

Reactive Intermediate Chemistry
.pdf526 NITRENES
phenylcarbene reacts more readily with electron pairs than does singlet phenylnitrene. The empty orbital of phenylcarbene can coordinate with pairs of electrons and react rapidly with C H bonds. This type of interaction and facile insertion is not possible with singlet phenylnitrene, which has an open-shell singlet state. Basically, phenylcarbene is a strong Lewis acid, whereas singlet phenylnitrene is not.
H |
pπ |
|
|
Ph |
pσ |
|
|
|
× |
H R |
H R |
The UV–vis spectrum of triplet phenylnitrene, obtained by brief photolysis of phenyl azide in glassy ether–pentane–alcohol is shown in Figure 11.2.
Pople–Parr–Pariser (PPP) calculations in 1986 indicated that the visible band of triplet phenylnitrene at 500 nm is the result of promotion of an electron from the
0.9 |
|
|
|
|
|
0.04 |
|
0.6 |
|
|
Oscillator strength |
Absorbance |
|
0.02 |
|
|
|
||
0.3 |
|
|
|
|
|
fx10 |
|
0.0 |
|
0.00 |
|
|
|
|
|
300 |
400 |
500 |
|
Wavelength (nm)
Figure 11.2. The absorption spectrum of triplet phenylnitrene in EPA glass at 77 K. The computed positions and oscillator strengths (f, right-hand axes) of the absorption bands are depicted as solid vertical lines. For very small oscillator strength, the value multiplied by 10 is presented ( f 10). [Reproduced with permission from N. P. Gritsan, Z. Zhu, C. M. Hadad, and M. S. Platz, J. Am. Chem. Soc. 1999, 121, 1202. Copyright # 1999 American Chemical Society.]

|
PHENYLNITRENE AND SIMPLE DERIVATIVES |
527 |
||
TABLE 11.1. Maxima (in nm) of the Most Intense Absorption |
|
|||
Bands in the Electronic Absorption Spectra of Substituted |
|
|||
Singlet and Triplet Phenylnitrenes (near UV and vis) |
|
|||
|
|
|
|
|
Substituent |
Singlet Nitrene |
Triplet Nitrenea |
|
|
|
|
|
|
|
4-F |
365 |
a |
|
|
4-Cl |
360 |
a |
|
|
4-Br |
361 |
a |
|
|
2,4,6-triBr |
395 |
326, 340 |
|
|
4-I |
328 |
a |
|
|
4-Me |
365 |
315 |
|
|
4-CF3 |
320 |
a |
|
|
4-COMe |
334 |
a |
|
|
4-Ph |
345 |
320 |
|
|
4-(40-azidophenyl) |
380 |
a |
|
|
2-Me |
350 |
a |
|
|
2,6-diMe |
350 |
297, 310 |
|
|
2,4,6-triMe |
348, 366 |
319 |
|
|
2-F |
342 |
294, 315 |
|
|
2,6-diF |
331, 342 |
313 |
|
|
2,3,4,5,6-pentaF |
330 |
315 |
|
|
2-CN |
382 |
328 |
|
|
2,6-diCN |
385, 405 |
341 |
|
|
a Spectrum was not detected—a.
singly occupied ps-AO into the p* system.86 The transition from the sp lone pair on nitrogen to a p* orbital is obscured by a p ! p* transition. On the other hand, the sp ! p* and p ! p* transitions are very well separated in perfluorophenyl nitrene. The sp ! ps transition at 308 nm of triplet phenylnitrene, which is very characteristic of triplet nitrenes has been observed in NH (336 nm), MeN (317 nm), and CF3N (342, 347.5, 354 nm). More advanced calculations performed by Gritsan et al.94 are in excellent agreement with the experimental results for phenylnitrene. Table 11.1 lists the absorption maxima of several triplet arylnitrenes.
