

Reactive Intermediate Chemistry
.pdf
638 |
NITRENIUM IONS |
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
H |
|
|
|
|
|
|
|
|
|
|
N |
|||
|
|
N |
||||||||
|
|
|
|
H |
|
|
||||
|
|
|
|
|
|
|||||
|
|
|
|
Ph |
|
|
||||
|
|
|
|
|
|
|||||
|
136 |
137 |
|
66 |
||||||
|
1637 |
1625 |
|
1604 |
||||||
|
1607 |
1607 |
|
1525 |
||||||
|
1567 |
1496 |
|
1478 |
||||||
|
1486 |
1422 |
|
1455 |
||||||
Figure 13.67. Selected Raman bands (in cm 1) for arylnitrenium ions.
Phillips and co-workers188 introduced the technique of TRRR to these studies. They reported detailed TRRR studies and DFT calculations on 2-fluorenynitrenium ion (136), the 4-biphenylylnitrenium ion (137), and the N,N-diphenylnitrenium ion (66, Fig. 13.67). The former two nitrenium ions were generated through azide photolysis in MeCN–H2O mixtures, and the latter nitrenium ion was generated via the N-aminopyridinium ion route. Some of the major Raman bands measured for these species appear in Figure 13.67. As with the TRIR experiments, excellent agreement is obtained between measured and DFT-calculated values.
5.4. Direct Detection of Intermediates in Nitrenium Ion Reactions
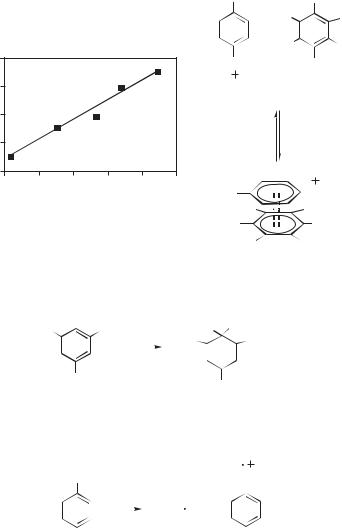
The LFP studies of the reaction of the N-methyl-N-4-biphenylylnitrenium ion with a series of arenes showed that no detectable intermediate formed in these reactions.162 The rate constants of these reactions correlated neither with the oxidation potentials of the traps (as would be expected were the initial step electron transfer) nor with the basicity of these traps (a proxy for their susceptibility toward direct formation of the sigma complex). Instead, a good correlation of these rate constants was found with the ability of the traps to form p complexes with picric acid (Fig. 13.68). On this basis, it was concluded the initial step in these reactions was the rapid formation of a p complex (140) between the nitrenium ion (138) and the arene (139). This was followed by s-complex formation and tautomerization to give adducts, or a relatively slow homolytic dissociation to give (ultimately) the parent amine.
In a subsequent study using diphenylnitrenium ion, several intermediates were detected. With 1,3,5-trimethoxybenzene or 1,3-dimethoxybenzene, the decay of the nitrenium ion occurred concurrently with the appearance of sigma adducts (141, Fig. 13.69).168 These were characterized on the basis of their absorption maxima and their behavior toward pyridine bases. On the other hand, when readily oxidized arenes, such as N,N-dimethylaniline were employed, the characteristic ion radicals were detected (Fig. 13.70).189
SPECTROSCOPIC AND KINETIC STUDIES OF NITRENIUM IONS |
639 |
|||||
|
Ph |
|
|
|
R1 |
|
|
|
|
|
R6 |
R2 |
|
Log k (trap) |
|
|
|
|
|
R3 |
|
|
|
R5 |
|||
|
|
|
|
|||
7.5 |
|
|
|
|
NMe |
R4 |
|
|
|
|
|
||
6.5 |
|
|
|
|
138 |
139 |
|
|
|
|
|
||
5.5 |
|
|
|
|
|
|
4.5 |
|
|
|
|
|
|
3.5 |
|
|
|
|
|
|
−0.2 |
0 |
0.2 |
0.4 |
0.6 |
0.8 |
NMe |
|
|
log K (picric acid) |
|
Ph |
||
|
|
|
R6 |
R1 |
||
|
|
|
|
|
||
|
|
|
|
|
R5 |
R2 |
|
|
|
|
|
R4 |
R3 |
|
|
|
|
|
|
140 |
Figure 13.68. Arene trapping correlates with p complexation.
