоты. Атомы углерода нумеруют, начиная с атома углерода карбоксильной группы. Непосредственно связанный с ним атом углерода обозначают как а, следующий как (3 и т. д. При составлении названия греческой буквой указывают атом углерода, с которым связана аминогруппа. Например,
Строение аминокислот правильнее представлять в виде внутренних солей типа:
Получение 1. Замена галогенов на аминогруппу в галогенозамещенных кислотах (метод Фишера).
2. Присоединение аммиака к Р-непредельным кислотам (против правила Мар-ковникова).
3. Восстановление нитрозамещенных кислот:
Химические свойства 1. Благодаря наличию двух функциональных групп для аминокислот характер ны как свойства, присущие аминам, так и свойства, присущие карбоновым кислотам. Поэтому они обладают одновременно и кислотными и основными свойствами. Как кислоты, аминокислоты образуют со спиртами сложные эфиры, а с металлами и основаниями-соли: NH2CH2COOC2H [NH2CH2COO ] 2Cu] Этиловый эфир медная соль аминоуксусной кислоты аминоуксусной кислоты Для аминокислот особенно характерно образование медных солей, обладающих специфической синей окраской. Эти вещества являются внутренними комплексными солями; в них атом меди связан не только с атомами кислорода, но и с атомами азота аминогрупп:
CH2―HN2―NH2―CH2
CO―O―Cu―O―CO
Кислотные свойства в моноаминокислотах выражены весьма слобо-аминокислоты почти не изменяют окраски лакмуса. Таким образом, кислотные свойства карбоксила в них значительно ослаблены. Как амины, аминокислоты образуют соли с кислотами, например: HCℓ∙NH2CH2COOH Но эти соли весьма непрочны и легко разлагаются. Таким образом, основные свойства аминогруппы в аминокислотах также значительно ослаблены. Аминокислоты можно алкилировать по аминогруппе. Алкилированием глицина получается метиламиноуксосная кислота-саркозин + NH3—CH2—C—O– + CH2I CH3NH2—CH2—C—O– + HI ║ ║ O O саркозин
которая в связанном виде содержится в некоторых белках. 2. Особые свойства аминокислот. Образование пептидной связи.
Аминокислоты — амфотерные соединения: они образуют соли с основаниями (за счет карбоксильной группы) и с кислотами (за счет аминогруппы). Ион водорода, отщепляющийся при диссоциации от карбоксила аминокислоты, может переходить к ее аминогруппе с образованием аммониевой группировки. Таким образом, аминокислоты существуют и вступают в реакции также в виде биполярных ионов (внутренних солей):
Этим объясняется, что растворы аминокислот, содержащих одну карбоксильную и одну аминогруппу, имеют нейтральную реакцию. Из молекул аминокислот строятся молекулы белковых веществ, или белков, которые при полном гидролизе под влиянием минеральных кислот, щелочей или ферментов распадаются, образуя смеси аминокислот. Белки — природные высокомолекулярные азотсодержащие органические соединения. Они играют первостепенную роль во всех жизненных процессах, являются носителями жизни. Белки содержатся во всех тканях организмов, в крови, в костях. Ферменты (энзимы), многие гормоны представляют собой сложные белки. Кожа, волосы, шерсть, перья, рога, копыта, кости, нити натурального шелка образованы белками. Белок, так же как углеводы и жиры, — важнейшая необходимая составная часть пищи. В состав белков входят углерод, водород, кислород, азот, и часто сера, фосфор, железо. Молекулярные массы белков очень велики — от 1500 до нескольких миллионов. Проблема строения и синтеза белков — одна из важнейших в современной науке. В этой области в последние десятилетия достигнуты большие успехи. Установлено, что десятки, сотни и тысячи молекул аминокислот, образующих гигантские молекулы белков, соединяются друг с другом, выделяя воду за счет карбоксильных и аминогрупп; структуру цепи такой молекулы можно представить так:
В молекулах белков многократно повторяются группы атомов -—СО—NH—; их называют амидными, или в химии белков— пептидными группами. Соответственно белки относят к при родным высокомолекулярным полиамидам или полипептидам. Все многообразие белков образовано 20 различными аминокислотами; при этом для каждого белка строго специфичной является последовательность, в которой остатки входящих в его состав аминокислот соединяются друг с другом. Найдены методы выяснения этой последовательности; в результате уже точно установлено строение некоторых белков. И самым замечательным достижением в этой области явилось осуществление синтеза из аминокислот простейших белков: как уже указывалось, в 50—60-х годах XX века синтетически получены гормон инсулин и фермент рибонуклеаза. Таким образом, доказана принципиальная возможность синтеза еще более сложных белков.
