

Reactive Intermediate Chemistry
.pdf778 ARYNES
2,6-isomer 112 and 2,3-isomer 113 are less stable than 111 by 11.7 and 23.2 kcal/ mol, respectively.123 Obviously, for the two-electron case the 1,3,5-topology (comparable to that in the trishomocyclopropenium cation) is favored over the 1,2,3-topology, while for the four-electron, three-center systems the destabilization is most pronounced in the second case. In addition to its remarkable stability, 111 also fulfills magnetic aromaticity criteria. At the same level of theory [MP4(SDTQ)/6-31G*//MP2/6-31G*þZPE] open-chain isomers 114 and 115 are less stable by 11.8 and 30.4 kcal/mol, respectively.123
|
|
C |
|
|
146.1 |
B |
|
135.9 |
N |
|
C |
C |
|
|
|
|
|
||
|
|
|
C |
204.4 |
C |
C |
C |
||
136.5 |
|
|
|
135.7 |
137.7 |
236.7 |
|||
|
200.3 |
|
203.6 |
193.9 |
|||||
|
C |
C |
|
C |
C |
C |
|||
|
|
|
|
C |
|||||
|
|
C |
145.6 |
|
137.2 |
C |
147.8 |
136.3 |
140.4 |
|
|
|
|
|
95.8 |
|
C |
||
|
111 |
94.4 |
|
117 |
102 |
90.7 |
|||
|
|
|
|
|
|||||
|
|
|
X |
|
X |
|
X = C+ : |
∆rH298 = −27.0 kcal/mol |
|
. |
. + |
|
. |
. + |
X = B: |
∆rH298 = −28.3 kcal/mol |
|||
|
|
|
|
|
|
|
X = N: |
∆rH298 = +3.0 kcal/mol |
|
Figure 16.6. The structures of 111 and 117 reveal substantial two-electron, three-center interaction, accompanied by stabilization, whereas destabilization in 102 is minimized by distortion towards a larger C3 N/C5 N distance (BLYP/cc-pVTZ).46 Bond lengths in picometers angles in degrees.
In contrast to the Jahn–Teller distorted, C2v symmetric 1,3,5-tridehydrobenzene (116) (cf. Section 6.1), cation 111 has D3h symmetry.123 According to BLYP calculations, the isoelectronic 3,5-borabenzyne (117) is even more stabilized relative to 13 than is 111 (Fig. 16.6). Both systems are strongly distorted to maximize the stabilizing two-electron three-center interaction, whereas 3,5-pyridyne circumvents destabilization to some degree by maximizing the C3 N/C5 N distances
(Fig. 16.6).83a In this context, it is interesting to mention the influence of protonation of the nitrogen atom in the pyridynes.114b Replacement of CH by N in
benzynes has a pronounced impact on the singlet states (rather than on the triplets), which is largely attenuated by protonation (Tables 16.5 and 16.6).114
The properties of 2,5-pyridyne (99) can once more be rationalized within the
through-bond coupling scheme. The singlet–triplet splitting in (99) is 6 kcal/ mol higher than in p-benzyne or in the protonated form (99 Hþ).114,124–126 A
detailed interpretation of these energy differences has been given by Kraka and Cremer.124 Anomeric delocalization of the lone pair at the nitrogen atom into the vicinal C C bonds results in shortening of the N C2 and N C6 bonds


780 ARYNES |
|
|
|
|
|
|
|
C |
137.4 |
136.5 |
C |
137.9 |
135.9 |
C |
137.7 |
C |
C |
C |
|
C |
C |
|
C |
|
142.2 |
138.5 |
|
141.8 |
135.9 |
|
142.0 |
C |
C |
N |
|
C |
N |
|
C |
C |
|
126.4 |
C |
138.7 |
132.4 |
C |
137.0 |
28 |
|
|
99 |
|
99-H+ |
|
|
Figure 16.7. The structures of p-benzyne (28), 2,5-pyridyne (99), and (99-Hþ), calculated at the UB3LYP/6-31G(d,p) level of theory.124 Bond lengths in picometers.
