

Metal-Catalysed Reactions of Hydrocarbons / 04-The Chemisorption of Hydrocarbons
.pdfTHE CHEMISORPTION OF HYDROCARBONS |
193 |
other than fcc(111) are not so popular (for Ni(100) see references 171, 172). These limitations, which will also doubtless be rectified in time, will lead us to consider the broader overview that students of catalysis need. Calculations have also been performed101 with a ten-atom Pt6Sn4 cluster analogous to the Pt10 but having tin at each apex, and with Pt16Sn3 in which the triangular face has tin at each corner. The tin atoms weaken somewhat the binding of di-σ and π ethene to platinum atoms held in the same way as before, but they affect that of ethylidyne much more. The results show electron transfer from tin to platinum.
It is noteworthy that both experimental and theoretical studies suggest that only one σ C––M bond can be formed and that C M bonds do not exist; species such as ethylidene (5) and ethylidyne (8) therefore require respectively two and three metal atoms: this is in keeping with the principle of maximum utilisation of surface free valencies. αα-Diadsorbed species such as ethylidene have frequently been postulated as intermediates in reaction of alkanes (see e.g. Chapter 6). Group theoretic arguments indicated that a C––M double bond if formed would have σ and π components.173
M bonds do not exist; species such as ethylidene (5) and ethylidyne (8) therefore require respectively two and three metal atoms: this is in keeping with the principle of maximum utilisation of surface free valencies. αα-Diadsorbed species such as ethylidene have frequently been postulated as intermediates in reaction of alkanes (see e.g. Chapter 6). Group theoretic arguments indicated that a C––M double bond if formed would have σ and π components.173
We return now to the broader prospect, and re-visit the question of the conditions under which the π and di-σ orms of ethene appear. DFT calculations are not yet sufficiently numerous to reveal the variations in C––C bond order and vibration frequency that are observed in practice (Section 4.4.2). The basic model suggests that the di-σ form will occur when there is substantial π back-bonding, and when this is only moderate the π form will result. This led Yagasaki and Masel8 to propose a correlation between interstitial electron density (i.e. that outside the atomic cores) and the occurrence of either form: this quantity emerges from basic theories of the metallic state, which start with the jellium model,108 and varies with Periodic Group number somewhat as sublimation heat (Section 1.11), although strangely its values in Groups 6 to 9 increase in the sequence: 2nd row<lst row <3rd row. Elements in these Groups (and nickel and platinum) have values greater than four, and generally favour formation of the σ state, while palladium and the Group 11 metals, having values between two and four, favour the π state. This correlation is however based on somewhat limited experimental evidence, and the unusual behaviour of palladium may find alternative explanations. This metal is in so many respects sui generis. The concept of electron density also helps to understand why the π state occurs along the ridges of stepped surfaces such as Pt(210); it is thought to be lower there than on terraces. A linear relation between both bond order and C––C vibration frequency with interstitial electron density for ethene on several single crystal surfaces and films has also been observed.
The first and very pictorial attempt to invoke the ideas of co-ordination chemistry and to apply them to chemisorption at metal surfaces58,174 was introduced in Section 1.23: it was based on identifying the character of orbitals emerging from various crystal planes, assuming that the arrangement within the bulk was maintained at the surface. This notion has been quite properly ridiculed as being

194 |
CHAPTER 4 |
overly simplistic (‘it is not accepted’8), but it has had its uses175,176 (‘Nevertheless there is a good correlation between the model and the experiment’). This last statement is based on a compilation of the available orbitals for each type of structure, and finding that these correlate with the tendency of several platinum surfaces to form bonds with them.8 Once again, however, the way in which the calculation is made remains quite unclear. The principal observation from all experimental studies of hydrocarbon chemisorption is however so obvious that it escapes attention. Adsorbed molecules and fragments occupy clearly defined crystallographic sites on metal surfaces; they form σ and π bonds to specific atoms, having well-defined strengths and lengths, and their spectroscopic characteristics are well simulated by organometallic complexes. Exceptions to this generalisation are rare, and are open to doubt.21 The rules of chemical bonding apply equally to chemisorption, and we are therefore dealing with chemistry in two dimensions. In some cases, species occupy sites that might have been predicted using chemical common sense; it does not need refined theory to see that ethylidyne, wanting to form three σ bonds to the surface, will bond at trigonal sites on the fcc(111) plane (except, curiously, and inexplicably, on Ni(111), where it does not form), rather than elsewhere. Ethene and ethyne in their adsorbed states are usually more or less in register with the surface lattice (the asymmetrical di-σ ethene on Pt(111) has been queried113), but there are differences in the orientations of the C––C axes and in the locations of the carbon atoms with respect to the Ni(111) surface lattice (Figure 4.6) that are not replicated by the DFT calculations on the Pt10 cluster referred to above. It will be interesting to see whether DFT calculations on Ni(111) produce the correct result. As noted in Section 1.2.2, surfaces having kinks possess chiral sites, and calculations177 have suggested that enantiomeric hydrocarbons will bond to kinks of different form with slightly different strengths. There is still a need for a sound molecular orbital account of all these interactions, one that offers explanations for the structures adopted, phrased in language understandable by chemists.