|
№39. Арены. Ароматические соединения (арены) — органические соединения с плоской циклической структурой, в которой все углеродные атомы создают единую делокализованную n-электронную систему, содержащую (4л+2) п-электронов.К ароматическим соединениям относятся прежде всег всего бензол С6Н6 и его многочисленные гомологи и производные.Ароматические соединения могут содержать в молекуле одно или несколько бензольных ядер (многоядерные ароматические соединения). Ароматические соединения с одним бензольным ядром Строение молекулы бензола
Главным представителем ароматических углеводородов является бензол С6Н6. Строение молекулы бензола чаще всего выражают формулой, предложенной немецким химиком А. Кекуле. Этой формулой пользуются и сейчас, но вкладывают в нее новый смысл. Согласно Кекуле, бензол—замкнутая система с тремя сопряженными двойными связями—циклогексатриен-1,3,5. Но эта формула, соответствуя элементному составу молекулы бензола, не отвечает многим его свойствам. Например, согласно формуле Кекуле бензол формально является непредельной системой. В то же время он вступает преимущественно в реакции замещения, а не присоединения. Формула Кекуле также не в состоянии объяснить равенства расстояний между углеродными атомами, что имеет место в реальной молекуле бензола. Кроме того, она не может объяснить и высокой устойчивости бензольного кольца.Если исходить из классической формулы Кекуле, у бензола должно быть два орто-томера:
Все углеродные атомы в молекуле бензола находятся в состоянии л/Р-гибридизации. Каждый из них связан тремя своими гибридными орбиталями с двумя такими же орбиталями двух соседних углеродных атомов и одной орбиталью атома водорода, образуя три о-связи (две связи С-С и одну С-Н-связь) . Четвертая, негибридизованная 2р-орбиталь атома углерода, ось которой перпендикулярна плоскости бензольного кольца, перекрывается с подобными орбиталями двух соседних углеродных атомов, расположенных справа и слева. Такое перекрывание происходит над и под плоскостью бензольного кольца. В результате образуется единая замкнутая ситема n-электронов. Возникшая при этом делокализация n-электронов является более полной, чем в бутадиене-1,3. Две области наибольшей п-элеюронной плотности расположены по обе стороны кольца, которые можно представить в виде двух соприкасающихся тел вращения, по форме напоминающих торы*. В них сконцентрирована вся л-электронная плотность, а в плоскости между ними — область двенадцати а-связей (С-С и С-Н).тПонятие «ароматичности» В органической химии хорошо известно и широко используется такое понятие, как ароматичность некоторых органических соединений. Термин «ароматичность» связан прежде всего с бензолом, его гомологами и многочисленными производными. Этот термин относится исключительно к структуре молекул этих веществ, их свойствам, но не имеет никакого отношения к их запаху. Правда, первые ароматические соединения имели, вероятно, приятный запах (некоторые натуральные эфиры, душистые смолы, например ладан и др.). Термин «ароматичность» сохранился, однако в настоящее время в него вкладывают другой смысл. Ароматичность — общий признак некоторых циклических органических соединений, обладающих совокупностью особых свойств. Главной особенностью таких соединений является равномерное распределение я-электронной плотности в плоской циклической молекуле. Наличие единой замкнутой системы п-элеюпронов в молекуле — основной признак ароматичности. Ароматические соединения подчиняются правилу Э. Хюккеля плоские моноциклические соединения, имеющие сопряженную систему я-электронов, могут быть ароматическими, если число этих электронов равно 4п+2 (где л = 0,1,2,3, 4 и т.д., т.е. число я-электронов в молекуле может быть 2, 6,10,14,18 )Гомологи бензола получают при его взаимодействии с алкилгалогенидами в присутствии галогенидов алюминия. Номенклатура.
Систематическое название всех ароматических углеводородов — арены, а бензола — бензен. Гомологи бензола рассматривают как замещенные бензола и цифрами указывают положение заместителей: Однако систематическая номенклатура допускает название «бензол», а для некоторых гомологов бензола—тривиальные названия: винилбензол (I) называют стиролом, метилбензол (II) — толуолом, ди-метилбензол (III) — ксилолом, изопропилбензол (IV) — кумолом, метоксибензол (V) — анизолом.
