

Molecular Heterogeneous Catalysis, Wiley (2006), 352729662X
.pdf326 Chapter 7
Figure 7.12. Proposed reaction mechanism for TNP hydrolysis catalyzed by lipophilic and hydrophilic zinc(II)–cyclen complexes[19].
It is important to remember that enzymes contain internally a hydrophobic cavity, but their exterior is hydrophilic, which makes the enzyme water soluble. This phaseproperty di erence has been mimicked by Kimura and Koike[19] to develop an artificial organophosphorus hydrolase. Their system is illustrated in Fig. 7.12.
The system is designed to hydrolyze tris(4-nitrophenyl)phosphate (TNP), an insecticide. Both hydrophobic and hydrophilic Zn2+–cyclen complexes were synthesized and tested for activity. The lipophilic Zn2+–cyclen complex forms a co-micellar phase with triton, which creates a hydrophobic environment near the catalyst reactive center. The hydrophobic center allows for the e cient attack at the reaction center, thus leading to substantially enhanced catalytic reactivity. The hydrophilic catalyst, on the other hand, prevents an e cient attack at the reaction center and hence shows no reactivity. The nearly 100-fold di erence in rate of the co-micellar catalyst system as compared with the aqueous system stems mainly from the solvability of TNP in the micellar phase and, to a partial extent, to the higher reactivity of Zn2+ due to the exclusion of H2O.
Additional reviews on biomimetic catalytic systems are available[20, 21]. Shilov[20] reviews transition-metal complex systems that have related activities to biocatalytic systems. The review by de Vos et al.[21] compares the reactivities of zeolite and layered hydroxide-based enzyme-mimicking systems.
7.6 Bio-Electrocatalytic and Chemocatalytic Reduction Reactions
7.6.1 Oxidation Catalysis
When the two oxygen atoms of O2 are incorporated into a product molecule, there must be a change in spin state. The reaction between triplet molecular oxygen and a singlet state
Mechanisms in Biocatalysis; Relationship with Chemocatalysis 327
reactant to produce singlet product molecules is spin forbidden. One way to overcome this spin forbidden event is if the reaction proceeds via free radicals. A transition-metal catalyst helps to overcome this spin forbidden event by an exchange of electrons between catalyst and oxygen molecules. Paramagnetic metal centers make the desired reaction towards singlet product molecules possible via a set of spin-conserving reaction steps. The spin state of the metal center is altered after reaction. A second major challenge in oxidation catalysis is to cleave the oxygen molecular bond. One has to distinguish between reactions in which one of the two oxygen atoms of the molecule is incorporated into the product molecule from reactions where both oxygen atoms are incorporated. In biocatalysis, enzymes that catalyze the former are called monooxygenases and those that catalyze the latter reaction are called dioxygenases. The monooxygenases use a co-reactant to remove the other oxygen atom so that the catalyst can be regenerated.
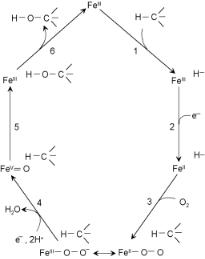
The heme-containing cytochrome P450 catalysts that hydroxylate CH bonds and are active as epoxidation catalysts are known to be monooxygenases. They utilize molecular oxygen as a source of two electrons to catalyze oxygen insertion into a CH bond. The overall electrocatalytic cycle for the oxidation reaction is shown in Scheme 7.1[22]
Scheme 7.1
Six steps occur in the cytochrome P450 reaction cycle. These steps appear to be universal for cytochromes P450 and are
1 Binding of substrate at the active site of low-spin hexacoordinate iron(III) form of the enzyme. This converts the low-spin Fe(III) into a high-spin pentacoordinate iron(III) enzyme. The substrate in Scheme 7.1 is represented simply by the C–H that is to be hydroxylated.
2 Addition of an electron to form the iron(II)-containing substrate form of the enzyme. 3 Addition of molecular oxygen to reduced the complex. Resonance forms exist for
ferrous-dioxygen and ferric-superoxide with the latter favored.
4Addition of a second electron. The addition of two protons to the O–O bond results in its cleavage and the subsequent elimination of water.
328 Chapter 7
5A radical-type hydrogen atom abstraction/oxygen rebound reaction occurs, the net e ect of which is hydroxylation and re-formation of the iron(III) form of the enzyme.