A recurring theme throughout this chapter has been the anomalous characteristics shown by palladium. Specifically, in comparison to its neighbours (Rh, Ni, Pt) it chemisorbs ethene much more weakly: both the DFT-calculated and experimental heats of adsorption (Tables 4.8 and 4.12) are lower, the π form is seen in all low-index faces, bond order estimates confirming this, and it desorbs unchanged on heating. Ethyne is also weakly adsorbed on palladium (Table 4.9). One other characteristic that palladium shares with nickel, but which is not highlighted in fundamental studies, is its propensity to form π -alkenyl species when three or more carbon atoms are present. The question now arises as to whether it is palladium or platinum in Group 10 that shows unexpected behaviour. It might be thought that the general similarities shown by metals in the second and third rows of the Periodic Classification would lead a near-identity of behaviour in palladium and platinum, but the curious discontinuous changes seen in Groups 11

THE CHEMISORPTION OF HYDROCARBONS |
195 |
TABLE 4.13. Calculated Hydrocarbon-Metal Bond Energies (kJ mol−1 ) for Ethene and Ethynyl Complexes (P = PPh3 )
Metal |
M0 P2 (C2 H4 ) |
M0 P2 (C2 H2 ) |
M0 (CO)4 (C2 H4 ) |
Ni |
159 |
189 |
163 |
Pd |
83 |
91 |
128 |
Pt |
88 |
104 |
164 |
|
|
|
|
and 12,178 which have been attributed (Section 1.1) to the operation of relativistic effects caused by heavy nuclei, may suggest that it is platinum that is the odd man out.
The importance of relativistic phenomena both in coordination complexes and in chemisorption has been reviewed.178,179 For complexes containing coordinated ethene or other unsaturated hydrocarbons, comparable quantitative information on all the Group 10 metals is extremely hard to come by, but calculations180 on various ethene and ethyne complexes (Table 4.13) performed by the non-local quasi-relativistic DF method are instructive. For each complex the bond energy is in the sequence: Ni > Pt > Pd; marked differences in the stabilities and reactivities of complexes of the type MIIP2(CH3) (M = Pd, Pt; P = PPh3) were also noted. In this context, it is never remarked that nearly all reactions homogeneously catalysed by metal salts or complexes, and metal-mediated reactions, involve elements from the first and second rows, and very rarely a third row element. Ruthenium, rhodium and palladium feature often; osmium, iridium and platinum hardly at all. This is because very generally the complexes of the third row elements are too stable to be reactive.
It was concluded178 that this is caused by the stabilisation of the 6s electron level relative to that of the 5d level, permitting the latter to be involved in chemical bonding. In the case of alkene and carbene complexes, and chemisorbed molecules, platinum therefore has a greater facility for π -back-bonding than with palladium, resulting in preference for di-σ rather than π bonding, with a consequential greater stability.181 This simple factor underscores much of the catalytic chemistry of these metals, and will be a recurring theme in subsequent chapters. The discontinuity that arises between palladium and platinum (and somewhat less clearly between rhodium and iridium) has often been noted, and interpreted in various theoretical frameworks: for example, ‘The enhanced stability of the π -bound complex on Pd(111) is associated with the smaller orbital exponent of the d-bands in palladium’,8 and ‘Relativistic augmented plane-wave calculations have shown the palladium d-band to have lower energy, and therefore overlap with the hydrocarbon’s π orbitals is poorer’.178 Which metal is out of line is therefore a matter of personal choice: what is important is to understand the causes of their characteristics (rerum cognoscere causas).