(Scheme 16.28).127 However, a 2,5-didehydropyridine intermediate could not be detected or trapped, which was attributed to the low barrier for ring opening of this species (a mechanism involving an aza-butalene structure was also not excluded).127
|
Ph |
. |
|
N |
|
|
Ph |
||
N |
110 °C |
N |
Ph |
|
|
|
|||
Me |
Me |
. |
Ph |
Ph |
|
Ph |
? |
|
Me |
(Z )-118 |
|
|
(Z )-119 |
|
|
|
|
Scheme 16.28. Rearrangement of C; N-dialkynylimine 118 to b-alkynyl acrylonitrile 119.127
One year later Chen and co-workers126 reported trapping experiments of protonated 2,5-pyridyne. Detectable hydrogen-abstraction rates were only observed under low pH conditions, which suggest the existence of a protonated 2,5-pyridyne derivative.126 Theoretical investigations show that—in contrast to p-benzyne 28 and enediyne (Z)-16 (Fig. 16.4)—2,5-didehydropyridine (99) is stabilized relative to
imine 120 (Fig. 16.8); the relative stability of nitrile 108 is also increased significantly.114,125,126 However, the barrier for ring-opening of 2,5-pyridyne (99–108)
was determined to be only 0.9 kcal/mol (cf. Ref. 125 for numerical details). In addition to the short lifetime, the large singlet–triplet separation accounts for the poor hydrogen-abstraction ability of 99, as discussed below. Protonation does not only
decrease the singlet–triplet gap significantly (see above), but also increases the barrier for retro-Bergman reaction of 99 Hþ to 108 Hþ to >10 kcal/mol.114,124–126
Interest in the aza-Bergman reaction and the influence of protonation on 2,5- pyridyne and its derivatives was (once more) directly stimulated by the search for less toxic antitumor drugs. One approach to increase the selectivity is to
decrease the reactivity of the biradical intermediate in hydrogen-abstraction reac- tions.59,66,124–126,128 In this regard, Chen has established a simple model that correlates the reactivity of singlet biradicals with their singlet–triplet gap EST.66,126,128
This model is based on the assumption that the reactivity of the triplet biradical is
|
|
HETEROCYCLIC ARYNES |
781 |
|
|
∆E (kcal mol−1) |
21.6 |
|
|
|
|
19.4 |
|
|
|
|
9.3 |
|
|
|
0.0 |
−1.4 |
|
|
|
|
|
|
|
|
|
−4.4 |
|
|
|
|
−5.3 |
|
|
|
X |
. |
|
|
|
|
|
|
|
|
|
X |
|
|
|
120 |
. |
|
|
|
|
99 |
|
|
|
|
|
−29.8 |
|
~ |
X |
X = NH+ |
|
|
|
|
|
||
|
|
|
−42.8 |
|
|
|
108 |
X = N |
|
|
|
|
|
|
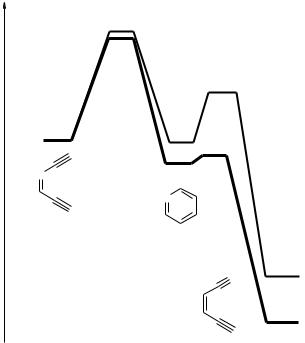
Figure 16.8. Calculated energy profile of the Bergman cyclization of imine 120 and its
protonated form 120 Hþ and of the subsequent retro-Bergman cyclization to nitrile 108
(108 Hþ).125
comparable to that of the monoradical, while effects stabilizing the singlet state have to be overcome in the transition state of the hydrogen abstraction. Accordingly, an increased singlet–triplet gap is expected to raise the barrier for hydrogen abstraction and (possibly) the selectivity of the reaction. An interesting method to estimate EST based on the isotropic hyperfine coupling constant of the radical center in the respective monoradical with the proton at the second center has been developed by Cramer and Squires.129 Since this approach provides reliable results with DFT methods it can be applied to larger systems such as potential antitumor drugs.
