

Reactive Intermediate Chemistry
.pdf758 ARYNES
which allowed identification of the frequencies of two totally symmetric (and therefore IR inactive) modes at 600 and 1000 cm 1, again in good agreement with the DFT-calculated spectrum.8
Recently, the assignment of the band at 980 cm 1 to 28 has been doubted based on new calculations (this band is shifted to 976 cm 1 if 28 is generated from 1,4- diiodobenzene (37), which is not unusual in the presence of iodine atoms. This shift may also be attributable to the change of the matrix host from argon to neon).70 On the other hand, ab initio calculations of the IR spectrum of 28 are complicated by the existence of orbital instabilities,71 the effect of which may (often) be negligible for first order properties (such as geometry and energy), but can result in severe deviations for second-order properties (vibrational frequencies, IR intensities).70,71
In |
further matrix isolation studies, cyclophanedione |
(38), was |
envisioned as |
a |
precursor, which upon long wavelength irradiation |
fragments |
quantitatively |
to p-xylylene and bisketene (39) (Scheme 16.14).72 In contrast to benzannellated derivative 32, however, the latter turned out to be photochemically stable under matrix isolation conditions.72
In 1968, Hoffmann et al.73 systematized the properties of the benzynes by formally separating the interaction of the radical centers into through-space and through-bond contributions. Specifically, this concept predicts that in p-benzyne the antisymmetric combination (b1u) of the formally nonbonding orbitals at C1
and C4 is lowered beyond the symmetric combination (ag). This idea has been confirmed by high-level calculations (Scheme 16.15).26,70 The partial occupation of the
antibonding C2 C3and C4 C5-s* orbitals results in elongation of these bonds, while the other C C bonds are shortened.70,73,74 This coupling of the electrons in
p-benzyne stabilizes the singlet state relative to the triplet. Experimentally a singlet
ground |
state with a singlet–triplet splitting of 3.8 |
|
0.5 kcal/mol was determined by |
|
22 |
|
|
||
NIPES. |
|
|
|
|
|
E |
|
|
|
|
|
b1u |
|
|
|
b1u |
|
|
b1u |
|
ag |
|
|
|
|
|
|
|
|
|
p |
|
|
|
|
|
b1u |
|
|
|
ag |
ag |
|
ag |
|
|
|
||
|
|
|
|
|
Scheme 16.15. Schematic MO representation of through-space and through-bond coupling in p-benzyne.73,74
SUBSTITUTED ARYNES |
759 |
This simple MO diagram forms the basis for understanding the structure and electronic properties of several derivatives of 28, which will become clear in the following sections. As in the case of m-benzyne, however, many aspects of p-ben- zyne chemistry are still poorly understood. The stereoelectronic factors influencing the thermal Bergman cyclization are a subject of ongoing and intense research;1,75 the photochemical cycloaromatization is even more difficult to rationalize. First attempts to investigate the intermolecular chemistry of 28 and its derivatives have been described only recently.76 Thus, p-benzynes will continue to fascinate theorists and experimentalists alike.
3. SUBSTITUTED ARYNES
3.1. o-Benzynes
Numerous nucleophilic aromatic substitution reactions take place by an elimination–addition mechanism via intermediate formation of arynes (cf. Scheme 16.2).77 In order to understand the regioselectivity of these reactions it is essential to take substituent effects into account.78 Several substituted didehydrobenzenes have been investigated theoretically.26w,79 A systematization of substituent effects with respect to structure and reactivity of 4 is anything but trivial, since the substituents act on the s- as well as the p-system, resulting in a complex interplay of inductive and resonance effects.79 Nevertheless, substituent effects can often be understood qualitatively in terms of an increase or decrease of the contribution of zwitterionic structures to the resonance hybrid.26w,79 Generally, effects are significantly more pronounced in 2,3-arynes (polarization of the triple bond) than in the 3,4-didehydro isomers.79 The interpretation of experimental reactivity data is often complicated by the fact that the attack of approaching nucleophiles is not solely charge controlled.26w It appears that more systematic (experimental and computational) studies are necessary for an extensive understanding of substituent effects on o-benzyne (4). Experimental data (such as C C stretching vibrational frequencies or gas-phase structures) to compare the theoretical results with are rare. Nonetheless, some examples have been described in the literature and will be discussed in this chapter.