196 |
CHAPTER 4 |
4.8. CHEMISORPTION OF ALKANES182–186
This is mercifully a relatively straightforward matter compared with the complexities attending the chemisorption of unsaturated hydrocarbons, at least as far as adsorbed structures go. At low temperatures, alkanes are physically adsorbed with a strength that depends on the size of the molecule, i.e. on the number of contacts it makes with the surface when lying flat. The adsorbed state of n-butane on Pt(111) at 98 K has been studied in some detail using LEED and molecular beam methods, changes in adsorption energy and mobility with coverage being noted.187 Frennet spoke182,184,185 of a non-dissociative chemisorption of methane that occurs below 273 K without evolution of hydrogen, but it is not clear how it would be bound to the surface and whether it would be in truly molecular form. With metal films above a critical temperature (Rh, 323 K; Ni, 373 K; Pd, 398 K), hydrogen evolution begins, and equilibrium is established between gaseous hydrogen and methyl, methylene and methylidyne radicals. Above 473 K, equilibrium is reached quickly if carbide formation is impossible, but with tungsten for example it may require more than two days. Detailed studies185 of the effects of methane and hydrogen pressures on rates and positions of equilibrium have led to the conclusion that the most likely mechanism for chemisorption is:
CH4 + H → CH3 + H2 |
(4.B) |
the reverse of which is an Eley-Rideal step. The forward process seems however also to need a number of vacant ‘sites’ around the hydrogen atom in order to accommodate the methyl radical, these forming a ‘free potential site’ or ‘landing site’. This mechanism has however not been translated into an easily visualised picture. Methane is in many ways an atypical alkane, and the general picture that emerges is of an initial breaking of a C––H bond with the formation of an alkyl radical; this may be followed, at a rate dependent on the temperature, by the loss of further hydrogen atoms, leading to an alkylidyne,187 or at higher temperatures to the complete disruption of the molecule.188
It is very much harder to chemisorb an alkane than hydrogen. The process is activated,189 and sticking coefficients are low, but this is attributed to an unfavourable entropy of activation.184 In atomic terms the electrons in the C––H bond to be broken are shielded from the metal atoms by the other hydrogen atoms, thus forbidding easy reaction. Higher alkanes chemisorb more easily because the stability of their precursor state is greater.
Much effort has been devoted to trying to decide the number of metal atoms that comprise a landing site for an alkane. As with so many mechanistic questions in heterogeneous catalysis, a definite answer cannot be obtained. As to the first question, it had been thought that use of alloy surfaces might provide an answer

THE CHEMISORPTION OF HYDROCARBONS |
197 |
since dilution of the active metal by an inactive should lead to decreases in the size of various ensembles of the former in a way that can be calculated, so that the dependence of rate or amount of adsorption upon composition should identify the minimum ensemble size required. However, this calculation is fraught with difficulties, which may explain the improbable numbers that the early work produced.
There have been a number of studies of the mechanism of chemisorption on single crystal surfaces, with particular attention being made to the type of energy needed to secure the breaking of the first C––H bond.183 Work performed during the 1970s revealed the importance of surface geometry: close-packed planes (Ni(111)190 and Ir(111)191) were unreactive, while the more open planes190,192 (e.g. Ni(100)) and stepped surfaces193 (e.g. Ir(110)(1 × 2)) were more reactive. Use of molecular beams194,195 has shown that energy stored in the surface is relatively unimportant, and that the more critical factor is the rate of vibrational activation of the C––H bond to be broken, using translational and other vibrational modes.4,196
On platinum and rhodium surfaces, however, sticking coefficients increase with temperature when the kinetic energy of the molecule is low, and they show activation energies of some 20–40 kJ mol−1. Sticking coefficients for methane were4,184 as low as 10−10 to 10−12. Impact-induced adsorption of methane and ethane produced by hammering physically-adsorbed molecules with other impinging molecules on Pt(111) and Pt25Rh75(111) surfaces suggested that two vibrational quanta in the C––H deformation mode were needed for adsorption to occur.197 Recent molecular beam work with methane on Ir(110), Ir(111) and Pt(111) has identified a new low translational energy pathway;198 adsorption of isobutane and of neopentane on Ir(110) has also been followed by this technique.199
Nickel and ruthenium ‘model’ catalysts have also been used. Adsorption of methane on small palladium particles supported on alumina is photo-assisted: UV radiation effects dissociation as well as desorption of physically-adsorbed molecules, the former process requiring larger particles than the latter.200
There have been a number of DFT calculations of C1 species on metal surfaces (see Further Reading section), producing quantitative estimates of binding energies, but few surprises.