In this context, it is noteworthy that the pH value of tumor cells (6.2–6.6) is lower than that of normal cells (7.5).7,130 This difference could be used to control
the Bergman cyclization of substituted and/or heterocyclic derivatives of 28 by protonation–deprotonation or to control the hydrogen abstraction depending on the pH value. In an exhaustive theoretical study Kraka and Cremer59 investigated a large number of substituted and heterocyclic enediynes and the corresponding benzynes and proposed potential candidates for future anticancer drugs. While simply replacing CH by N allows pH-dependent control of the Bergman cyclization
782 ARYNES
to some extent, calculations by these authors indicate, that the proton affinity of 99 is not sufficient to achieve efficient protonation under physiological conditions.59 Whether the investigation of p-arynes, as very briefly outlined in the foregoing sections, is going to facilitate the development of novel antitumor drugs is not foreseeable at the moment, and undue optimism is certainly not justified. Nevertheless, the concerted efforts of many research groups have contributed to the refinement of the experimental and theoretical tools of Physical-Organic chemistry, and it is no exaggeration to state that arynes are prototypical examples for the interplay of
experiment and theory in modern chemistry.
6. RELATED SYSTEMS
6.1. Tridehydrobenzynes
In contrast to benzynes and benzdiynes (cf. Section 6.2), knowledge of tridehydroarenes is scarce. According to calculations by Bettinger et al.26x 1,2,4-tridehy- drobenzene (121) and 1,3,5-tridehydrobenzene (122) are less stable than the 1,2,3- isomer 123 by 4.0 and 11.8 kcal/mol, respectively. In contrast to didehydrophenyl cation 111 (Section 5), 122 possesses C2v symmetry; the calculated C1 C3 and C1 C5 distances are different (194 and 230 pm, respectively, at the CCSD/DZP level of theory).26x For the 1,2,3-isomer 123 a significantly shorter C1 C3-distance ( 170 pm) is predicted by various methods.
. |
. |
. |
|
|
|
|
|
||
. |
. |
. |
. |
. |
|
. |
|
||
121 |
|
122 |
123 |
|
Cs |
|
C2v |
C2v |
|
Formula 16.13
6.2. Benzdiynes
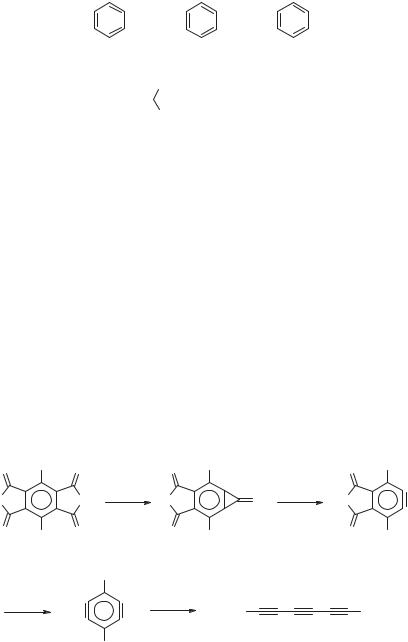
Polar carbon-rich species such as hexapentaeneylidene (124)131 have been identi-
fied in interstellar clouds,132 which has attracted some attention and interest in C6H2 compounds.26x,133,134 The existence of 124 in interstellar space is remarkable, since
this structure is 50 kcal/mol [CCSD(T)/cc-pVTZ] less stable than hexatriyne (125),133 which, however, is nonpolar and therefore difficult to observe radioastronomically. At the same level of theory the three isomeric benzdiynes 126–128 are
higher in energy than 125 by 39.4, 46.3, and 56.2 kcal/mol, respectively.133
The C1 C3 distance of 176 pm in 12826x,133,134 is rather low, which has been attributed to strong s-allylic interaction as in the case of m-benzyne 13 (Section 2.2).26x Like o-benzyne (4), 127, and 128 possess C C bonds with

784 ARYNES
irradiation converts 130 into bis(trifluorormethyl)-hexatriyne (131). The difluoro derivative of 128 has been prepared and characterized in a similar fashion.137 The overall reaction scheme is more complex in this case and two different ketene