In 1997, Karney and Borden68 predicted that the rearrangement of 35s to ketenimine 30 proceeds in two steps, via the azirine intermediate 29. The initial reaction should be thought of as simply the cyclization of a 1,3 biradical as discussed for singlet vinylnitrene. Singlet phenylcarbene is not a biradical, and therefore has a larger barrier to cyclization than does singlet phenylnitrene. Because the bimolecular reactions of singlet phenylcarbene are very fast in solution, its thermal cyclization to a benzocyclopropene (see the discussion on the interconversion of pyridylcarbene and phenylnitrene, Section 5.9) occurs only in the gas phase at high temperature.95
Since the ring opening of 29 to the didehydroazepine 30 is very rapid, the first step of the unimolecular rearrangement of singlet phenylnitrene 33s is rate determining. The calculated potential surface is shown in Figure 11.3. Thus, it is not surprising that 29 has so far evaded spectroscopic detection although the IR


PHENYLNITRENE AND SIMPLE DERIVATIVES |
529 |
Schrock and Schuster98 confirmed and extended this work using LFP techniques in 1984. Schuster and co-workers99 later employed flash photolysis with IR detection to confirm that ketenimine 30 is the reactive intermediate present on the microsecond time scale.
In 1997, two groups simultaneously reported that LFP of phenyl azide 32 or
compounds 34 and 35 produces a previously undetected transient with lmax ¼ 350 nm and a lifetime of 1 ns at ambient temperature.100,101 The transient decays
at the same rate that cyclic ketenimine 30 is formed, implying that the newly detected transient is singlet phenylnitrene 33s.
O |
|
O |
C |
|
N S |
N |
|
|
|
Me |
|
|
|
|
34 |
35 |
Me |
|
Laser flash photolysis (266 nm) of phenyl azide in pentane at 233 K produces a transient absorption spectrum with two sharp bands with maxima at 335 and 352 nm (Fig. 11.4). Spectrum 1 was measured, point by point, 2 ns after the laser pulse. In later work, the spectrum of 33s was reinvestigated and an additional very weak, long wavelength absorption band at 540 nm was observed (Spectrum 2).94 The transient spectrum of Figure 11.4 was assigned to singlet phenylnitrene in its lowest open-shell electronic configuration (1A2).
The electronic absorption spectrum of 33s in the 1A2 state was calculated at the CASPT2 level and is in good agreement with the transient spectrum (Fig. 11.5). The two lowest excited singlet states of 33s are both of A1 symmetry and electronic transitions into these states are predicted to occur at 1610 and 765 nm. However, they are forbidden by electric dipole selection rules and thus neither of these transitions has been detected. In the absorption spectrum of 33s, the only intense absorption band is located 350 nm, 40 nm from the intense 308-nm band in 33t. This band has a long tail out to 450 nm and displays some fine structure that may be associated with the vibrations of the phenyl ring in 33s. The strongest absorption band in 33s, predicted at 368 nm by CASPT2, is due to the transition to the 21B1 exited state. The main configuration involved in this transition is similar to that of 23B1 state, that is, it arises by promotion of an electron from the sp lone-pair orbital on nitrogen to the singly occupied nitrogen ps orbital.
The electronic absorption spectra of 33s and 33t are very similar (Figs. 2 and 4), but all of the calculated and experimental bands of 33s exhibit a red shift compared to those of 33t. This result is very reasonable because both these species have very similar open-shell electronic configurations (3A2 and 1A2). The absorption maxima of several singlet arylnitrenes are given in Table 11.1.
5.3. Dynamics of Singlet Phenylnitrene
The decay of 33s was monitored in pentane at 350 nm over a temperature range of 150–270 K, which allowed direct measurement of kISC and determination of

530 NITRENES
|
0.20 |
|
|
|
|
|
0.15 |
1 |
|
|
|
|
|
|
0.03 |
|
|
Absorbance |
0.10 |
|
|
0.02 |
Oscillator strength |
|
|
|
|
||
|
0.05 |
|
|
0.01 |
|
|
|
|
|
2 |
|
|
|
|
|
x10 |
|
|
0.00 |
|
|
|
|
|
300 |
400 |
500 |
600 |
|
Wavelength (nm)
Figure 11.4. Transient spectrum of singlet phenylnitrene produced upon LFP of phenyl azide. Spectrum 1 was recorded 2 ns after the laser pulse (266 nm, 35 ps) at 233 K. Longwavelength band (2) was recorded with an optical multichanal analyzer at 150 K (with a 100-ns window immediately after the laser pulse, 249 nm, 12 ns). The computed positions and oscillator strengths ( f , right-hand axes) of the absorption bands are depicted as solid vertical lines. For very small oscillator strength, the value multiplied by 10 is presented ( f 10). [Reproduced with permission from N. P. Gritsan, Z. Zhu, C. M. Hadad, and M. S. Platz, J. Am. Chem. Soc. 1999, 121, 1202. Copyright # 1999 American Chemical Society.]