|
|
|
|
MeO |
|
OMe |
|
H |
|
|
|
|
NPh2 |
||
Ph2N+ |
|
|
|
|
|
|
|
MeO |
|
|
|
|
|
|
OMe |
|
|
|
|
|
|
OMe |
|
|
|
|
|
|
|
|
λmax = 330 nm |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
OMe |
|||||
|
|
|
|
|
|
|
|
|
|
141 |
|
||||
Figure 13.69. Sigma complex detected with diphenylnitrenium ion.
|
|
|
|
|
|
NMe2 |
|
|
|
|
|
|
|
NMe2 |
Ph2N + |
|
|
|
|
|
|
|
Ph2N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
λmax = 660 nm |
λmax = 460 nm |
|||||
Figure 13.70. Radicals detected from electron-transfer reaction.
These results lead to a general mechanism for the reaction nitrenium ions with aromatic compounds (Fig. 13.71). Initial encounter leads to a p-complex (141). The latter is converted into isomeric s complexes (142–144) which, in turn, either tautomerize to give stable adducts (145–147) or else dissociate to give radicals. The relative rates of these processes depend on the reactivities of the nitrenium ion and the arene. With less reactive nitrenium ions the p-complex is relatively long lived. With more reactive nitrenium ions the p complex forms in a low steady-state
 NR' R
NR' R
 H
H


 N
N
THE ROLE OF ARYLNITRENIUM IONS IN DNA DAMAGING REACTIONS |
641 |
O
NN NH
NH
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
N NH2 |
|
|||||||
|
|
||||||||||||
|
|
|
|
R |
|
|
|
|
|
||||
|
O |
|
|
|
|
O |
|||||||
|
|
H |
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||
N |
NH |
|
|
N |
NH |
||||||||
|
|
||||||||||||
|
|
||||||||||||
|
N |
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
N |
N NH2 |
N |
N N |
|
|
|
|||||||||
|
|
|
R |
|
|||||||||
R |
H |
|
|
|
|
||||||||
|
|
|
|
|
|||||||||
NH2 |
|
|
|
|
|
||||||||
|
|
|
|
|
|
||||||||
|
148 |
|
|
|
|
|
|
|
149 |
|
|||
Figure 13.72. Reaction of naphthylnitrenium ion with guanosine.
Boche and co-worker190 carried out detailed studies of the decomposition reactions of various N-hydroxylamine esters in the presence of DNA and deoxyguanosine, and characterized the adducts that resulted from these reactions. Thus, the nitrenium ion derived from O-acetyl-N-(2-aminofluorene) added to DNA to give an adduct joining the C8 position of the base with the nitrenium ion nitrogen. Similar experiments carried out with the precursor of 2-naphthylnitrenium ion gave N2ortho 148 and C8–N adducts 149 as shown in Figure 13.72.96
With regard to DNA damage, the most extensively studied arylnitrenium ions are the 2-fluorenenylnitrenium ion and the 4-biphenylylnitrenium ion. The toxicity of these species is explained by the fact that they are relatively long lived in water (>1 ms) yet react with DNA components at nearly the diffusion limit. Novak and Kennedy191 carried out extensive competitive trapping studies using N-acetyl-4- biphenylylnitrenium ion with various nucleosides. The pyrimidines, cytidine, uracidine, and thymidine did not show any reaction with this nitrenium ion (i.e., reaction with water was faster). On the other hand, gaunosine and its derivatives trapped this nitrenium ion with rate constants in excess of 109 M 1 s 1. Other purines, adenine, and inosine showed about a tenth of the reactivity of guanosine.