6 Product dissociates from the active site.
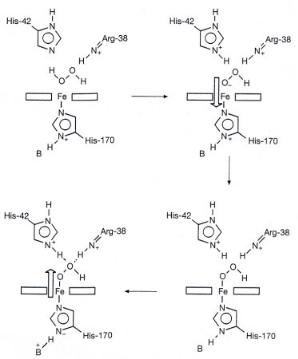
Biochemical oxidation is part of a catalytic system in which electron and proton transport steps are coupled together in the generation of the specific oxygen atom necessary to carry out the specific selective oxidation reaction. Reactions that require hydrogen peroxide will similarly incorporate only one of the oxygen atoms into the desired product; the coproduct is H2O. The decomposition of H2O2 occurs through a tetrahedral transition state stabilized by intermittent hydrogen bond formation and proceeds with proton and electron transfer. This is illustrated in Fig. 7.13. Note also the changes in the hydrogen bonds between the imidazolinium attached to Fe and the near basic groups.
Figure 7.13. The decomposition of hydrogen peroxide by cytochrome P450 catalyst.
Dioxygenases form the second class of biochemical oxidation systems. They often oxidize hydrocarbons selectively to carboxylic acids. These catalysts proceed through low valence states of the catalytic metal center and through radical-type elementary steps. The metal centers donate electrons to the oxygen molecule, so as to assist oxygen bond cleavage.
Various heterogeneous analogues of dioxygenases exist such as the (M1−x 2+ AlxPO4) zeolite redox systems discussed in Chapter 4 and the (BiOx)2.(MoO3)y solid-state catalyst used to oxidize propylene to acrolein (see also Section 5.6.1). In this latter example, the two oxygen atoms generated by the dissociation of molecular oxygen at an Mo center,
Mechanisms in Biocatalysis; Relationship with Chemocatalysis 329
di use through the solid oxide material and then react such that one oxygen atom is used to produce H2O, the hydrogen source being propylene, and the other inserts to give acrolein.
The Wacker reaction, introduced in Chapter 2, page 25, is another example of a chemocatalytic system that acts as a dioxygenase. In the homogeneous Wacker reaction catalyzed by the Pd0/Pd2+; Cu+/Cu2+ couple the overall reaction is
H2C=CH2 + 12 O2 −→ CH3CHO
Mechanistically the reaction proceeds through the key reaction step
C2H4 + PdCl4 2− + H2O −→ CH3CHO + Pd0 + 2H+ + 4Cl−
The Pd2+ reaction center generates an oxygen atom through the activation of water to form an OH intermediate that subsequently inserts into ethylene. Molecular oxygen is then used to regenerate the Pd2+ through a redox cycle with Cu+/Cu2+. In the overall reaction the two atoms of O2 become “incorporated” into acetaldehyde.
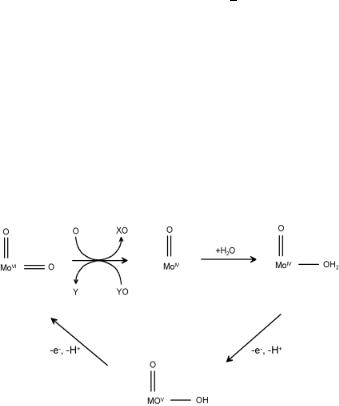
The biochemical oxomolybdooxidase enzymes operates similarly to the Pd2+ system with the important di erence that reoxidation now proceeds electrochemically. As in the Wacker system, the selective oxygen atom is generated by the oxidation of water. A possible catalytic cycle for the sulfite oxidase system[24] is shown in Fig. 7.14.
Figure 7.14. Possible catalytic cycle for sulfite (X) oxidase and dimethyl sulfoxide (DMSO) (Y) reductase[24].
The overall reaction is
R + H2O −→ RO + 2H+ + 2e−
The coupling of oxidation catalysis in the biosystems with electrochemical systems enables such reactions to proceed under mild conditions. The electrochemical potential is adjusted to overcome the high activation barrier for generation of an oxygen atom from H2O.
Propylene epoxidation over metallic Cu and ethylene epoxidation over Ag are two unique heterogeneous chemocatalytic systems which have no biochemical analogue. The epoxidation of ethylene over the Ag catalyst, which is briefly discussed here, was initially
330 Chapter 7
thought to proceed via the formation of an Ag superoxide or peroxide intermediate which would be the analogue of a monooxygenase system[25]. One oxygen atom was thought to react with ethylene to give epoxide while the second O atom would be removed from the surface through combustion of ethylene. The maximum selectivity towards epoxide would then be only 6–7. The reaction proceeds through a low valency silver state.