accurate barriers to cyclization. The lifetime of 33s at 298 K in CH2Cl2 was found to be 0.6 ns.94
The formation of the products [cyclic ketemimine (30) and/or triplet nitrene (33t)] was monitored at 380 nm. The decay of singlet phenylnitrene and the growth of the products are exponential and can be analyzed to yield an observed rate constant kOBS. An Arrhenius treatment of these data (open circles), is presented in Figure 11.5. The magnitude of kOBS decreases with decreasing temperature until170 K, whereupon it reaches a value of 3:2 106 s 1. Below this temperature, kOBS remains constant.94 The breakpoint in the Arrhenius plot is 180–200 K, which is in exactly the same temperature range in which the solution phase chemistry changes from the trapping of ketenimine 30 with diethylamine to the dimerization of 33t.86 Thus, the low-temperature data of Figure 11.5 were associated with kISC, the rate constant for intersystem crossing of singlet to triplet phenylnitrene, and the high temperature data with kR, the rate constant for rearrangement of 33t.
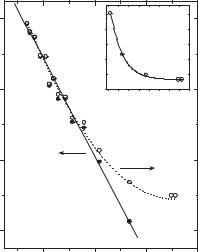
PHENYLNITRENE AND SIMPLE DERIVATIVES |
531 |
20 |
–1 |
0.02 |
|
|
|
|
s |
|
|
|
|
|
–9 |
|
|
|
|
|
, 10 |
0.01 |
|
|
|
|
OBS |
|
|
|
|
|
|
|
|
|
|
|
k |
|
|
|
|
18 |
|
0.00 |
|
|
|
) |
|
5.0 |
5.5 |
6.0 |
6.5 |
ISC |
|
||||
|
|
1000/T |
OBS |
||
-k |
|
|
|||
|
|
|
|
||
OBS |
|
|
|
|
k |
k |
|
|
|
|
ln |
ln ( |
|
|
|
|
|
16 |
|
|
|
|
|
14 |
|
|
|
|
|
|
4 |
5 |
6 |
|
7 |
|
|
1000/T |
|
|
|
Figure 11.5. Arrhenius treatment of the kOBS data (open circles) and kR data (filled circles) for singlet phenylnitrene deduced upon assuming that kISC is independent of temperature. Insert: temperature dependence of kOBS data. [Reproduced with permission from N. P. Gritsan, Z. Zhu, C. M. Hadad, and M. S. Platz, J. Am. Chem. Soc. 1999, 121, 1202. Copyright
# 1999 American Chemical Society.]
The temperature-independent rate constant observed at low temperature (3:2 106 s 1) is the rate constant for ISC to the triplet ground state (kISC). The rate constant kOBS is equal to kR þ kISC, where kR is the absolute rate constant for rearrangement for singlet phenylnitrene to benzazirine (29). As kOBS ¼ kR þ kISC, it is possible to deduce values of kR as a function of temperature and to obtain its associated Arrhenius parameters. Indeed, an Arrhenius plot of kR ¼ kOBS kISC is linear (Fig. 11.5, solid circles) with an activation energy for rearrangement of Ea ¼ 5:6 0:3 kcal/mol and preexponential factor A ¼ 1013:1 0:3 s 1.94
The calculated barrier for the cyclization of 33t to 29 is 6 kcal/mol after taking into consideration that the CASPT2 method overestimates the energy difference between openand closed-shell states by 3 kcal/mol.68 The correspondingly adjusted predicted 6 kcal/mol barrier is in nearly exact agreement with the experimental value (5:6 0:3 kcal/mol).94
5.4. Intersystem Crossing Rates (kISC)
Singlet phenylnitrene undergoes ISC three orders of magnitude more slowly than arylcarbenes.59,94 There are at least three reasons why arylcarbenes do ISC much
faster than singlet phenylnitrene. The rate of a radiationless transition increases as the energy separation between the two states goes to zero.102 The calculated

532 NITRENES
gas-phase singlet–triplet splitting of phenylcarbene (PC) is 2–4 kcal/mol;71 whereas in phenylnitrene it is 18 kcal/mol.91–93 Carbenes are divalent and have a bending mode with which to couple singlet and triplet surfaces, a vibration that is, of course, lacking in monovalent nitrenes. However, the most important factor is probably the electronic structure of the respective singlet intermediates. Singlet phenylcarbene has a closed-shell electronic structure with one filled and one empty nonbonding orbital. In such an ‘‘ionic’’ singlet, spin–orbit coupling (SOC) is a particularly effective mechanism of intersystem crossing.102–106 Singlet phenylnitrene, on the other hand, is an open-shell covalent singlet. The SOC is forbidden in this case and is ineffective in promoting ISC.
