

-
Электрофильный катализ
Электрофильный катализ осуществляется кислотами Льюиса. Электрофильными катализаторами в органических реакциях могут выступать нейтральные молекулы, соли металлов, ионы и некоторые другие соединения, например:
Соли - ZnCl2, AlCl3, FeCl3, TiCl4, SnCl4, BF3;
Ионы металлов - Li+, Ag+, Hg+;
Молекулы и ионы - SO3, P2O5, R+, NO2+
Роль электрофильного катализатора заключается в активации реагента и аналогична роли протона в кислотном катализе. Взаимодействие электрофильного катализатора с реагентом-основанием заключается в образовании донорно-акцепторной связи за счет пары электронов реагента-основания, занимающей вакантную орбиталь одного из атомов катализатора. Электрофильные катализаторы оказываются гораздо более эффективными, чем протонные кислоты при активировании таких слабоосновных реагентов, как галогены, алкилгалогениды, ангидриды и галогенангидриды, эпоксиды и т.п. Образование донорно-акцепторного комплекса либо сильно поляризует связь в молекуле реагента, либо приводит к полному ее разрыву с образованием катиона:
 (2.65)
(2.65)
![]()
Образовавшийся катион или поляризованная молекула легко вступает в дальнейшее химическое превращение:
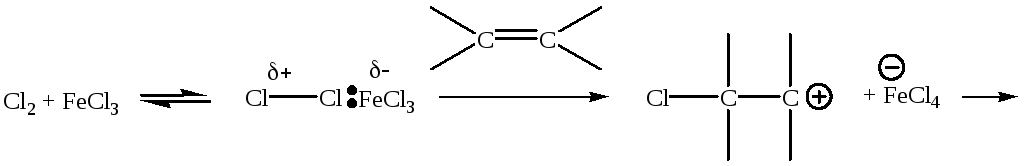
 (2.66)
(2.66)

В некоторых случаях кислота Льюиса выполняет каталитические функции, активируя не сам реагент, а протонную кислоту, являющуюся сокатализатором:
HF + SbF5 HSbF6 HF + BF3 HBF4 (2.67)
Такие комплексы оказываются значительно более эффективными донорами протона (увеличение кислотности), чем исходные кислоты и относятся к разряду суперкислот (см. раздел Суреркислоты). В частности, комплекс HFTaF5 обладает настолько высокой кислотностью, что отщепляет гидрид-анион от углеводородов с образованием карбокатиона:
+
RCH2-CH3 + HFTaF5 RCH-CH3 + H2 + TaF6- (2.68)
Галогениды металлов (AlCl3, FeCl3, TiCl4, SnCl4, BF3) разлагаются при взаимодействии с кислород- или азотсодержащими органическими соединениями и водой либо образуют с ними очень прочные и малореакционноспособные комплексы. Поэтому галогениды металлов обычно не катализируют реакции с их участием.
-
Основный катализ
Основный катализ осуществляется веществами со свободной или лабильной электронной парой электронов, которые могут быть как анионами, так и нейтральными молекулами (HO-, RO-, NH2-, R3N: и др.). При этом реагент должен обладать кислотными свойствами (по отношению к катализатору), и активирование реагента осуществляется за счет отрыва от него протона или образования водородной связи:
![]() или
или ![]() (2.69)
(2.69)
Причем в активации реагента могут принимать участия все формы оснований, образующиеся в реакционной массе, в том числе и ионы лиата, образующиеся по протолитической реакции катализатора с растворителем:
SH + B- S- + BH (2.70)
После активации реагента (уравнение (2.69)) превращения могут протекать по двум вариантам:
-
Мономолекулярная реакция. Из продуктов превращений на последующих стадиях регенерируется катализатор. Например:
 (2.71)
(2.71)
2)Бимолекулярная реакция. Участие активированного реагента в качестве нуклеофила в реакции со вторым реагентом. Эти реакции близки по механизму к нуклеофильному катализу (схема (2.27)). Отличие состоит в том, что при основном катализе протекает еще одна стадия в каталитическом цикле - отщепление катализатором протона от реагента с образованием сопряженного основания реагента, играющего далее роль нуклеофильного катализатора, но с обязательной регенерацией основного катализатора в конце каталитического цикла. Например в реакции альдольной конденсации:
![]() (2.72)
(2.72)