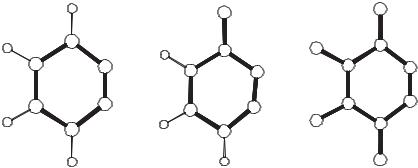
The perfluorinated o-benzyne (40) could be isolated in cryogenic matrices by photolysis of the corresponding phthalic anhydride.80 According to CASSCF calculations the singlet–triplet gap of tetrafluoro-o-benzyne is larger than that of 4 by several kilocalories per mole. The fluorine substituents only have a minor influence on the length of the C C bond. Interestingly, a nonplanar C2 symmetrical structure is found with DFT methods (BLYP, B3LYP) for the triplet state, whereas CASSCF predicts a planar C2v symmetrical geometry.80 In a careful matrix isolation study, Radziszewski et al.81 identified the C C stretching vibration of 40 at 1878 cm 1. Despite the similar C C bond lengths, the chemistry of 40 differs significantly from that of 4, the former being much more electrophilic and reactive. Thus, in contrast to 4, compound 40 reacts readily with thiophene.82
760 |
ARYNES |
|
|
|
|
|
|
|
|
|
|
|
F |
|
|
F |
|
|
|
|
|
|
|
|
|
|
|
140.7 |
137.9 |
140.0 |
|
137.8 |
|
139.8 |
136.9 |
|
141.9 C |
138.7 |
141.2 |
C |
138.6 |
F |
141.1 C |
137.8 |
|
|
|
|
|
|
|
||
|
C |
C |
C |
|
C |
|
C |
C |
140.1 |
|
124.1 |
|
140.9 |
124.1 |
|||
|
139.8 |
|
124.5 |
|||||
140.8 |
|
125.3 |
|
141.8 |
125.3 |
|||
|
140.5 |
|
125.6 |
|||||
|
C |
C |
|
|
C |
C |
||
|
C |
|
C |
|
||||
|
|
|
|
|
F |
|
|
|
|
C |
|
141.0 C |
136.7 |
C |
|
||
|
|
|
40 |
|
||||
|
4 |
|
142.2 |
|
137.5 |
|
|
|
|
|
|
41 |
|
|
|
F |
|
~ |
|
|
~ |
|
|
|
~ |
|
ν(C1C2) = 2017cm−1 |
ν(C1C2) = 1999cm−1 |
|
ν(C1C2) = 2022cm−1 |
|||||
~ |
|
|
~ |
|
|
|
~ |
|
ν(C1C2) = 1941cm −1 |
ν(C1C2) = 1925cm −1 |
|
ν(C1C2) = 1941cm −1 |
|||||
~ |
|
|
~ |
|
|
|
~ |
|
ν(C1C2) = 1846cm−1 |
ν(C1C2) = 1866cm−1 |
|
ν(C1C2) = 1878cm−1 |
|||||
Figure 16.2. Calculated structures and C C stretching vibrational frequencies of 4, 40, and
41. Normal print: B3LYP/cc-pVTZ data; in italics: BLYP/cc-pVTZ, bold print: experimental data.81,83a Bond lengths in picometers.
3-Fluoro-o-benzyne (41) has also been isolated in cryogenic matrices by photolysis of the corresponding phthalic anhydride. The C C stretching vibration of 41
has been observed at 1866 cm 1.81 Compared to the parent, the triple bond length is only modestly increased according to DFT calculations (Fig. 16.2).81,83 Note, how-
ever, that the observed frequency shifts upon fluorine substitution in 4 are not reproduced at the B3LYP or BLYP level of theory (Fig. 16.2).81,83 Thus, the performance
of different DFT functionals to account for the properties of o-arynes still remains a matter of debate,26y and casts some doubts on the results of certain DFT studies.