McClelland used LFP methods to measure rate constants for the reactions of
various substituted 4-biphenylylnitrenium ions with deoxyguanosine and found that they fell mostly in the narrow range of 1.0–2.2 109 M 1 s 1.129,131,183 An
exception was the highly stabilized 40-methoxy derivative that was trapped at 3:6 107 M 1 s 1. The 2-fluorenyl derivatives also showed somewhat attenuated reactivity, with rate constants with deoxyguanosine of 2–9 108 M 1 s 1.131
With free bases or nucleosides in solution, the main adduct arises from a net coupling of the C8 on gaunosine with the nitrenium ion nitrogen. This result is interesting for two reasons. First, most reactive nucleophiles tend to add to the ring carbons of the arylnitrenium ion rather than the nitrogen. In some cases, highly reactive arenes have been observed to give a mixture of regioisomers, including N adducts;



644 NITRENIUM IONS
proteins. The nitrenium ion 156 is trapped by deoxyguanosine to give a C8 adduct (157) analogous to those observed for the carbocyclic nitrenium ions. An adduct connecting the guanine’s N2 with the ring carbon of the heterocycle (158) is also observed. Interestingly, this nitrenium ion also decays, presumably through hydride abstraction, to give the 155, along with some dimeric products. It was also found that similar reactions could occur through the less reactive conjugate base of this nitrenium ion.
7. CONCLUSION AND OUTLOOK
The past two decades have witnessed remarkable progress in the understanding of nitrenium ions. It is now widely agreed that these intermediates have a discrete existence. Several examples have been detected and characterized by direct means, including LFP. Numerous decay pathways for singlet-state nitrenium ions have been established both through product studies as well as by direct detection. The vast majority of studies have focused on the behavior of singlet arynitrenium ions. In a few cases, the chemical reactions of triplet arylnitrenium ions have been characterized, although few triplets have been studied by direct means. More information about the reactivity of alkyl and other non-arylnitrenium ions would be desirable.
Density functional-based theoretical methods have been demonstrated to give highly accurate predictions of nitrenium ions structures, singlet–triplet energy gaps, and vibrational spectra. Future challenges in the area of theoretical predictions include the accurate modeling of nitrenium ion reactions and reaction rates.
More recently, time-resolved vibrational spectroscopy has been applied to nitrenium ions. These techniques hold much promise for future research as they can provide more detailed structural information about nitrenium ions. In addition, the vibrational frequencies measured in this way can be used to assess the accuracy of theoretical calculations.
Nitrenium ions have been applied to the synthesis of macrocycles and other medicinally interesting compounds. The most successful reactions have been in cases where the nitrenium ion is covalently tethered to its intended target. Further efforts aimed at modulating the selectivity of these intermediates would increase their synthetic utility.
ADDITIONAL READING
R.A. Abramovitch and R. Jeyaraman, ‘‘Nitrenium Ions,’’ in Azides and Nitrenes, E. F. V. Scriven, Ed., Academic Press, New York 1984, p. 297.
M. Novak and S. Rajagopal, ‘‘N-Arylnitrenium Ions,’’ Adv. Phys. Org. Chem. 2001, I36, 167.
R.A. McClelland, ‘‘Flash Photolysis Generation and Reactivities of Carbenium Ions and Nitrenium Ions,’’ Tetrahedron, 1996, 52, 6823.
REFERENCES 645
D. E. Falvey, ‘‘Photochemical Generation and Studies of Nitrenium Ions,’’ in Organic, Physical and Materials Photochemistry, V. Ramamurthy and K. S. Schanze, Eds., Marcel Dekker, New York, 2000, pp. 249–284.
F.F. Kadlubar, ‘‘DNA Adducts of Carcinogenic Amines,’’ in DNA Adducts Identification and Biological Significance, K. Hemmink, A. Dipple, D. E. G. Shugar, F. F. Kadlubar, D. Segerback, and H. Bartsch, Eds., University Press, Oxford, UK, 1994, pp. 199–216.