Ароматические радикалы имеют общее название — арилы (Аг). Радикал С6Н5 - называют фенилом (от старого названия бензола —«фен»), С6Н4 — фениленом, С6Н5-СН2 -- бензилом, бензилиденом. Изомерия. Общая формула гомологов бензола СЛН2n_6. Все шесть атомов водорода в молекуле бензола одинаковы и при замещении одного из них на один и тот же радикал образуется одно и то же соединение. Поэтому однозамещенный бензол изомеров не имеет. При замещении двух атомов водорода на метальные группы образуются три изомера — ксилолы, которые отличаются друг от друга расположением заместителей в кольце:
Вместо буквенного обозначения (орто-, мета-, пара-, или сокращенно: о-, м-, л-) можно пользоваться цифровым: 1,2-, 1,3-, 1,4-. Изомеры могут отличаться характером заместителей:
Некоторые изомеры отличаются как строением заместителей, так и их положением в кольце. Например, углеводород состава С8Н10 имеет четыре изомера: один — этилбензол и три-диметилбензолы:
Получение Основными природными источниками ароматических углеводородов являются каменный уголь и нефть. Гомологи бензола можно получать синтетическим путем.Получение из каменного угля .При сухой перегонке каменного угля (при 1000-1200°С) образуются несколько продуктов: коксовый газ, кокс. Из смолы- фенол, нафталин. Получение из нефти, синтез ароматических углеводородов.Физические свойства. Низшие члены гомологического ряда бензола- жидкости с запахом. Ароматичес-е углеводор-ы не растворимы в воде, но растворяются во многих органических растворителях. Химические свойства.Основными химическими реакциями, в которых участвует бензольное кольцо, являются замещение, присоединение и окисление. Учитывая строение ароматических углеводородов, следует отметить, что для них наиболее характерными будут реакции электрофильного замещения (SЕ). 1. Реакции электрофильного замещения. К ним относятся реакции галогенирования, нитрования, сульфирования, реакции алкилирования и ацилирования (реакции Фриделя-Крафтса) и др. Все они протекают практически по одному механизму. 2. Реакции присоединения. Эти реакции протекают с большим трудом. Только в особых условиях (высокая температура, облучение УФ-светом, катализатор) бензол может проявлять слабо выраженный непредельный характер и присоединять водород или галогены. Например, в присутствии катализатора (платина, никель или палладий) бензол вступает в реакцию гидрирования с образованием циклогексана
3. Реакции окисления. Бензол довольно устойчив к действию окислителей. Так, бензол, в отличие от обычных непредельных углеводородов не обесцвечивает раствор перманганата калия. Только сильные окислители (например, кислород воздуха при высокой температуре в присутствии У2О5) могут привести к разрыву бензольного кольца с образованием малеиновой кислоты или малеинового ангидрида
Понятие о многоядерных углеводородах. Ароматические соединения, содержащие в своих молекулах несколько бензольных ядер, называют многоядерными. В зависимости от способа соединения этих ядер между собой различают соединения с неконденсированными и конденсированными бензольными ядрами. Канцерогенные вещества -бластомогенные вещества, канцерогены, карциногены, химические соединения, способные при воздействии на организм вызывать рак. Известно несколько сот К. в., принадлежащих к разным классам химических соединений. Так, к сильным К. в. относятся некоторые полициклические углеводороды с группировкой фенантрена в молекуле, азокрасители, ароматические амины, нитрозамины и др. алкилирующие соединения. К. в. найдены в составе некоторых промышленных продуктов, в воздухе, загрязнённом промышленными выбросами, в табачном дыме и др. Первые представления о существовании К. в. относятся к 18 в., когда случаи возникновения у английских трубочистов рака кожи были поставлены в связь с её систематическим загрязнением каменноугольной смолой и сажей. Впоследствии из смолы были выделены К. в. — 3,4-бензпирен и др. полициклические углеводороды. До внедрения соответствующих мер профилактики у работавших в анилинокрасочной промышленности, подвергавшихся воздействию К. в. (бета-нафтиламин, бензидин, 4-аминодифенил), нередко возникал рак мочевого пузыря. Канцерогенное действие связывают с химической активностью и электронным строением определённой части молекулы К. в. («область К»), ответственной за образование комплексов с определёнными компонентами клетки (по-видимому, нуклеиновыми кислотами и некоторыми белками).