5.5. para-Substituted Derivatives of Phenylnitrene
Values of kISC for singlet para-substituted phenylnitrenes are given in Table 11.2.107 The ISC rate constant for singlet p-bromo phenylnitrene is about seven times larger than that of parent 33s and the p-fluoro and p-chloro analogues. This difference is easily attributable to a small heavy atom effect. The heavy atom effect of iodine is even larger than that of bromine, as expected, and raises the ISC rate by more than a factor of 20, relative to parent 33s.
A very large acceleration in kISC is observed with p-methoxy and dimethylamino
substituents (Table 11.2). This observation is consistent with the solution-phase photochemistry of p-methoxy and p-dimethylaminophenyl azides.107–111 The Me,
CF3, acetyl, fluoro, and chloro substituents are not sufficiently strong p donors or acceptors to significantly influence the size of kISC (Table 11.2), but the strong p
TABLE 11.2. Kinetic Parameters of Para-Substituted Singlet Arylnitrenes in Pentanea
Para-X |
t295K (ns) |
kISC ( 106 s 1) |
Ea (kcal/mol) |
Log A (s 1) |
||
H |
1 |
3.2 |
0.3 |
5.6 0.3 |
13.1 0.3 |
|
Me |
1 |
5.0 |
0.4 |
5.8 0.4 |
13.5 0.2 |
|
CF3 |
1.5 |
4.6 0.8 |
5.6 0.5 |
12.9 0.5 |
||
C(O)Me |
5.0 |
8 |
3 |
5.3 0.3 |
12.5 0.3 |
|
F |
0.3 |
3.5 |
1.4 |
5.3 0.3 |
13.2 0.3 |
|
Cl |
1 |
3.9 |
1.5 |
6.1 0.3 |
13.3 0.3 |
|
Br |
b3 |
17 4 |
4.0 b |
0.2 |
11.4 b 0.2 |
|
I |
<1 |
72 |
10 |
b |
|
b |
OMe |
>500 |
7.2 0.8 |
13.5 0.6 |
|||
CN |
8 4 |
6 |
2 |
|||
Ph c |
15 2 |
12 |
1 |
6.8 b |
0.3 |
12.7 b 0.3 |
NMed |
0.12 |
8300 |
200 |
b |
|
b |
NO2 |
<20 |
>50 |
|
|
|
|
aSee Ref. 95.
b
Not measured. cIn toluene. dIn benzene.

534 NITRENES
An o-cyano group has little influence on kISC, but two o-cyano groups slightly accelerate intersystem crossing.107–110 Singlet arylnitrenes with electronwithdrawing groups in the para-position have little influence on the rate constant of ISC.
An o-methyl group accelerates intersystem crossing relative to singlet p-tolyl nitrene. Two o-methyl groups are more effective than one at accelerating intersystem crossing. Singlet 2,4,6-trimethylphenylnitrene undergoes intersystem crossing about as readily as 2,6-dimethylphenylnitrene. These results are consistent with the general trend that electron donating groups (methyl, methoxy, dimethylamino) accelerate intersystem crossing. These groups increase the zwitterionic character of the singlet nitrene relative to the parent system, which facilitates a spin–orbit coupling mechanism of intersystem crossing.
5.7. Cyclization to Azirines
The cyclic ketenimine 30 is the major trappable reactive intermediate present in solution when phenyl azide (at moderate concentrations) is decomposed photolytically at 298 K. The rate of decay of singlet phenylnitrene 33s is equal to the rate of formation of the cyclic ketenimine. The first step, cyclization to benzazirine (29) is rate determining, and is followed by fast electrocyclic ring opening to cyclic ketenimine 30.