3.2. m-Benzynes
Substituted m-arynes have been extensively studied computationally.79 One important parameter for understanding substituent effects on the structure and reactivity
of 13 is the distance between the radical centers. Despite the flat potential between 13 and 18 (Section 2.2),44–46 the effect of substitution on the structure of m-benzyne
seems to be rather small.79 The data obtained with different theoretical methods could again be interpreted in terms of varying weights of mesomeric structures. Two resonance structures may be drawn for the bicyclic structure 18; apart from the anti-Bredt form 18A, a charge separated structure 18B contributes to the resonance hybrid. Substituents, which stabilize the cyclopropenyl cation or allyl anion unit are expected to favor the bicyclic structure and to decrease the distance between the radical centers.79 Accordingly, calculations show that donor substituents (NH2, SiH3) decrease, acceptor substituents (CN, NH3þ) increase the C2 C6 distance in 2,6-didehydro isomers slightly. In 2,4-isomers the substituents interact with a negative partial charge, and therefore donor substituents result in increased


762 ARYNES
|
H |
|
|
F |
|
|
F |
. |
. |
|
. |
. |
|
. |
. |
H |
H |
F |
|
F |
F |
|
F |
|
H |
|
|
F |
|
|
H |
|
13 |
|
|
42 |
|
|
43 |
|
F |
|
|
F |
|
|
F |
. |
. |
|
. |
. |
|
. |
. |
F |
F |
F |
|
F |
F |
|
F |
|
CF3 |
|
|
SiMe3 |
|
|
I |
|
44 |
|
|
45 |
|
|
46 |
Formula 16.3
a1 symmetrical vibration in the range 1765–1824 cm 1 and a b2 symmetrical ring stretching vibration between 1551 and 1605 cm 1. Substituents at C5 are hardly involved in these vibrations (as is evident, e.g., from the neglegible H/D shift between 43 and its 5-deuterated isotopomer). Therefore changes in the frequencies arise mainly from electronic effects of the substituents on the ring system. Thus, these vibrations may be used as a probe for substituent effects on 2-fluoro-m- benzynes.85
In summary, substituent effects on the structure and electronic properties of m- benzyne are rather small in many cases. One aspect of forthcoming investigations may be the search for derivatives of 13, in which the distance of the radical centers is reduced drastically, which should lead to markedly different properties and reactivity.
3.3. p-Benzynes
Experimental data on substituted p-benzynes are rare. Only the perfluorinated derivative 47 has been the subject of a recent matrix isolation study. Irradiation of 1,4- diiodotetrafluorobenzene (48) in solid neon at 3 K generates p-didehydrotetrafluoro- benzene (47) in good yield.86 According to calculations, this derivative has a singlet ground state. However, the lowest triplet state is only 0.5 kcal/mol higher in energy (CASPT2/cc-pVDZ). Definitive experimental evidence for the ground state of 47 is not available yet, since the calculated IR spectra of the singlet and triplet states are essentially identical, and an ESR spectrum of the photolysis products in solid neon could not be measured so far.86 Interestingly, longer wavelength irradiation (260– 320 nm) converts 47 into the ring-opened 1,3,4,6-tetrafluorohex-3-ene-1,5-diyne (49, Scheme 16.16). This observation is particularly remarkable, since the high

SUBSTITUTED ARYNES |
763 |
|
I |
|
|
I |
|
F |
F |
254 nm |
F |
|
F |
Ne, 3 K |
|
||||
|
|
|
|
|
|
F |
F |
Ne, 7 K |
F |
. |
F |
|
I |
|
|
|
|
|
48 |
|
|
|
|
|
|
|
254 nm |
|
Ne, 7 K |
|
|
|
Ne, 3 K |
|
|
|
|
|
|
|
|
|
F |
|
|
. |
|
F |
|
260−320 nm |
F |
F |
|
|
|
Ne, 3 K |
|
. |
|
F |
|
|
F |
F |
|
|
|
|
|
|
|
|
F |
|
|
47 |
|
|
49 |
|
|
|
Scheme 16.16. Photochemistry of 48 in a neon matrix at 3 K.86
electronegativity of the fluorine substituents destabilizes the enediyne relative to the biradical, so that the ring opening of 47 is endothermic by 8 kcal/mol [UB3LYP/6- 311þþG(d,p)], and the barrier for retro-Bergman cyclization is significantly increased (37.5 kcal/mol) relative to that of the parent system 28.86
Substituent effects on geometry and singlet–triplet splitting in p-didehydro- arenes were investigated in several theoretical studies and can in most cases be rationalized in terms of amplification or attenuation of through-bond coupling. Complete H/F exchange leads to an increase in EST for the o- and m-benzyne, but decreases the energy gap for the para-derivative 47. This effect has been
explained by a stabilization of the bonding MO (symmetric combination of the nonbonding orbitals, S) by interaction with s*(C F) orbitals.86,87 As shown in
Figure 16.3, this interaction increases the gap between the highest occupied (HOMO) and lowest unoccupied molecular orbitals (LUMO) for 40 and 42, but decreases the orbital energy difference in 47. (Note, however, that the relationship between state energy differences and orbital energy differences is all but trivial.) A similar argumentation has been used by Cramer and Johnson79 to rationalize the calculated singlet–triplet splittings of several singly substituted p-benzynes. For s-withdrawing substituents (X ¼ NH2, CN, OH), it has been shown that the reduction of EST is the result of a stabilization of the triplet state rather than destabilization of the singlet ground state according to the calculated biradical stabilization energies (BSE). The MO derived from symmetric combination of the nonbonding orbitals (S), which is empty in the singlet, but singly occupied in the triplet state, is stabilized by mixing with the empty s*(Cipso X) orbital in all cases.79


 − CO, CO
− CO, CO O
O
766 ARYNES
Several naphthynes have already been investigated in the early 1970s. Gru¨tzmacher and Lohmann90 generated some derivatives by pyrolysis of different precursors and measured the ionisation potentials of the pyrolysis products. At about the same time, Lohmann91 investigated the photochemistry of the isomeric naphthalenedicarboxylic anhydrides 61 and 62 using LFP and found that dimerization of the 2,3- naphthyne (57) is significantly faster than that of 4. The 1,2-isomer 50, on the other hand, hardly dimerizes at all.