№51. дисахариды.
Строение. Олигосахариды (от греч. oligos — немногочисленный, незначительный) — сложные углеводы, имеющие сравнительно невысокую молекулярную массу и схожие с моносахаридами свойства. Они состоят из остатков моносахаридов, связанных между собой гликозид-ными связями. При гидролизе из олигосахарида образуется несколько молекул моносахаридов (от 2 до 10). В зависимости от их числа различают дисахариды (биозы), гидролизующиеся на две молекулы монозы, трисахариды (триозы), гидролизующиеся на три молекулы монозы и т.д. Наибольшее значение имеют дисахариды, которые являются природными веществами. Так как большинство биоз состоит из гексоз, то их общая формула имеет вид С12Н22 О11. Дисахариды можно рассматривать как гликозиды циклического моносахарида, в молекуле которого место алкила занято остатком другого моносахарида: Смотри стр.341 Артеменко
С другой стороны, это соединение можно представить как простой эфир, в молекуле которого кислородный атом соединяет два остатка моносахаридов: Смотри стр.341 Артеменко
Эти остатки могут быть одинаковыми или разными. Связь между остатками моноз образуется с помощью двух гидро-ксильных групп — по одной от каждой молекулы монозы. Однако характер этих гидроксилов может быть разным. Если одна из молекул монозы всегда предоставляет для образования такой связи полуацеталь-ный (гликозидный) гидроксил, то другая молекула монозы участвует или полуацетальным гидроксилом (в этом случае образуется гликозид-гликозидная связь), или любым другим (обычным) гидроксилом (образуется гликозид-гликозная связь). В последнем случае в молекуле диса-харида сохраняется один полуацетальный гидроксил. Такой дисахарид способен из циклической формы переходить в таутомерную ей альдегидную (цепную) форму, которая обладает восстанавливающими свойствами. Эти дисахариды называют восстанавливающими. Если же дисахарид образуется за счет двух полуацетальных гидро-ксилов (по одному от каждой молекулы монозы), то он обладает устойчивым циклическим строением и не проявляет восстанавливающих свойств. Такие дисахариды называют невосстанавливающими.
Получение. Дисахариды обычно образуются при частичном гидролизе полисахаридов: Физические свойства. Дисахариды — твердые или сиропообразные вещества, хорошо растворимые в воде. Обладают сладким вкусом. Химические свойства. Химические свойства невосстанавливающих дисахаридов определяют гидроксильные группы, а восстанавливающих — еще и альдегидная или кетонная группа. Дисахариды легко гидролизуются при нагревании с водными растворами минеральных кислот или при действии ферментов. Это приводит к разрыву глико-зидной связи и образованию моносахаридов: Смотри стр.342 Артеменко
Сахароза (свекловичный, или тростниковый сахар) — белые кристаллы с т. пл. 184,5°С. Хорошо растворима в воде. Распространена в природе. Получают из сахарной свеклы или сахарного тростника. Молекула сахарозы состоит из остатков двух различных моноз — α-D-глюкозы (в пиранозной форме) и p-D-фруктозы (в фуранозной форме), которые связаны между собой \,2'-гликозид-гликозидной связью: Смотри стр.343 Артеменко
В молекуле сахарозы гликозидная связь образовалась за счет обоих полуацетальных гидроксилов глюкозы и фруктозы. Отсутствие полу-ацетального гидроксила в молекуле приводит к тому, что сахароза не имеет таутомерной (альдегидной) формы и поэтому не обладает восстанавливающими свойствами. При гидролизе (кислотном или ферментативном) сахароза расщепляется на две молекулы — D-глюкозу и D-фруктозу: Смотри стр.343 Артеменко