species of yet unknown constitution are involved in the photochemistry of the anhydride.137
The parent systems 127 and 128 are not accessible via this route.137,138 Photolysis of 1,2,3,4- and 1,2,4,5-benzenetetracarboxylic dianhydride with different wavelengths invariably results in formation of hexatriyne 125.137 Attempts to isolate 1,2,5,6- and 2,3,6,7-tetradehydronaphthalenes (naphthdiynes) by photolysis of the corresponding naphthalenetetracarboxylic dianhydrides yield only acetylenic compounds, which suggests that naphthdiynes are formed, but rapidly decompose under photochemical conditions.139
6.3. Cyclo[6]-carbon
Aside from fullerene research, carbon clusters have always attracted much interest among scientists.140 Linear cumulene-like C6 (D1h) 132 is well known from computational studies as well as experimentally.141 Cyclic C6, which will be considered as the extreme case of a dehydrobenzene here, has a long and varied history.142 In particular, two cyclic isomers possessing D3h and D6h symmetry (133 and 134) have been discussed.142 According to the most recent calculations, 133 (1A01) is lowest in
energy; 134 (1A1g) and 132 (3 g ) being less stable by 8 and 10 kcal/mol, respectively.142e
|
. |
. |
|
133 |
132 |
134 |
Dooh |
|
D6h |
D3h |
|
Formula 16.15
The IR spectrum of 133 was obtained by laser vaporization of graphite and subsequent condensation of the reaction products in solid argon at 10 K. However, only
the most intense mode at 1695 cm 1 could be detected.143 The antisymmetric stretching vibration of the linear isomer 132 is observed at 1952 cm 1.141b,141e
The assignment could be corroborated by measuring the spectra of isotopically labeled compounds. In a more recent theoretical work, the UV spectra of 133 and 134 were calculated,144 but experimental data are lacking so far.
7. CONCLUSIONS AND OUTLOOK
The interest in arynes has changed the status of these molecules from laboratory curiosities to well-established reactive intermediates. Yet, our knowledge of this
|
|
|
|
CONCLUSIONS AND OUTLOOK 785 |
||||
|
C |
|
C |
|
C |
142.1 |
C |
138.2 |
C |
|
C |
C |
C |
|
C |
|
C |
141.9 |
281.3 |
144.2 |
147.4 |
271.5 |
150.2 |
141.5 |
||
C |
|
C |
C |
C |
|
C |
|
C |
138.1 |
C 142.4 |
C |
134.6 C 139.1 |
C |
|
|||
|
|
60 |
|
|
|
52 |
|
|
|
139.5 C |
140.3 |
|
138.7 C |
140.6 |
|||
|
C |
|
|
C |
||||
C |
|
|
C |
|
|
|||
|
C |
|
140.2 |
|
C |
|
140.6 |
|
|
|
|
|
|
||||
C |
230.7 |
150.9 |
C |
C |
203.1 |
146.5 |
|
C |
141.0 |
C |
|
|
138.6 |
C |
C |
|
|
C |
|
C |
|
|
|
C |
||
|
141.1 |
C |
|
142.2 |
|
|||
|
|
|
|
|
C |
|
|
|
|
135 |
|
|
|
136 |
|
|
|
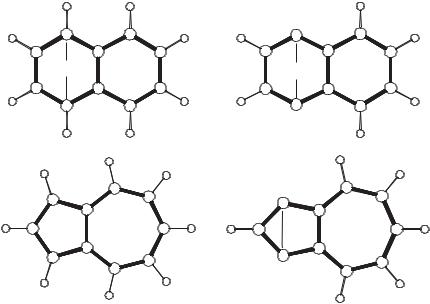
Figure 16.9. Calculated structures of azulene 135 and 1,3-azulyne 136 compared to naphthalene 60 and 1,4-naphthyne 52.46
class of compounds is still incomplete and many aspects of their chemistry remain to be investigated in more detail. This lack pertains in particular to the mechanisms of intraand intermolecular reactions and rationalizing the influences of heteroatoms or substituents on the structure and reactivity of arynes. This knowledge will contribute to our understanding of basic processes in a large variety of fields, spanning synthesis, biological activity of cytostatics, heterogenous catalysis, combustion processes, and the chemistry of interstellar clouds.