4.9. THE FINAL STAGE: CARBONACEOUS DEPOSITS201–203
This termed is frequently deployed in the literature to describe ill-defined and unwelcome substances deposited during hydrocarbon reactions on metals, leading to loss of catalytic activity. The work on hydrocarbon adsorption which we have reviewed has shown that, in almost every case, adsorbed species when heated decompose first to C1 species (e.g.  CH2 or
CH2 or  CH), and that these then lose their hydrogen atoms to form carbon atoms, which on Ir(100) have been shown by LEED (and DFT) to reside in fourfold sites in a (2 × 2) overlayer. Further heating may lead to their polymerisation to graphite or to a diamond-like structure.204 Carbon
CH), and that these then lose their hydrogen atoms to form carbon atoms, which on Ir(100) have been shown by LEED (and DFT) to reside in fourfold sites in a (2 × 2) overlayer. Further heating may lead to their polymerisation to graphite or to a diamond-like structure.204 Carbon
198 |
CHAPTER 4 |
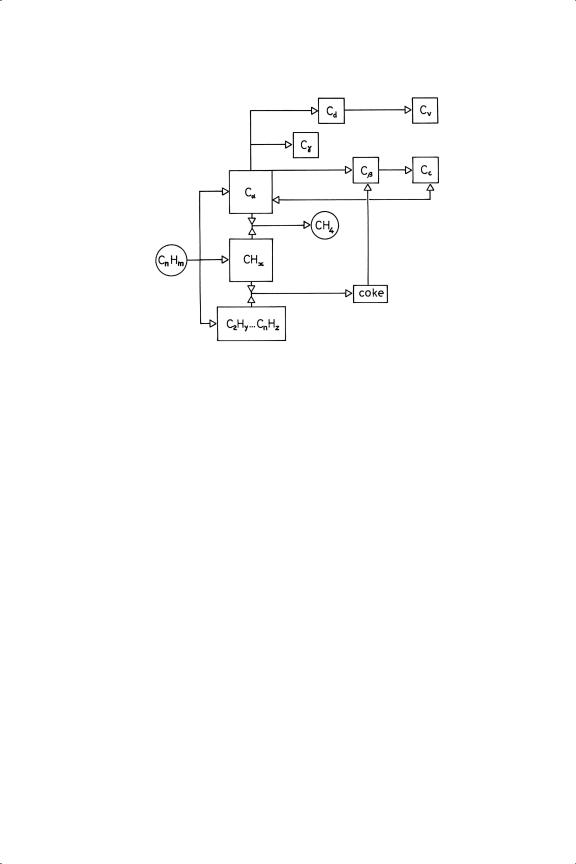
Figure 4.12. Forms of carbon involved in the decomposition of the hydrocarbon Cn Hm and modes of their transformation: Cα , carbidic (atomic) carbon; Cd ; dissolved carbon; Cβ , amorphous carbon; Cc , graphitic carbon; Cγ , metal carbide; Cv , filamentary carbon.
atoms on Pd(100) affected the reactivity of the surface towards other molecules such as ethene.49
A number of techniques have been used to examine the structure and reactivity of carbon deposits on technical catalysts (e.g. 13C NMR205). Hydrogen treatment may convert surface carbon to methane if it is not too polymerised, but oxidation is usually more successful in removing it, although it may have other undesired consequences. Several different forms of ‘carbon’ are recognised (see Figure 4.12). Carbon atoms may dissolve into the surface and into the bulk, forming non-stoichiometric interstitial compounds,11 or, especially with the base metals of Groups 8 to 10, stoichiometric carbides. These metals, particularly nickel, will catalyse the formation of filamentary whiskers if exposed continuously to a hydrocarbon vapour at high temperature: they constitute carbon ‘nanotubes’ which have a buckminsterfullerene-type structure. Homologation of C1 species to higher alkanes will be mentioned in Chapter 13.
REFERENCES
1.H. Hopf, Classics in Hydrocarbon Chemistry, Wiley-VCH: Chichester, 2000.
2.J.H. Sinfelt, Surf. Sci. 500 (2002) 923.
3.F. Zaera, Surf. Sci. 500 ( 2002) 947.
4.F. Zaera, Chem. Rev. 95 (1995) 2654.

THE CHEMISORPTION OF HYDROCARBONS |
199 |
5.B.E. Bent, Chem. Rev. 96 (1996) 1361.
6.V. Ponec and G.C. Bond, Catalysis by Metals and Alloys, Elsevier: Amsterdam (1995).
7.G.C. Bond, Catalysis by Metals, Academic Press: London (1962).
8.E. Yagasaki and R.I. Masel in: Specialist Periodical Reports: Catalysis, Vol. 11 (J.J. Spivey and S.K. Agarwal, eds.), Roy. Soc. Chem. (1994), p. 16.
9.N. Sheppard and C. de la Cruz, Adv. Catal. 42 (1998) 181.
10.G.A. Somorjai and K.R. McCrea, Adv. Catal. 45 (2000) 385.
11.Z. Karpinski,´ personal correspondence.
12.R.N. Reifsnyder and H.H. Lamb, Catal. Lett. 40 (1996) 155.
13.O. Beeck, A.E. Smith and A. Wheeler, Proc. Roy. Soc. A177 (1941) 159.
14.O. Beeck and A.W. Ritchie, Discuss. Faraday Soc. 8 (1950) 159.
15.G.A. Somorjai, J. Molec. Catal. A: Chem. 107 (1996) 1.
16.N. Sheppard, Ann. Rev. Phys. Chem. 39 (1988) 589.
17.N. Sheppard and C. de la Cruz, Adv. Catal. 41 (1996) 1.
18.N. Sheppard and J. Erkelens, Appl. Spectroscopy 38 (1984) 471.
19.H.A. Pearce and N. Sheppard, Surf. Sci. 59 (1976) 205.
20.A.M. Bradshaw, Surf. Sci. 331–333 (1995) 978.