В других случаях не происходит полного отщепления протона, а нуклеофильность реагента повышается за счет образования водородной связи с основанием-катализатором:
 (2.73)
(2.73)
Специфический основный катализ
Специфический основный катализ распространен гораздо меньше, чем специфический кислотный. Его схема включает быстрые протолитические реакции реагента (RH) с основанием-катализатором (B) и лиат-ином (S-), образующимся по равновесию с катализатором:
Образование лиат-иона:
(2.74)
B + SH BH+ + S-
Активирование реагента (быстро):
RH + B BH+ + R- RH + S- SH + R-
Далее следует медленное превращение в продукты (РН), которое может быть как мономолекулярным, так и с участием второго реагента, после чего происходит регенерация катализатора:
Мономолекулярно R- Р- + Z
(2.75)
С участием Y R- + Y Р-
Регенерация катализатора Р- + BH+(SH) РН + B(S-)
Скорость реакций, протекающих по такой схеме, однозначно определяется рН (или Н_) реакционной массы, а кинетические закономерности реакций имеют тот же вид, что и для специфического кислотного катализа. Так для реакций, протекающих по схеме (2.74)-(2.75) нетрудно вывести кинетические уравнения, аналогичные уравнениям(2.44) и (2.52), включающее зависимость константы каталитической реакции от h_:
для мономолекулярного маршрута:
![]() (2.76)
(2.76)
при реакции со вторым реагентом (YH):
 (2.77)
(2.77)
Общий основный катализ
Закономерности общего основного катализа аналогичны закономерностям общего кислотного. Общий основный катализ наблюдается, если стадия отрыва протона является лимитирующей, а дальше идут быстрые реакции превращения активированных молекул. В общем виде механизм реакций можно представить следующей схемой:
медленно: (2.78)
![]()
быстро:
![]()
Образовавшийся на первой (лимитирующей) стадии активированный реагент (R-) далее быстро превращается в продукт реакции (P) по мономолекулярной реакции или при участии второго реагента (Y). В соответствии с данной схемой скорость реакции лимитируется протолитической реакцией и кинетическое уравнение имеет вид:
r = k1[RН][В] (2.79)
При этом, если катализатор присутствует в разных формах (Вi), то каждой из них соответствует своя константа скорости (ki), определяемая активностью данной формы катализатора, и в общем виде уравнение (2.79) можно представить так:
![]() (2.80)
(2.80)
Рассмотренная схема характерна для реакций, в которых на первой стадии происходит медленный разрыв С-Н связи. Примером служат основно-каталитические реакции енолизации кетонов:
![]() (2.81)
(2.81)
Общий основный катализ проявляется не только в случае разрыва С-Н связи. Известно множество примеров, когда лимитирующей оказывается стадия передачи протона между атомами катализатора и реагента. Это происходит, когда перенос протона сопровождается синхронным разрывом или образованием новой связи. Этому обычно предшествует быстрая стадия координации кислоты и основания за счет образования водородной связи. Общая схема для таких случаев выглядит так:
Образование водородной связи (быстро):
(2.82)
![]()
Лимитирующая стадия:
мономолекулярная
![]()
бимолекулярная
![]()
Примером бимолекулярной реакции, протекающей по такой схеме, служит основно-каталитическое взаимодействие производных карбоновых кислот с аминами (реакция (2.73)).
Связь каталитической активности с природой катализатора при общем основном катализе.
Количественную связь активности основного катализатора при общем катализе установил Бренстед. Уравнение, связывающее величину экспериментально найденной константой скорости kэф с константой основности (KВ) катализатора по форме аналогично такой же зависимости для общего кислотного катализа (уравнение (2.64)):
kэф = GBKB (2.83)
Это уравнения применимо к одной и той же реакции при катализе структурно подобными о основаниями. Величина характеризуют чувствительность данной реакции к изменениям основности катализатора. Значение ее лежит в интервале 0 < Эмпирический коэффициент GВ – индивидуален для каждого типа реакций.