Despite the long history, there are still many branches of aryne chemistry that are totaly unexploited. Not even one five-membered aryne has so far been characterized spectroscopically. Do we expect to find something new in this direction? Are the concepts that serve well to clarify the structures and electronic properties of sixmembered arynes sufficient to understand the properties of the five-membered intermediates as well? The structures of azulene 135 and 1,3-azulyne 136, calculated at the (U)BLYP/cc-pVTZ level of theory, are shown in Figure 16.9. Whereas the central bond in 1,4-naphthyne (52) is longer than that in naphthalene (60) because of through-bond coupling,88 the opposite is found for the isomeric azulyne.46 A detailed discussion of this (at first-glance counterintuitive) finding will be given elsewhere; here this example may suffice to demonstrate that surprises cannot be excluded, turning from sixto five-(or seven-)membered systems. Just as the concept of through-bond–through-space interactions, introduced by Hoffmann et al.73,74 in 1968, had profound influence on almost all branches of chemistry, arynes will continue to be a source of inspiration for theorists and experimentalists alike.
786 ARYNES
SUGGESTED READING
H.H. Wenk, M. Winkler, and W. Sander, ‘‘One Century of Aryne Chemistry,’’ Angew. Chem. Int. Ed. Engl., 2003, 42, 502.
For a nice review on heterocyclic arynes, see M. G. Reinecke, ‘‘Hetarynes.’’ Tetrahedron, 1982 38, 427.
H.C. van der Plas,‘‘The Chemistry of Triple Bonded Groups,’’ in Supplement C of The Chemistry of Functional Groups, S. Patai and Z. Rappoport, Eds., Wiley-Interscience, New York, 1982.
For synthetic aspects of aryne chemistry, see C. Grundmann, ‘‘Arene und Arine,’’ in HoubenWeyl, Methoden der Organischen Chemie, Vol. 5, C. Grundmann, Ed., Thieme, Stuttgart,
1981.
H. Heaney, ‘‘Chemistry of Highly Halogenated Arynes,’’ Fortschr. Chem. Forsch. 1970, 16, 35.
For a summary of early work, see R. W. Hoffmann, Dehydrobenzene and Cycloalkynes; Academic Press, New York, 1967.
REFERENCES
1.H. H. Wenk, M. Winkler, and W. Sander, Angew. Chem. Int. Ed. Engl. 2003, 42, 502.
2.R. Stoermer and B. Kahlert, Ber. Dtsch. Chem. Ges. 1902, 35, 1633.
3.A. Lu¨ttringhaus and G. Saaf, Justus Liebigs Ann. Chem. 1930, 592, 250.
4.(a) J. D. Roberts, H. E. Simmons, L. A. Carlsmith, and C. W. Vaughan, J. Am. Chem. Soc. 1953, 75, 3290; (b) J. D. Roberts, D. A. Semenow, H. E. Simmons, and L. A. Carlsmith,
J.Am. Chem. Soc. 1956, 78, 601, 611.
5.(a) R. Huisgen and H. Rist, Naturwissenschaften 1954, 41, 358; (b) R. Huisgen and
H.Rist, Ann. Chem. 1955, 594, 137; (c) R. Huisgen and R. Knorr, Tetrahedron Lett. 1963, 1017.