21.T. Gieβel, R. Terborg, O. Schaff, R. Lindsay, P. Baumg¨artel, J.T. Hoeft, K.-M. Schindler, S. Bao, A. Theobald, V. Fernandez, A.M. Bradshaw, D. Chrysostomou, T. Cabe, D.R. Lloyd, R. Davis, N.A. Booth and D.P. Woodruff, Surf. Sci. 440 (1999) 125.
22.J.-H. Kang, R.L. Toomes, J. Robinson, D.P. Woodruff, O. Schaff, R. Terburg, R. Lindsay, P. Baumg¨artel and A.M Bradshaw, Surf. Sci. 448 (2000) 23.
23.W.A. Brown, R. Kose and D.A. King, Chem. Rev. 98 (1998) 797.
24.Q.-F. Ge, R. Kose and D.A. King, Adv. Catal. 45 (2000) 207.
25.N. Cardona-Martinez and J.A. Dumesic, Adv. Catal. 38 (1992) 149.
26.C.D. Makowka, C.P. Slichter and J.H. Sinfelt, Phys. Rev.B 31 (1985) 5663.
27.C.P. Slichter, Ann. Rev. Phys. Chem. 37 (1986) 25.
28.K. Sakaie, C.P. Slichter and J.H. Sinfelt, J. Magnet. Res. A 119 (1996) 235.
29.J.E. Demuth, H. Ibach and S. Lehwald, Phys. Rev. Lett. 40 (1978) 1044.
30.J.E. Demuth, IBM J. Res. Develop. 22 (1978) 265.
31.J.E. Demuth, Phys. Rev. Lett. 40 (1978) 409.
32.J.E. Demuth and D.E. Eastman, J. Vac. Sci. Technol. 13 (1976) 283.
33.T.E. Fischer, S.R. Kelemen and H.P. Bonzel, Surf. Sci. 64 (1997) 157.
34.J.E. Demuth, Chem. Phys. Lett. 45 (1977) 12.
35.J.E.Demuth and D.E. Eastman, Phys. Rev. B 13 (1976) 1523.
36.J.E. Demuth, Proc. 7th. Internat. Vac. Congr. and 3rd. Internat. Conf. Solid Surfaces, Vienna (1977), p. 779.
37.W. Ranke and W. Weiss, Surf. Sci. 465 (2000) 317.
38.R. Mason, M. Textor, Y. Iwasawa and I.D. Gay, Proc. Roy. Soc. A 354 (1977) 171.
39.I.D. Gay, M. Textor, R. Mason and Y. Iwasawa, Proc. Roy. Soc. A 356 (1977) 25.
40.G. Brod´en, T. Rhodin and W. Capehart, Surf. Sci. 61 (1976) 143.
41.D.R. Lloyd, C.M. Quinn and N.V. Richardson, Solid State Comm. 23 (1977) 141.
¨
42. J. Grewe, U. Erfurk¨ and A. Otto, Langmuir 14 (1998) 696.
43. W. Krasser and A.J. Renouprez, Solid State Commun. 41 (1982) 231. 44. R.L. Burwell Jr., Chem. Eng. News 44 (1966) 56.
45. E.L. Muetterties, T.N. Rhodin, E. Band, C.F. Brucker and W.R. Pretzer, Chem. Rev. 79 (1979) 91.
46. S. Carr` and R. Ugo, Inorg. Chim. Acta Rev. 1 (1967) 49.
47. C.E. Anson, N. Sheppard, D.B. Powell, B.R. Bender and J.R. Norton, J. Chem. Soc. Faraday Trans. 90 (1994) 1449.

200 |
CHAPTER 4 |
48.C.E. Anson, N. Sheppard, D.B. Powell, J.R. Norton, W. Fischer, R.L. Keiter, B.F.G. Johnson, J. Lewis, A.K. Bhattcharrya, S.A.R. Knox and M.L. Turner, J. Am. Chem. Soc. 116 (1994) 3058.
49.I. Ratajczykowa, Surf. Sci. 152/153 (1985) 627.
50.L.L. Kesmodel, R.C. Baetzold and G.A. Somorjai, Surf. Sci. 66 (1977) 299.
51.N. Sheppard and C. de la Cruz, Catal. Today 70 (2001) 3.
52.J. Evans and G.S. McNulty, J. Chem. Soc. Dalton Trans. (1983) 639.
53.J. Evans and G.S. McNulty, J. Chem. Soc. Faraday Trans. 80 (1984) 79.
54.C.E. Anson, B.F.G. Johnson, J. Lewis, D.B. Powell, N. Sheppard, A.K. Bhattacharya, B.R. Bender, R.M. Bullock, R.T. Hembre and J.R. Norton, J. Chem. Soc. Chem. Comm. (1989) 703.
55.F. Zaera and N. Bernstein, J. Am. Chem. Soc. 116 (1994) 4881.
56.F. Solymosi, Catal. Today 28 (1996) 191.