The alternative mechanism, which is now generally accepted, is the dioxygenase analogue. Selective reaction occurs through Ag in a high valency state, generated by the overoxidation of the Ag surface. This has an optimum selectivity at conditions where the oxygen adatom to Ag atom ratio (Oat/Ags) is equal to 2. Half of the oxygen atoms that belong to the surface-layer atoms are subsurface. Oxygen atom insertion into the ethylene bond occurs by its reaction with an AgO+ species. In principle now the two oxygen atoms from molecular O2 can be inserted, which gives a theoretical selectivity of 100%.
7.7 Reduction Catalysis
The catalytic reduction of a particular intermediate involves the addition of hydrogen. Just as in oxidation catalysis, there is the issue of whether one or two hydrogen atoms from H2 are incorporated into the substrate through identical intermediate hydrogen atoms. In heterogeneous catalysis, this question translates into whether dissociation occurs homolytically or heterolytically. On a transition-metal surface H2 dissociation generates two equivalent hydrogen atoms such as we have seen in Chapter 3. As discussed in Chapter 4, however, H2 can dissociate heterolytically: H2 → H− + H+.
The presence of H+ at the cation site of a cation-exchange zeolite will become the acidic proton on the zeolite lattice. The presence of H−, on the other hand, will take on the form of an MH species. We discussed such a reaction for the Zn2+ ion-exchanged zeolite. The reaction is driven by the strong Lewis base nature of the negatively charged zeolite oxygen atoms.
In biochemistry there are two ways to hydrogenate or to dehydrogenate. In one reaction path, an H− ion is transferred from NADH or NADPH to the substrate molecule that is to be reduced and the additional hydrogen atom is added as a proton. Such a step occurs, for instance, in the reduction of glutamate to proline. Cations such as Zn2+ or Mg2+ play a role in enzymes catalyzing such reactions.
The reactions can also be of the acid–base type induced by the electrostatics of the enzyme cavity.
A schematic description of such a concerted reaction is shown in Scheme 7.2.
An important representative of the reaction route through equivalent hydrogen atoms is that for the reduction of N2 to NH3 in the nitrogen fixation reaction. The nitrogenase enzyme catalyses the overall reaction:
N2 + 10H+ + 10e− −→ H2 + 2NH3 + H2
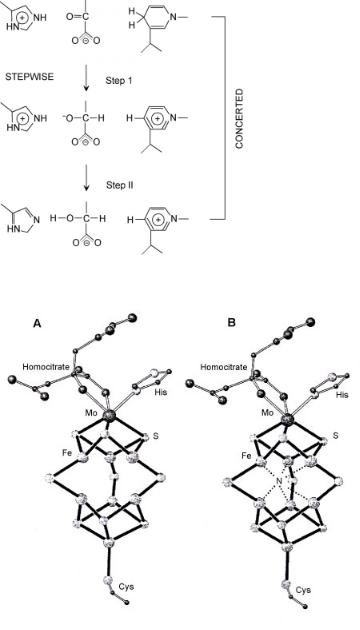
The reaction proceeds with co-evolution of hydrogen. Nitrogenase has been studied extensively and appears to contain three di erent kinds of Fe/Mo/S clusters distributed over two proteins. The actual nitrogen reduction takes place at the Fe7MoS8 cluster shown in Fig. 7.15.
It has recently been discovered that structure (A), which was widely believed to be the active cluster, was incorrectly identified. Structure (B), which contains a nitrogen atom attached to six Fe atoms, is the actual structure of the active nitrogen reducing cluster.
Mechanisms in Biocatalysis; Relationship with Chemocatalysis 331
Scheme 7.2 The stepwise and concerted reactions[26] carried out by lactate hydrogenase.
Figure 7.15. Two models of the FeMo cofactor of the nitrogen-fixing enzyme nitrogenase[27].
We will discuss available theoretical results for this reduction reaction and report on experimental studies of a recently discovered homogeneous reaction system. We subsequently conclude this section with an analysis of the heterogeneous catalyst used to synthesize ammonia industrially from N2 and H2 over Fe.
332 Chapter 7
All theoretical models used so far to study the activation of N2 have been based on the structure shown in Fig. 7.14A. It is not clear whether structure (B) is an intermediate or actually the catalytically reactive system that initiates the reaction by the adsorption of an N2 molecule. It is generally believed that the seven Fe atoms provide the binding
site for the N2 molecule. We summarize here the general theoretical understanding of this system[28].