P. G. Gassman, ‘‘Nitrenium Ions,’’ Acc. Chem. Res. 1970, 3, 26.
REFERENCES
1.C. J. Cramer, F. J. Dulles, and D. E. Falvey, J. Am. Chem. Soc. 1994, 116, 9787.
2.A. P. Dicks, A. Ahmad, R. D’Sa, and R. A. McClelland, J. Chem. Soc. Perkin Trans. 2
1999, 1.
3.R. H. Wiley and J. Moffat, J. Am. Chem. Soc. 1955, 77, 1703.
4.G. Boche, P. Andrews, K. Harms, M. Marsch, K. S. Rangappa, M. Schimeczek, and
C.Willecke, J. Am. Chem. Soc. 1996, 118, 4925.
5.M. Robert, A. Neudeck, G. Boche, C. Willecke, K. S. Rangappa, and P. Andrews, New J. Chem. 1998, 1437.
6.H.-W. Wanzlick and E. Schikora, Chem. Ber. 1961, 94, 2389.
7.A. J. Arduengo, III, R. L. Harlow, and M. Kline, J. Am. Chem. Soc. 1991, 113, 361.
8.R. A. Abramovitch and B. A. Davies, Chem. Rev. 1964, 64, 149.
9.P. T. Lansbury, in Nitrenes, W. Lwowski, Ed., Wiley-Interscience, New York, 1970, pp. 405–418.
10.P. G. Gassman, Acc. Chem. Res. 1970, 3, 26.
11.R. A. McClelland, Tetrahedron 1996, 52, 6823.
12.D. E. Falvey, J. Phys. Org. Chem. 1999, 12, 589.
13.D. E. Falvey, in Organic, Physical, and Materials Photochemistry, V. Ramamurthy and
K.Schanze, Eds., Marcel Dekker, New York, 2000, pp. 249–284.
14.M. Novak and S. Rajagopal, Adv. Phys. Org. Chem. 2001, 36, 167.
15.R. A. Abramovitch and R. Jeyaraman, in Azides and Nitrenes: Reactivity and Utility,
E.F. V. Scriven, Ed., Academic Press, Orlando, FL, 1984, pp. 297–357.
16.F. F. Kadlubar, in DNA Adducts Identification and Biological Significance, K. Hemmink,
A.Dipple, D. E. G. Shugar, F. F. Kadlubar, D. Segerback, and H. Bartsch, Eds., University Press, Oxford, UK, 1994, pp. 199–216.
17.R. C. Garner, C. N. Martin, and D. B. Clayson, in Chemical Carcinogens, Vol. 1, 2nd ed.,
C.E. Searle, Ed., American Chemical Society, Washington, DC, 1984, pp. 175–276.
18.J. A. Miller, Cancer Res. 1970, 30, 559.
19.G. R. Hoffman and R. P. P. Fuchs, Chem. Res. Toxicol. 1997, 10, 347.
20.H. A. J. Schut and E. G. Snyderwine, Carcinogenesis 1999, 20, 353.
21.R. A. Abramovitch, H. H., Gibson, Jr., T. Nguyen, S. Olivella, and A. Sole´, Tetrahedron Lett. 1994, 35, 2321.
22.R. A. Abramovitch, J. M. Beckert, P. Chinnasamy, H. Xiaohua, W. Pennington, and
A.R. V. Sanjivamurthy, Heterocycles 1989, 28, 623.
646NITRENIUM IONS
23.Y. Kikagawa and M. Kawase, J. Am. Chem. Soc. 1984, 106, 5728.
24.R. A. Abramovitch, X. Ye, W. T. Pennington, G. Schimek, and D. Bogdal, J. Org. Chem. 2000, 65, 343.
25.D. J. Wardrop and A. Basak, Org. Lett. 2001, 3, 1053.