57.K.C. McGee, M.D. Driessen and V.H. Grassian, J. Catal. 157 (1995) 730.
58.G.C. Bond, Discuss. Faraday Soc. 41 (1966) 200.
59.G.A. Ozin, Acc. Chem. Res. 10 (1977) 21.
60.R.M. Atkins, R. Mackenzie, P.L. Timms and T.W. Turney, J. Chem. Soc. Chem. Comm. (1975) 764.
61.R. Ugo, in: Proc. 5t h . Internat. Congr. Catal. (J.W. Hightower, ed.), North Holland: Amsterdam (1972) 19.
62.T.E. Felter and W.H. Weinberg, Surf. Sci. 103 (1981) 265.
63.M.A. Chesters, C. De La Cruz, P. Gardner, E.M. McCash, J.D. Prentice and N. Sheppard, J. Elec. Spec. Rel. Phen. 54/55 (1990) 739.
64.N.N. Greenwood and A. Earnshaw, Chemistry of the Elements, 2nd edition, ButterworthHeinemann: Oxford (1997).
65.F.A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, 4th edition, Wiley: Chichester (1980).
66.G.C. Bond, J. Molec. Catal. A: Chem. 156 (2000) 1.
67.G.C. Bond, Platinum Metals Rev. 44 (2000) 146.
68.Y.-T. Wong and R. Hoffmann, J. Chem. Soc. Faraday Trans. 86 (1990) 4083.
69.A. Cassuto, Mane Mane, J. Jupille, G. Tourillon and P. Parent, J. Phys. Chem. 96 (1992) 5987.
70.J.-F. Fan and M. Trenary, Langmuir 10 (1994) 3649.
71.E.M. Stuve and R.J. Madix, J. Phys. Chem. 89 (1985) 105.
72.B.E. Bent, C.M. Mate, C.-T. Kao, A.J. Slavin and G.A. Somorjai, J. Phys. Chem. 92 (1988) 4720.
73.G.H. Hatzikos and R.I. Masel, Surf. Sci. 185 (1987) 479.
74.C. Egawa, Surf. Sci. 454–456 (2000) 222.
75.S.B. Moshin, M. Trenary and H.J. Robota, J. Phys. Chem. 92 (1988) 5229.
76.R. Raval, Surf. Sci. 331–333 (1995) 1.
77.B. Kruger¨ and C. Benndorf, Surf. Sci. 170 (1976) 704.
78.D. Stacchiola, G. Wu, M. Kaltchev and W.T. Tysoe, Surf. Sci. 486 (2001) 9.
79.M. Nishijima, J. Yoshinobu, T. Sekitani and M. Onchi, J. Chem. Phys. 90 (1989) 5114.
80.L. Hammer, T. Hertlein and K. Muller,¨ Surf. Sci. 178 (1976) 693.
81.E. Cooper and R. Raval, Surf. Sci. 331–333 (1995) 94.
82.D. Stacchiola, L. Burkholder and W.T. Tysoe, Surf. Sci. 511 (2002) 215.
83.D. Stacchiola, S. Azad, L. Burkholder and W.T. Tysoe, J. Phys. Chem. 105 (2001) 11233.
84.E. Yagasaki, A.L. Blackman and R. I. Masel, J. Phys. Chem. 94 (1990) 1066.
85.Sh. Shaikhutdinov, M. Heemeir, M. Baumer, T. Lear, D. Lennon, R.J. Oldman, S.D. Jackson, and H.-J. Freund, J. Catal. 200 (2001) 330.
86.N. Sheppard, N.R. Avery, B.A. Morrow and R.P. Young, in: Chemisorption and Catalysis, Institute of Petroleum: London (1971), p. 135.
87.J.D. Prentice, A. Lesiunas and N. Sheppard, J. Chem. Soc. Chem. Comm. (1976) 76.

THE CHEMISORPTION OF HYDROCARBONS |
201 |
88.M.K. Ainsworth, M.R.S. McCoustra, M.A. Chesters, N. Sheppard and C. de la Cruz, Surf. Sci. 437 (1999) 9.
89.H. Steiniger, H. Ibach and S. Lehwald, Surf. Sci. 117 (1982) 685.
90.J. Kubota, S. Ichihara, J.N. Kondo, K. Domen and C. Hirose, Surf. Sci. 337–338 (1996) 634; langmuir 12 (1996) 196.
91.R.D. Cortright and J.A. Dumesic, J. Catal. 157 (1995) 576.
92.R.G. Windham, M.E. Bartram and B.E. Koel, J. Phys. Chem. 92 (1988) 2862.
93.R.G. Windham, B.E. Koel and M.T. Paffet, Langmuir 4 (1988) 1113.
94.E. Yagasaki and R.I. Masel, J. Am. Chem. Soc. 112 (1990) 8746; Surf. Sci. 222 (1989) 430.