Nitrogen can initially adsorb end-on or side-on. Nitrogen binding is a ected by protonation and the addition of an electron to generate a bridging SH group. Fe then experiences a weaker Fe–S interaction and, hence, can bind more strongly with the substrate N2 molecule. Successive hydrogen transfers (protonation and electron donation) to nitrogen lead to unstable intermediates such as NNH, HNNH and HNNH2 (Fig. 7.16).
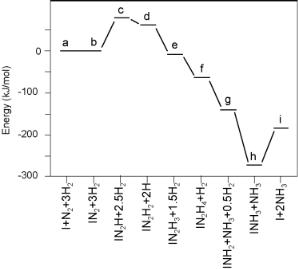
Thermodynamics becomes favorable only once a three-electron transfer occurs. The first stable irreversibly bonded intermediate is hydrazine. Adsorbed NH3 is the most stable state. It will desorb as NH4 + .
Figure 7.16. The calculated binding energies of the intermediates for hydrogenation of N2 [28b].
It is of interest to compare the reaction energy diagram for the N2 reduction by the “Fe7MoS8” cluster with those for the heterogeneous Fe-based catalyst that are used industrially and operates at a reaction temperature at around 400◦C. The reaction energy diagram constructed from experimental results for the industrial Fe–metal surface is shown in Fig. 7.17.
The relative energies of the reaction intermediates are shown to be completely di erent. On iron, the initial step is not the hydrogenation of N2, but N2 dissociation. Compared with the adsorbed Natom state, the formation of NHads , NH2ads and NH3 are all thermodynamically endothermic. The industrial catalyst is promoted with oxides such as potassium oxides, which are thought to lower the activation energy Fe in the rate-limiting step, which is the dissociative adsorption of N2.

Mechanisms in Biocatalysis; Relationship with Chemocatalysis 333
Figure 7.17. Schematic energy profiles for the ammonia synthesis on a promoted iron catalyst with energies in kJ/mol. Adapted from G. Ertl[29].
Figure 7.18. (a) Rates of ammonia synthesis over five iron single-crystal surfaces with di erent orientations: (111), (211), (100), (210), and (110). (b) Schematic representations of the idealized surface structures of the (111), (211), (100), (210), and (110) orientation of iron single crystals. The coordination of each surface atom is indicated. Adapted from Spencer et al.[30b].
As shown in Fig. 7.18, the ammonia synthesis reaction is highly structure sensitive. The most reactive face of iron is the Fe(111) surface plane[30]. The di erent phases tested
in the reactivity study are shown in Fig. 7.18. The preference for this surface in the hydrogenation of nitrogen implies that the barrier for the activation of N2 on this surface is the lowest[31]. Interestingly, on this surface, the site responsible for N2 dissociation consists of seven Fe atoms, as in the enzyme “Fe7MoS8” cofactor. It illustrates that heterogeneous catalyst can also contain unique sites that are responsible for maximal substrate activation.
334 Chapter 7
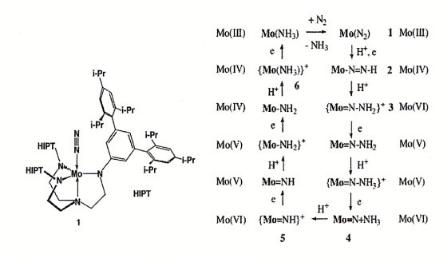
Figure 7.19. Proposed intermediates in the reduction of dinitrogen at an [HIPTN3 N]Mo (Mo) center through the stepwise addition of protons and electrons[32].
The concept that the low-temperature nitrogen fixation reaction requires electron transfer to nitrogen followed by subsequent protonation steps has been exploited to design biomimetic single-center homogeneous organometallic catalysts based on Mo. Yandulov and Schrock[32] designed di erent Mo complexes with tetradentate triamidoamine ligands that allowed for N2 reduction using a mildly acidic molecule and reductant in heptane. The reaction sequence they propose is shown in Fig. 7.19, which is similar to earlier proposals for reduced Mo systems[33].
Unique to this biomimetic system over that of the previously reported [Fe7MoS8] cofactor systems is the inequivalence between the reduction of the first and second nitrogen atoms. The finding of the nitride atom in the new crystal structure of the Fe7MoS8 cluster may indicate that such a pathway may also occur in the nitrogenase system.