6.(a) G. Wittig, Naturwissenschaften 1942, 30, 696; (b) G. Wittig and G. Harborth, Ber. Dtsch. Chem. Ges. 1944, 77, 306, 316; (c) G. Wittig and L. Pohmer, Chem. Ber. 1956, 89, 1334. (d) G. Wittig, Pure Appl. Chem. 1963, 7, 173. For a general overview about Wittig’s achievements, see: (e) R. W. Hoffmann, Angew. Chem. Int. Ed. Engl. 2001, 40, 1411.
7.See, for example: (a) K. C. Nicolaou and W.-M. Dai, Angew. Chem. Int. Ed. Engl. 1991, 30, 1387; (b) K. C. Nicolaou and A. L. Smith, Acc. Chem. Res. 1992, 25, 497; (c) D. B. Borders and T. W. Doyle, Eds., Endiyne Antibiotics as Antitumor Agents, Marcel Dekker, New York, 1995; (d) W. K. Pogozelski and T. D. Tullius, Chem. Rev. 1998, 98, 1089; (e)
Z.Xi and I. H. Goldberg, in Comprehensive Natural Products Chemistry, Vol. 7, D. H. R. Barton and K. Nakanishi, Eds., Pergamon, Oxford, 1999, (f) S. E. Wolkenberg and D. L. Boger, Chem. Rev. 2002, 102, 2477.
8.W. Sander, Acc. Chem. Res. 1999, 32, 669.
9.(a) L. K. Madden, L. V. Moskaleva, S. Kristyan, and M. C. Lin, J. Phys. Chem. 1997, 101, 6790; (b) L. V. Moskaleva, L. K. Madden, and M. C. Lin, Phys. Chem. Chem. Phys. 1999, 1, 3967; and references cited therein.
10.(a) K. Johnson, B. Sauerhammer, S. Titmuss, and D. A. King, J. Chem. Phys. 2001, 114, 9539; (b) S. Yamagishi, S. J. Jenkins, and D. A. King, J. Chem. Phys. 2002, 117, 819; and literature cited therein.
REFERENCES 787
11.W. E. Bachman and H. T. Clarke, J. Am. Chem. Soc. 1927, 49, 2089.
12.I. P. Fisher and F. P. Lossing, J. Am. Chem. Soc. 1963, 85, 1018.
13.(a) R. S. Berry, G. N. Spokes, and M. Stiles, J. Am. Chem. Soc. 1962, 84, 3570. (b) R. S. Berry, J. Clardy, and M. E. Schafer, J. Am. Chem. Soc. 1964, 86, 2738.
14.(a) O. L. Chapman, K. Mattes, C. L. McIntosh, J. Pacansky, G. V. Calder, and G. Orr, J. Am. Chem. Soc. 1973, 95, 6134; (b) O. L. Chapman, C. C. Chang, J. Kolc, N. R.
Rosenquist, and H. Tomioka, J. Am. Chem. Soc. 1975, 97, 6586.
15.I. R. Dunkin and J. G. MacDonald, J. Chem. Soc. Chem. Commun. 1979, 772.
16.C. Wentrup, R. Blanch, H. Briehl, and G. Gross, J. Am. Chem. Soc. 1988, 110, 1874.
17.(a) J. W. Laing and R. S. Berry, J. Am. Chem. Soc. 1976, 98, 660; (b) N.-H. Nam and
G.E. Leroi, Spectrochim. Acta A 1985, 41, 67.
18.D. G. Leopold, A. E. Stevens-Miller, and W. C. Lineberger, J. Am. Chem. Soc. 1986, 108, 1379.
19.A. C. Scheiner, H. F. Schaefer, III, and B. Liu, J. Am. Chem. Soc. 1989, 111, 3118.
20.J. G. G. Simon, N. Muenzel, and A. Schweig, Chem. Phys. Lett. 1990, 170, 187.