95.C.T. Campbell, Ann. Rev. Phys. Chem. 41 (1990) 775.
96.E.M. Stuve and R.J. Madix, J. Phys. Chem. 89 (1985) 3183.
97.M. Bowker, J.L. Gland, R.W. Joyner, X.-Y. Li, M.M. Slinko and R. Whyman, Catal. Lett. 25 (1994) 293.
98.J.C. Bertolini, A. Cassuto, Y. Jugnet, J. Massardier, B. Tardy and G. Tourillon, Surf. Sci. 349 (1996) 88.
99.G. Papoian, J.K Nørskov and R. Hoffmann, J. Am. Chem. Soc. 122 (2000) 4129.
100.Y.-L. Tsai, C. Xu and B.E. Koel, Surf. Sci. 385 (1997) 37.
101.J.-Y. Shen, J.M. Hill, R.M Watwe, B.E. Spiewak and J.A. Dumesic, J. Phys. Chem. B 103 (1999) 3923.
102.C. Becker, T. Pelster, M. Tanemura, J. Breitbach and K. Wandelt, Surf. Sci. 433–435 (1999) 822.
103.N.R.M. Sassen, A.J. den Hartog, F. Jongerius, J.F.M. Aarts and V. Ponec, Faraday Discuss. Chem. Soc. 87 (1989) 311.
104.G.A. Somorjai and G. Rupprechter, J. Chem. Educ. 75 (1998) 171.
105.G.A. Somorjai, Ann. Rev. Phys. Chem. 45 (1994) 721.
106.R.W. Joyner in: Specialist Periodical Reports: Catalysis, Vol. 5 (G.C. Bond and G. Webb, eds.),
Roy. Soc. Chem. (1982), p. 1.
107.S. Titmuss, A. Wander and D.A. King, Chem. Rev. 96 (1996) 1291.
108.R.I. Masel, Principles of Adsorption and Reaction on Solid Surfaces, Wiley: New York (1996).
109.C. Kemball, Catal. Rev. 5 (1971) 33.
110.G. Webb in: Specialist Periodical Reports: Catalysis, Vol. 2 (C. Kemball and D.A. Dowden, eds.),
Roy. Soc. Chem. (1978), p. 145.
111.G.A. Somorjai, Surface Science: An Introduction, Butterworth-Heinemann: New York (1992).
112.M.A. Van Hove and G.A. Somorjai, J. Molec. Catal. A: Chem. 131 (1998) 243.
113.R. Doll,¨ C.A. Gerken, M.A. Van Hove and G.A. Somorjai, Surf. Sci. 374 (1997) 151.
114.H. Ibach, H. Hopster and B. Sexton, Appl. Phys. 14 (1977) 21.
115.R.C. Pitkethly in: Chemisorption and Catalysis, Inst. Petroleum: London (1971), p.98.
116.A.B. Anderson, J. Chem. Phys. 65 (1976) 1729.
117.A.B. Anderson, J. Am. Chem. Soc. 100 (1978) 1153.
118.N. Sheppard and J.W. Ward, J. Catal. 15 (1969) 50.
119.R.M. Watwe, B.E. Spiewak, R.D. Cortright and J.A. Dumesic, J. Catal. 180 (1998) 184.
120.S. Bao, Ph. Hofmann, K.-M. Schindler, V. Fritzsche, A.M. Bradshaw, D.P. Woodruff, C. Casado and M.C. Ascencio, Surf. Sci. 307–309 (1994) 722.
121.C.J. Baddeley, A.F. Lee, R.M. Lambert, T. Gieβel, O. Schaff, V. Fernandez, K.-M. Schindler, A. Theobald, C.J. Hirschmugl, R. Lindsay, A.M. Bradshaw and D.P. Woodruff, Surf. Sci. 400 (1998) 166.
122.S. Bao, K.-M. Schindler, Ph. Hoffman, V. Fritsche, A.M. Bradshaw, and D.P. Woodruff, Surf. Sci. 291 (1993) 295.
123.Chen Xu, J.W. Peck and B.E. Koel, J. Am. Chem. Soc. 115 (1993) 751.

202 |
CHAPTER 4 |
124.S. Bao, R. Lindsay, M. Polcik, A. Theobald, T. Giessel, O. Schaff, P. Baumg¨artel, A.M. Bradshaw,
R.Terborg, N.A. Booth and D.P. Woodruff, Surf. Sci. 478 (2001) 35.
125.F.S. Thomas, N.S. Chen. L.P. Ford and R.I. Masel, Surf. Sci. 486 (2001) 1.
126.N.C. Chen, L.P. Ford and R.J. Masel, Catal. Lett. 56 (1998) 105.
127.G. Held, M.P. Bessent, S. Titmuss and D.A. King, J. Chem. Phys. 104 ( 1996) 11305.