ВЫСОКОМОЛЕКУЛЯРНЫЕ (НЕСАХАРОПОДОБНЫЕ) ПОЛИСАХАРИДЫ Высокомолекулярные (несахароподобные) полисахариды — природные вещества, представляющие собой продукты конденсации большого числа молекул моносахаридов. Общая формула полисахаридов (С6Н10 05)n. Несахароподобные полисахариды построены подобно дисахаридам. Входящие в их состав остатки моносахаридов циклической формы соединены друг с другом гликозид-гликозидной связью, в образовании которой принимают участие полуацетальныи гидроксил одной молекулы моносахарида и четвертый гидроксил второй молекулы. Основными представителями несахароподобных полисахаридов являются крахмал и целлюлоза. Крахмал (греч. amylum) — самый распространенный в природе полисахарид, играющий роль резервного вещества многих растений. Он является главным компонентом картофеля, риса, пшеницы и кукурузы. Основное количество крахмала получают из клубней картофеля. Крахмал представляет собой смесь полисахаридов двух типов: растворимой в воде амилозы — внутренняя часть крахмального зерна (20-30%) и нерастворимого амилопектина — оболочка крахмального зерна (70-80%). Эти полисахариды построены из остатков cc-D-глюкозы, связанной между собой а-(1,4')-гликозид-гликозными связями: Смотри стр.346 Артеменко
У крахмала имеется очень чувствительная реакция: раствор иода вызывает синее окрашивание. Эту реакцию дает амилоза. Крахмал — невосстанавливающий углевод. При гидролизе крахмала (при нагревании в присутствии минеральных кислот или при действии фермента — амилазы) образуются различные промежуточные продукты Смотри стр.346 Артеменко
Растворимый крахмал — частично гидролизованный полисахарид. Его молекулярная масса несколько меньше, чем у обычного крахмала. Растворимый крахмал растворяется в горячей воде, с иодом дает синее окрашивание. Декстрины — менее сложные, чем крахмал, полисахариды. Они являются продуктами неполного гидролиза крахмала. В отличие от крахмала декстрины — восстанавливающие сахара. Они растворяются в холодной воде и иодом окрашиваются от фиолетового до желтого цвета. Промышленный способ получения декстринов — нагревание крахмала до 180-200°С. Например, процесс хлебопечения состоит в превращении нерастворимого крахмала в растворимые и гораздо легче усваиваемые организмом декстрины. Блестящая поверхность накрахмаленного белья после глажения горячим утюгом также объясняется образованием декстринов. Крахмал — ценный пищевой продукт. Применяется он и в химической промышленности. Например, кислотный гидролиз крахмала (при кипячении) служит промышленным методом получения глюкозы. Крахмал является сырьем для производства этилового и н-бутилового спиртов, ацетона, молочной и лимонной кислот, глицерина и других продуктов. Он используется для проклеивания бумаги и картона, производства декстринов и клеев. Ацилированный крахмал применяют для приготовления покрытий и загустителей. Алкильные производные крахмала используют в качестве пластификаторов и клеев. Целлюлоза, или клетчатка (от лат. cellula — клетка) — главная составная часть оболочек растительных клеток, выполняющая функции конструкционного материала. Целлюлоза в чистом виде обычно не встречается, но волокна хлопчатника (очищенная вата) и фильтровальная бумага могут служить образцом практически чистой целлюлозы (до 96%). Для выделения целлюлозы в чистом виде используют различные методы. Один из них — сульфитный. Он состоит в предварительном измельчении и последующей «варке» древесины при 135-145°С под давлением (0,7-0,8 МПа) с гидросульфитом кальция Ca(HSO3)2. Все вещества, сопутствующие целлюлозе, переходят при этом в раствор («сульфитный щелок»). Целлюлоза представляет собой полисахарид, который состоит из остатков P-D-глюкозы. В отличие от крахмала эти остатки связаны между собой β-(1,4-гликозид-гликоз-ными связями. Казалось бы, такое незначительное отличие в строении макромолекулярных цепей целлюлозы и крахмала не должно сильно сказываться на их физических и химических свойствах. Целлюлоза очень устойчива к механическим и химическим воздействиям. Она нерастворима в воде, спирте, эфире, ацетоне и других растворителях. Хорошо растворяется в концентрированном растворе хлорида цинка и в реактиве Швейцера. Целлюлоза не обладает восстанавливающими свойствами и труднее, чем крахмал, подвергается гидролизу. Но при длительном нагревании целлюлозы с минеральными кислотами, например серной, можно получить промежуточные продукты, вплоть до D-глюкозы Смотри стр.349 Артеменко
Химические свойства целлюлозы определяются главным образом гидроксильными группами. В каждом остатке глюкозы содержится три гидроксила Смотри стр.349 Артеменко