In summary, protonation and electron transfer steps in biochemical systems are typically rate limiting steps. Biomimetic chemocatalytic systems have been designed based on ideas of incorporating specific functions that could carry out similar chemistry.
7.8 Enzyme Mechanistic Action Summarized
Fischer[3] suggested at the end of the 19th century that unique activity of enzymes is related to the need for reactant molecules to fit optimally in the enzyme cavity. This is the lock and key molecular recognition model. Later Koshland[4] postulated the concept of induced fit; the enzymes assume shapes that are complementary to that of the substrate after the substrate is bound.
Pauling[34] suggested in 1948 a strategy for developing enzyme cavities that stabilize the transition state of the rate-limiting step. This can be recognized as the need for a substrate to have an optimum interaction with the enzyme. We have seen that the stabilization of pretransition-state structures is usually the essential step that stabilizes transition states.
Mechanisms in Biocatalysis; Relationship with Chemocatalysis 335
Transition-state or pretransition-state structures must attain maximum free energy stabilization. The induced changes of the enzyme pocket, the concerted bond cleavage and the formation of bonds within the enzyme cavity, within reacting reagents and between substrate and enzyme lead to the optimum stabilization of pretransition-state structures. Enthalpy gains and entropy counteracts. This sometimes leads to anti-lock and key behavior when enthalpy advantages are too small to be overcome by entropy loss. The altered shape of the enzyme cavity after reaction decreases the optimum fit and, thus helps the desorption of the product. Hence the flexibility of the enzyme–protein framework is an intrinsic and an essential feature for its high activity.
References
1.L. Stryer, Biochemistry, Freeman, New York (1995)
2.(a) S. Natsch, Thesis, M¨unster, 2002;
(b) T.A. Steitz, R.J. Flathericky, W.F. Anderson, C.M. Anderson, J. Mol. Biol. 104,
197 (1976);
(c) P.R. Kuser, S. Kranchenco, O.A.C. Antunes, I. Polikarpov, J. Biol. Chem. 275, 20814 (2000)
3.E. Fischer, Ber. Dtsch. Chem. Ges. 27, 2984 (1894)
4.D.E. Koshland, in The Enzymes, Vol. 1, P.D. Boyer, H. Hardy (eds.), Academic Press, New York, p305 (1959)
5.W. Kla ke, personal private communications
6.I.A. Rose, Biochemistry, 36, 12346 (1997); I.A. Rose, Biochemistry, 37, 17651 (1998)
7.G.I. Liktenstein, New Trends in Enzyme Catalysis and Biomimetic Chemical Reactions, Kluwer, Dordrecht (2003);
P.D. Boyer, Biokhimiya, 10, 1312 (2001);
M. Loog, D.O. Morgan, Nature, 434, 104 (2005)
8.W.S. Allison, Acc. Chem. Res. 31, 819 (1998)
9.R.L. Cross, Annu. Rev. Biochem. 50, 681 (1981)
10.I. Ogilvie, R. Aggeler, R.A. Capaldi, J. Biol. Chem. 272, 16652 (1997)
11.F.M. Harold, The Vital Force, Freeman New York (1986)
12.(a) E.A. Erikson, P.M. Jones, A. Liljan, Proteins, 4, 283 (1980;
(b) D.N. Silverman, S. Lindskog, Acc. Chem. Res. 21, 30 (1988)
13.D. Lu, G.A. Voth, J. Am. Chem. Soc. 120, 4006 (1998)
14.G. Wul , Chem. Rev. 102, 1 (2002)
15.V.T. D’Souza, M.L. Bender, Acc. Chem. Res. 20, 146 (1987)
16.J.M. Lehn, Angew. Chem. Int. Ed. Engl. 27, 89 (1988)
17.M.W. Hisselni, J.M. Lehn, J. Am. Chem. Soc. 109, 7047 (1987)
18.C.J. Walter, H.L. Anderson, J.K.M. Sanders, J. Chem. Soc. Chem. Commun. 458
(1991);
L.G. Mackay, R.S. Wylie, J.K. Sanders, J. Am. Chem. Soc. 116, 3141 (1994)
19.E. Kimura, T. Koike, in Bioinorganic Catalysis, J. Reedijk, E. Bouwman (eds.), Marcel Dekker, New York, p. 33 (1999)
20.A.E. Shilov, Metal Complexes in Biomimetic Chemical Reactions, CRC Press, Boca
Raton, 1997;
A.E. Shilov, CaTTech, 5, 72 (1999)