For most aryl azides, the rate constants of singlet nitrene decay and product formation (triplet nitrene and/or ketenimine) are the same. Thus, in all these phenylnitrenes cyclization to substituted benzazirines is the rate-limiting step of the process of isomerization to ketenimine, as is the case for the parent phenylnitrene. The only known exception, o-fluorophenylnitrene, will be discussed in the next section.
The activation parameters for cyclization of para-substituted singlet phenylnitrenes are presented in Table 11.2.107–110 It is readily seen from that table that polar
substituents such as para Me, CF3, halogen, and acetyl have little influence on kR.
This noneffect is not very surprising given that theory predicts that singlet phenylnitrene has an open-shell electronic structure.91,92,106 Cyclization of singlet phenyl-
nitrene only requires that the nitrogen atom bends out of the molecular plane, so
that the singly occupied s nonbonding molecular orbital (NBMO) can interact with the singly occupied p NBMO.68,106 Azirine formation may therefore simply
be regarded as the cyclization of a quinoidal 1,3-biradical, which originally has two orthogonal, antiparallel spins. Thus, polar effects are not anticipated.
Two para-substituents, phenyl112 and cyano depress kR and retard the rate of cyclization significantly (Table 11.2).110 p-Phenyl and p-cyano are both radical stabilizing substituents. These conjugative substituents reduce the spin density on the carbon ortho to the nitrene nitrogen. The reduced spin density at carbons ortho to the nitrogen lowers the rate at which the 1,3-biradical cyclizes. The effect with p-cyano and p-biphenyl singlet phenylnitrene is quite dramatic. The lifetimes of these singlet nitrenes at ambient temperature are 8 and 15 ns, respectively, and the activation barriers to cyclization are 7.2 and 6.8 kcal/mol, respectively.

PHENYLNITRENE AND SIMPLE DERIVATIVES |
535 |
Sundberg, et al.113 demonstrated that simple alkyl substituents direct cyclization away from the substituent.
N |
1N |
N |
Me slow H |
Me rapid |
Me |
H |
A single o- or p-methyl substituent has no influence on the rate of cyclization of the singlet tolylnitrene to the favored azirine.108 The methyl group has no bystander effect on benzazirine formation. Cyclization of 2,6-dimethylphenyl or 2,4,6-tri- methylphenylnitrenes necessarily proceeds toward a carbon bearing a substituent. A steric effect raises the barrier to cyclization by 1.5–2.0 kcal/mol, in excellent agreement with the predictions of Karney and Borden.114 The steric effect extends the lifetime of 2,6-dimethylphenylnitrene at ambient temperature to 13 ns in Freon-113 and of 2,4,6-trimethylphenylnitrene to 8 ns, in the same solvent (Table 11.4).108
A cyano group is a smaller substituent than methyl, thus cyclization toward and away from a cyano-substituted carbon should be more evenly balanced. Consistent with this hypothesis, Lamara et al.115 found that singlet o-cyanophenylnitrene (36s) undergoes ring expansion to afford not only 37, the product formed by cyclization away from the cyano substituent, but also 38, the product formed by cyclization toward the cyano group. Similar results have been found in the ring expansion of singlet o-acetylphenylnitrene.116
|
1N |
|
|
N |
H |
N |
|
CN |
|
CN |
|
CN |
||
|
H |
|
+ |
|
|
|
|
|
|
|
36s |
37 |
|
38 |
TABLE 11.4. Summary of Kinetic Results for Singlet
Methyl-Substituted Phenylnitrenesa
Substituent |
t(295) (ns) |
log A (s 1) |
Ea (kcal/mol) |
Solvent |
|
b |
13.2 0.2 |
5.8 0.4 |
|
4-Methyl |
1b |
C5H12 |
||
2-Methyl |
1 |
12.8 0.3 |
5.3 0.4 |
C5H12 |
2,6-Dimethyl |
12 1 |
13.0 0.3 |
7.0 0.3 |
C6H14 |
2,6-Dimethyl |
13 1 |
12.9 0.3 |
7.5 0.5 |
CF2ClCFCl2 |
2,4,6-Trimethyl |
8 1 |
13.4 0.4 |
7.3 0.4 |
CF2ClCFCl2 |
aSee Ref. 96.
bLifetime estimated by extrapolation of the data to 295 K.