21.J. G. Radziszewski, B. A. Hess, Jr., and R. Zahradnik, J. Am. Chem. Soc. 1992, 114, 52.
22.P. G. Wenthold, R. R. Squires, and W. C. Lineberger, J. Am. Chem. Soc. 1998, 120, 5279.
23.(a) P. G. Wenthold and R. R. Squires, J. Am. Chem. Soc. 1994, 116, 6401. For earlier work concerning the enthalpy of formation of 4, see: (b) P. G. Wenthold, J. A. Paulino, and R. R. Squires, J. Am. Chem. Soc. 1991, 113, 7414 (106 3 kcal/mol); (c) J. M. Riveros, S. Ingemann, and N. M. M. Nibbering, J. Am. Chem. Soc. 1991, 113, 1053 (105 3 kcal/mol); (d) Y. Guo and J. J. Grabowski, J. Am. Chem. Soc. 1991, 113, 5923 (105 5 kcal/mol).
24.A. M. Orendt, J. C. Facelli, J. G. Radziszewski, W. J. Horton, D. M. Grant, and J. Michl,
J.Am. Chem. Soc. 1996, 118, 846.
25.(a) R. Warmuth, Angew. Chem. Int. Ed. Engl. 1997, 36, 1347; (b) R. Warmuth, Eur. J. Org. Chem. 2001, 423; (c) R. Warmuth and J. Yoon, Acc. Chem. Res. 2001, 34, 95.
26.From the numerous studies up to 1998 the following are mentioned here as examples: (a)
E.Haselbach, Helv. Chim. Acta 1971, 54, 1981; (b) M. J. S. Dewar and W.-K. Li, J. Am. Chem. Soc. 1974, 96, 5569; (c) W. Thiel, J. Am. Chem. Soc. 1981, 103, 1420; (d) M. J. S.
Dewar, G. P. Ford, and C. H. Reynolds, J. Am. Chem. Soc. 1983, 105, 3162; (e) C. W. Bock, P. George, and M. Trachtman, J. Phys. Chem. 1984, 88, 1467; (f) M. J. S. Dewar and T.-P. Tien, J. Chem. Soc. Chem. Commun. 1985, 1243; (g) L. Radom, R. H. Nobes, D. J. Underwood, and L. Wai-Kee, Pure Appl. Chem. 1986, 58, 75; (h) K. Rigby, I. H. Hillier, and M. A. Vincent, J. Chem. Soc. Perkin Trans. 2 1987, 117; (i) I. H. Hillier, M. A. Vincent, M. F. Guest, and W. von Niessen, Chem. Phys. Lett. 1987, 134, 403; (j) A. C. Scheiner, and H. F. Schaefer, III, Chem. Phys. Lett. 1991, 177, 471; (k) R. Liu, Z. Xuenfeng, and P. Pulay, J. Phys. Chem. 1992, 96, 259; (l) H. U. Sutter and T.-K. Ha, Chem. Phys. Lett. 1992, 198, 259; (m) A. Nicolaides and W. T. Borden, J. Am. Chem. Soc. 1993, 115, 11951; (n) S. G. Wierschke, J. J. Nash, and R. R. Squires, J. Am. Chem. Soc. 1993, 115, 11958; (o) P. B. Karadahov, J. Gerrat, G. Raos, D. L. Cooper, and M. Raimondi, Isr. J. Chem. 1993, 33, 253; (p) E. Kraka and D. Cremer, Chem. Phys. Lett. 1993, 216, 333; (q) E. Kraka and D. Cremer, J. Am. Chem. Soc. 1994, 116, 4936;
(r) R. Lindh and B. J. Persson, J. Am. Chem. Soc. 1994, 116, 4963; (s) R. Lindh, T. J. Lee, A. Bernhardsson, B. J. Persson, and G. Karlstro¨m, J. Am. Chem. Soc. 1995, 117, 7186;
(t) J. J. Nash and R. R. Squires, J. Am. Chem. Soc. 1996, 118, 11872; (u) R. Liu,