Matrix isolation of 2,3-didehydronaphthalene (57) was described for the first time by Weltner and co-workers92 and has been the subject of a recent investigation by Sato et al.93 Intermediates 50 and 57 are accessible photolytically from the anhydrides 61 and 62, respectively, when the photolysis conditions are selected carefully.93 Noteworthy again is the difference of both isomers compared with 4. Whereas—in analogy to the parent system (cf. Scheme 16.4)–50 is photochemically carbonylated to cyclopentadienylideneketene (63) (albeit irreversibly, in contrast to 4), photocarbonylation of 57 to 64 is not observed at all (Scheme 16.17).93 This finding was attributed to the different stability of the ketenes. The C C stretching vibrations of 50 and 57 could not be detected experimentally because of their low intensity. The calculated frequencies (4: 1953 cm 1; 50: 1994 cm 1; 57: 1922 cm 1; B3LYP/6-31G**) follow the same trend as the bond lengths (4: 125.1 pm; 50: 124.1 pm; 57: 126.0 pm; B3LYP/6-31G** ).
¨ 90b
In their early contribution, Grutzmacher and Lohmann identified diethynyl-
benzene (65) instead of 1,3-didehydronaphthalene (51) as the pyrolysis product of 1,3-dinitro- and 1,3-dibromonaphthalene, based on the high ionization potential measured (8.96 0.02 eV). Obviously, a rearrangement analogous to the ring opening of m-benzyne takes place for the annellated derivative as well (Schemes 16.10 and 16.18).
Br |
. |
NO2 |
|
|
|
900 °C |
900 °C |
|
− 2 Br |
. − 2 NO2 |
NO2 |
Br |
|
|
|
51 |
|
|
900 °C |
|
65
Scheme 16.18. Pyrolysis with mass spectrometric detection of 1,3-dibromo- and 1,3- dinitronaphthalene indicates that 51 is not thermally stable at 900 C. The measured ionization potential suggests that the ring opened product 65 is formed under these conditions (cf. Scheme 16.10).90b

ANNELLATED ARYNES |
767 |
A derivative of 1,3-naphthyne was postulated by Billups et al.94 as an intermediate of the dehydrohalogenation of the dichlorocarbene adduct of 2-bromoindene with potassium tert-butoxide; however, the complex product mixture did not allow the reaction mechanism to be conclusively elucidated (Scheme 16.19).
Cl |
. |
|
|
H |
|
|
Cl |
||
Cl |
|
Cl |
||
|
|
|
||
|
|
|
|
|
Br |
|
. |
|
H |
|
|
|
||
|
|
|
|
(7%) |
t-BuOK THF |
|
|
|
+ 9 additional products |
|
|
|
|
|
Cl |
|
|
|
|
Br |
|
|
Cl |
|
or Cl
Cl
Scheme 16.19. 2-Chloro-1,3-didehydronaphthalene was postulated as an intermediate of a complex dehydrohalogenation reaction by Billups et al.94 (Tetrahydrofuran ¼ THF.)
In a broader sense, 1,8-naphthyne (56) may also be regarded as a derivative of m- benzyne, since the radical centers are separated by the same number of bonds as in 51.
. |
. |
. |
|
. |
|
51 |
|
56 |
Formula 16.4
However, whereas 1,3-naphthyne (51) displays a strong preference for the singlet state (17.3 kcal/mol, comparable to that of m-benzyne at the same level of theory), singlet and triplet states are almost degenerate in the 1,8-isomer ( EST ¼ 0.9 kcal/ mol).88 The ionization potential (IP) of 56 (7.92 eV) is lower than that of naphthalene (8.26 eV), while the IPs of most other naphthynes are higher.90 Attempts to generate 56 by irradiation of 1,8-naphthalenedicarboxylic anhydride in an argon matrix were unsuccessful.95
The properties of the benzannellated p-benzyne (52) are very similar to that of the parent system 28. Both vicinal C C bonds are elongated due to through-bond coupling of the unpaired electrons (see Fig. 16.9, Section 7 for a calculated