Несмотря на химическую инертность, целлюлоза под воздействием некоторых химически активных реагентов способна образовывать многие производные за счет гидроксильных групп. Например, при обработке целлюлозы концентрированным раствором щелочи получают щелочную целлюлозу Смотри стр.349 Артеменко
Ксантогенат целлюлозы служит для получения вискозного волокна. Его получают продавливанием через фильеру* щелочного раствора ксантогената в раствор серной кислоты. При этом целлюлоза регенерируется и одновременно образуются тонкие нити Смотри стр.349 Артеменко
Если вискозу, пластифицированную глицерином, продавливать через узкие прорези в осадительный раствор, то получают тонкий прозрачный лист — целлофан. Большое значение в промышленности имеют простые и сложные эфиры целлюлозы. Из простых эфиров особое значение получили метил-, этил- и бутилцеллюлоза. Они получаются при действии галогеналкилов на щелочную целлюлозу. Смотри стр.350 Артеменко
При действии на целлюлозу минеральных или органических кислот образуются сложные эфиры целлюлозы. В зависимости от числа гидроксильных групп в глюкозном звене, вступивших в реакцию этерификации, образуются различные эфиры: моно-, ди- и тринитрат целлюлозы. Смесь моно- и динитрата целлюлозы называют коллоксилином, а тринитрат целлюлозы — пироксилином. На основе нитратов целлюлозы (нитроцеллюлозы) получают различные пороха. Наиболее распространены среди них пироксилиновые пороха, применяемые в ствольном оружии. Нитраты целлюлозы служат для синтеза нитролаков, нитрокраски и эмалей. Из нитратов целлюлозы, пластифицированной камфорой, был получен первый (1869) термопласт — целлулоид. Однако из-за горючести его производство постоянно сокращается. При взаимодействии целлюлозы с уксусной кислотой (в присутствии серной кислоты) или уксусным ангидридом образуется сложный эфир —ацетат целлюлозы. Наибольшее значение приобрел полный эфир — триацетат целлюлозы При этерификации щелочной целлюлозы монохлоруксусной кислотой (или ее солью) образуется водорастворимый эфир целлюлозы — карбоксиметилцеллюлоза (КМЦ) Смотри стр.351 Артеменко
Будучи поверхностно-активным веществом, КМЦ используется в качестве добавки для повышения моющего действия синтетических моющих средств.
|
||||||||||||||||||||||||||||











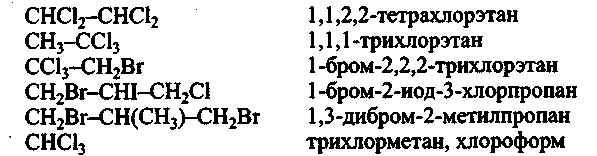






 Средство
к электрону
Еср
количественно
выражается в кДж/моль или эВ/моль.
Сродство к электрону зависит от
положения элемента в периодической
системе. Наибольшие значения сродства
к электрону имеют галогены, кислород,
сера, наименьшие и даже отрицательные
значения ее — элементы с электронной
конфигурацией j2
(He,
Be,
Mg,
Zn),
с полностью или наполовину заполненными
/ьподоболочками (Ne,
Аг, Кг, N,
P,
As).
Средство
к электрону
Еср
количественно
выражается в кДж/моль или эВ/моль.
Сродство к электрону зависит от
положения элемента в периодической
системе. Наибольшие значения сродства
к электрону имеют галогены, кислород,
сера, наименьшие и даже отрицательные
значения ее — элементы с электронной
конфигурацией j2
(He,
Be,
Mg,
Zn),
с полностью или наполовину заполненными
/ьподоболочками (Ne,
Аг, Кг, N,
P,
As). Средство
к электрону
Еср
количественно
выражается в кДж/моль или эВ/моль.
Сродство к электрону зависит от
положения элемента в периодической
системе. Наибольшие значения сродства
к электрону имеют галогены, кислород,
сера, наименьшие и даже отрицательные
значения ее — элементы с электронной
конфигурацией j2
(He,
Be,
Mg,
Zn),
с полностью или наполовину заполненными
/ьподоболочками (Ne,
Аг, Кг, N,
P,
As).
Средство
к электрону
Еср
количественно
выражается в кДж/моль или эВ/моль.
Сродство к электрону зависит от
положения элемента в периодической
системе. Наибольшие значения сродства
к электрону имеют галогены, кислород,
сера, наименьшие и даже отрицательные
значения ее — элементы с электронной
конфигурацией j2
(He,
Be,
Mg,
Zn),
с полностью или наполовину заполненными
/ьподоболочками (Ne,
Аг, Кг, N,
P,
As).




 +
+