26.D. J. Wardrop and W. Zhang, Org. Lett. 2001, 3, 2353.
27.E. M. Genies and M. Lapowski, J. Electroanal. Chem. 1987, 236, 189.
28.G. Zotti, N. Comisso, G. D’Aprano, and M. Leclerc, Adv. Mater. 1992, 4, 749.
29.R. Holze, Collect. Czech. Chem. Commun. 2000, 65, 899.
30.Y. Wei, R. Hariharan, and S. Patel, Macromolecules 1990, 23, 758.
31.W. Liu, J. Kumar, S. Tripathy, K. J. Senecal, and L. Samuelson, J. Am. Chem. Soc. 1999, 121, 71.
32.W. Liu, A. L. Cholli, R. Nagarajan, J. Kumar, S. Tripathy, F. F. Bruno, and L. Samuelson,
J. Am. Chem. Soc. 1999, 121, 11345.
33.Y. Ding, A. B. Padias, and H. K., Hall, Jr., J. Polym. Sci. Part A: Chem. 1999, 37, 2569.
34.F. Lux, Polymer Rev. 1995, 35, 2915.
35.Y. Wei, X. Tang, and Y. Sun, J. Polym. Sci. Part A: Chem. 1989, 27, 2385.
36.E. Bamberger, Chem. Ber. 1894, 27, 1548.
37.E. Bamberger and J. Lagutt, Chem. Ber. 1898, 31, 1500.
38.E. Bamberger, Liebigs Ann. Chem. 1925, 441, 297.
39.H. E. Heller, E. D. Hughes, and C. K. Ingold, Nature (London) 1951, 168, 909.
40.P. G. Gassman and R. L. Cryberg, J. Am. Chem. Soc. 1969, 91, 2047.
41.P. G. Gassman and R. L. Cryberg, J. Am. Chem. Soc. 1969, 91, 5176.
42.P. G. Gassman and G. A. Campbell, J. Am. Chem. Soc. 1971, 93, 2567.
43.P. G. Gassman, G. A. Campbell, and R. C. Frederick, J. Am. Chem. Soc. 1972, 94, 3884.
44.P. G. Gassman and G. A. Campbell, J. Am. Chem. Soc. 1972, 94, 3891.
45.C. H. Gudmundsen and W. E. McEwen, J. Chem. Soc. 1957, 79, 329.
46.P. G. Gassman, K. Uneyama, and J. L. Hahnfeld, J. Am. Chem. Soc. 1977, 99, 647.
47.J. C. Koziar and D. O. Cowan, Acc. Chem. Res. 1978, 11, 334.
48.N. J. Turro, Modern Molecular Photochemistry, Benjamin/Cummings: Menlo Park, CA, 1978.
49.R. V. Hoffman and D. J. Poelker, J. Org. Chem. 1979, 44, 2364.
50.R. V. Hoffman, A. Kumar, and G. A. Buntain, J. Am. Chem. Soc. 1985, 107, 4731.
51.R. V. Hoffman and N. B. Christophe, J. Org. Chem. 1988, 53, 4769.
52.U. Svanholm and V. D. Parker, J. Am. Chem. Soc. 1974, 96, 1234.
53.D. Serve, J. Am. Chem. Soc. 1975, 97, 432.
54.P. Haberfield and M. DeRosa, J. Chem. Soc. Chem. Commun. 1985, 1716.
55.V. J. Barclay, I. P. Hamilton, and P. Jensen, J. Chem. Phys. 1993, 99, 9709.
56.C. J. Cramer, F. J. Dulles, J. W. Storer, and S. E. Worthington, Chem. Phys. Lett. 1994, 218, 387.
57.O. Dopfer, D. Roth, and J. P. Maier, Chem. Phys. Lett. 1999, 310, 201.
58.C. Gonzales, A. Restrepo-Cossio, M. Ma´rquez, K. B. Wiberg, and M. D. Rosa, J. Phys. Chem. A. 1998, 102, 2732.