128.D. Schaff, V. Fernandez, Ph. Hofmann, K.-M. Schindler, A. Theobald, V. Fritsche, A.M. Bradshaw,
R.Davis and D.P. Woodruff, Surf. Sci. 348 (1996) 89.
129.J. Winterlin, Adv. Catal. 45 (2000) 131.
130.S. Chang, Chem. Rev. 97 (1997) 1083.
131.L. Vattone, Y.Y. Yeo, R. Kose, and D.A. King, Surf. Sci. 447 (2000) 1.
132.R. Kose, W.A. Brown and D.A. King, Chem. Phys. Lett. 311 (1999) 109.
133.W.A. Brown, R. Kose and D.A. King, J. Molec. Catal. A: Chem. 141 (1999) 21.
134.B.E. Spiewak, R.D. Cortright and J.A. Dumesic, J. Catal. 176 (1998) 405.
135.M.A. Natal-Santiago, S.G. Podkolzin, R.D. Cortright, and J.A. Dumesic, Catal. Lett. 45 (1997) 155.
136.F.B. Passos, M. Schmal and M.A. Vannice, J. Catal. 160 (1996) 118.
137.J.-Y. Shen, J.M. Hill, R.M. Watwe, S.G. Podkolzin and J.A. Dumesic, Catal. Lett. 60 (1999) 1.
138.M.A. Chesters and G.A. Somorjai, Surf. Sci. 52 (1975) 21.
139.K.A. Davis and D.W. Goodman, J. Phys. Chem. B 104 (2000) 8557.
140.A. Guerrero, M. Reading, Y. Grillet, J. Rouquerol, J.P. Boitiaux and J. Cosyns, Z. Phys. D 12
(1989) 583.
ˇ
141. S. P´alfi, W. Lisowski, M. Smutek and S. Cerny,´ J. Catal. 88 (1984) 300. 142. Z. Pa´al, E. Ful¨ op¨ and D. Marton, React. Kinet. Catal. Lett. 38 (1989) 131. 143. Po-Kang Wang, C.P. Slichter and J.H. Sinfelt, Phys. Rev. Lett. 53 (1984) 82. 144. G.A. Martin, Catal. Rev.-Sci. Eng. 30 (1988) 518.
145. P.W. Selwood, J. Am. Chem. Soc. 79 (1957) 3346. 146. G.A. Somorjai, Adv. Catal. 26 (1977) 2.
147. A. Bao, Ph. Hofmann, K.-M. Schindler, V. Fritsche, A.M. Bradshaw, C. Casado and M.C. Asensio,
J. Phys. Condens. Matter 6 (1994) L93.
148. E. Yagasaki and R.I. Masel, J. Am. Chem. Soc. 112 (1990) 8746.
149. L.P. Ford, H.L. Nigg, P. Blowers and R.I. Masel, J. Catal. 178 (1998) 163. 150. L.P. Ford, P. Blowers and R.I. Masel, J. Vac. Sci. Technol. A 17 (1999) 1705. 151. C.-H. Hwang, C.-W. Lee, H. Kang and C.M. Kin, Surf. Sci. 490 (2001) 144. 152. G.A. Somorjai, Catal. Lett. 12 (1992) 17.
153. U. Starke, A. Barbieri, N. Materer, M.A. Van Hove and G.A. Somorjai, Surf. Sci. 286 (1993) 1. 154. Po-Kang Wang, C.P Slichter and J.H. Sinfelt, Phys. Rev. Lett. 55 (1985) 2731.
155. D. Stacchiola, H. Molero and W.T. Tysoe, Catal. Today 65 (2001) 3.
156. T.P. Beebe Jr., M.R. Albert, and J.T. Yates Jr., J. Catal. 96 (1985) 1. 157. G. Blyholder and L. Orji, Ads. Sci. Technol. 4 (1987) 1.
158. N. Sheppard, personal correspondence.
159. P.R. Marshall, G.S. McDougall and R.A. Hadden, Topics in Catal. 1 (1994) 9. 160. I. Jungwirthov´a and L.L. Kesmodel, Surf. Sci. 470 (2000) L39.
161. P.A.P. Nascente, M.A. Van Hove and G.A. Somorjai, Surf. Sci. 253 (1991) 167. 162. J.A.Gates and L.L. Kesmodel, Surf. Sci. 124 (1983) 68.
163. L.L. Kesmodel, G.D. Waddill and J.A. Gates, Surf. Sci. 136 (1984) 464.
164. N.R.M. Sassen, A.J. den Hartog, F. Jongerius, J.F.M. Aars and V. Ponec, Faraday Discuss. Chem. Soc. 87 (1989) 311.
165. A. Clark, The Chemisorption Bond, Academic Press: New York (1974).
166. P.J. Hiett, F. Flores, P.J. Grout, N.H. March, A Martino-Rodero and G. Senatore, Surf. Sci. 140 (1984) 400.