

Metal-Catalysed Reactions of Hydrocarbons / 10-Hydrogenation of the Aromatic Ring
.pdf10
HYDROGENATION OF THE AROMATIC RING
PREFACE
The aromatic ring is the third type of unsaturated structure, the hydrogenation of which we need to consider. Although it is possible for its hydrogen atoms to be substituted for deuterium, the possible products of the reduction of benzene are effectively but two in number, viz. cyclohexene and cyclohexane: somewhat special conditions of catalyst type and reaction conditions are needed to procure the former, so that commonly cyclohexane is the only product observed. In comparison with say the reactions of the butenes or ethyne, the hydrogenation of benzene is simplicity itself, and for this reason it has been the choice of many who have wished for a quick and easy way of assessing the activity of a catalyst. Very simple analytical techniques are all that one needs, and this helps to explain the attractiveness of this class of reaction before the advent of gas-chromatography.
The straightforwardness of the reaction does not however extend to its interpretation. The mode of chemisorption of the ring has long been debated, and several different structures have been advanced. One is reminded of the student who was required to answer correctly only one of two questions in an oral examination. Question: ‘What is the colour of blue litmus?’ Answer: ‘Red’. This was incorrect. Question: ‘What is the structure of benzene?’ Answer: ‘The Lord only knows’. This was thought to be correct, so the student was passed.
The hydrogenation of alkyl-substituted benzenes leads us into areas of stereochemistry that are related to those we have visited in Chapter 7; and the hydrogenation of fused or condensed aromatic ring systems likewise has fascinating stereochemical consequences also akin to those discussed in Section 7.5.3. The reduction of these molecules has mainly been conducted by organic chemists, and mechanistic aspects have scarcely been examined.
437

438 CHAPTER 10
10.1. INTRODUCTION
10.1.1. Scope
The single aromatic ring as it exists in benzene is stabilised by the simultaneous involvement of the three sets of π electrons; this can be represented as resonance between canonical structures in which double-bonds are differently located, the net effect being that all the six C––C bonds are the same, and are intermediate in length between the formal singleand double-bonds (0.139 nm). The electron system is therefore hard to disrupt, and much of aromatic chemistry depends upon the greater reactivity of the C––H bonds, which can be manipulated in many different ways without the ring being affected. Hetero-aromatic molecules containing nitrogen or sulfur atoms are also resonance-stabilised, but they will not be considered here. Despite its stability, benzene and other aromatic molecules are quite strongly chemisorbed on metal surfaces, although whether this involves loss of the resonance energy by uncoupling the electron system, or leaving it intact as in for example bis(benzene)chromium, is something that will engage our attention in Section 10.13.
The size of the resonance energy in benzene can be estimated as follows. The heat of hydrogenation of benzene to cyclohexane is 206 kJ mol−1, and that of the hypothetical non-resonating cyclohexatriene can be estimated as three times that of Z -2-butene, namely, 359 kJ mol−1: the resonance energy is thus 150 kJ mol−1. The heat liberated on hydrogenating 1,3-cyclohexadiene is 232 kJ mol−1, so its production from benzene would be endothermic by 24 kJ mol−1; it is therefore an unlikely product. Cyclohexene (− HH = 120 kJ mol−1) is therefore the only possible product besides cyclohexane. The attachment of alkyl groups to the benzene ring lowers the heat of hydrogenation only very slightly, due to a small delocalisation of the ring electrons.
The temperature-dependence of the thermochemical parameters has important consequences for the study of ring hydrogenation. The free-energy change for
—O |
|
− |
G—O |
= |
98 kJ mol−1) increases rapidly |
forming cyclohexane form benzene ( |
|
|
|||
with temperature, so that G |
becomes zero and the equilibrium constant unity |
||||
at 560 K. At temperatures above about 473 K there is therefore a significant reverse reaction, the occurrence of which complicates kinetic analysis. The position of equilibrium in this sensitive range naturally depends upon the reactant ratio. It thus becomes clear that the dehydrogenation of cyclohexane or cyclohexene can be studied at comparatively modest temperatures, these reactions therefore being feasible for industrial-scale use (see Chapter 12). Its analogue with C5 and C7 rings, or indeed with linear alkanes, is hardly possible, because without the pull of resonance stabilisation much higher temperatures are needed to obtain useful amounts of the products, and at these temperatures the molecules will suffer total disruption to carbon and hydrogen.

HYDROGENATION OF THE AROMATIC RING |
439 |
Since the hydrogenation of benzene under most circumstances affords cyclohexane as the only product, the reaction has proved attractive as an easy means of assessing catalytic activity; it can be followed manometrically in a static reactor, or, in a flow-system, where product analysis may be carried out very simply. Many of the studies described in the literature antedate the arrival of gas chromatography, and the reaction is still amenable to study in laboratories deprived of this facility. It may of course be conducted either in the vapour or liquid phase. One of the earliest studies of the behaviour of bimetallic (Ni-Cu) systems was made in 1934 using benzene hydrogenation,1 and a review by Hilton A. Smith published in 1957 on the hydrogenation of all types of aromatic compounds, including hetero-aromatic, on all kinds of catalyst was supported by no fewer than 465 references.2 In what follows, therefore, a certain degree of selection has to be applied, and attention will be focussed on more recent publications.
On surveying the literature it becomes clear that certain themes emerge regularly, and that features of the reactions are sometimes common to benzene and its alkyl-substituted derivatives. The hydrogenation of toluene and the xylenes is often studied alongside that of benzene, with beneficial consequences, and it is therefore not appropriate to deal with each molecule individually. After a short introduction to the earlier literature (Section 10.2.1), the kinetics and mechanisms of these reactions in the low-temperature regime will be considered (Sections 10.2.2 and 10.2.3), and then attention is turned to the inversion of rate as temperature is raised: this interesting phenomenon has been widely studied (Section 10.2.4). The reaction of the aromatic ring with deuterium leads to simultaneous exchange and addition, so they can also be examined side-by-side (Section 10.2.5). Other general themes can be identified: these include particle-size effects (Section 10.2.2) and the use of bimetallic catalysts (Section 10.2.6). Features specific to alkyl-substituted benzenes are dealt with in Section 10.3, and the hydrogenation of multiple ring systems in Section 10.4.
10.1.2. Industrial Applications of Benzene Hydrogenation3
Benzene is the raw material for the two most important routes for the manufacture of polymers that are collectively referred to as Nylon. In the first route, cyclohexane is oxidised to adipic acid (HOOC––(CH2)4––COOH), and for this reason the full hydrogenation of benzene has commanded much attention. Because of the large heat of reaction, the process is best performed in the slurry phase, using an unsupported catalyst such as Raney nickel: the process heat is removed by vaporisation of the cyclohexane, part of which is returned to the feed, which typically contains 20% benzene + 80% cyclohexane. Reaction conditions are 30–50 atm. hydrogen and temperatures of 453–503 K. In the second route, the starting compound is cyclohexene, and for some time efforts have been made to effect the partial hydrogenation of benzene to this molecule (see Section 10.2.7).

440 |
CHAPTER 10 |
Separation from unreacted benzene is easier than with cyclohexane, because an azeotrope is not formed. The quality of diesel fuels is improved by removal of aromatics, and hydrogenation has been explored as a means of eliminating them: sulfur-tolerant catalysts are needed in this application.4−9 The combination of palladium + platinum is sometimes found to be more thiotolerant than either separately,10 although this is not the case with model (Pt + Pd)/γ -Al2O3 in the hydrogenation of tetralin.11
10.2.KINETICS AND MECHANISM OF AROMATIC RING HYDROGENATION
10.2.1. Introduction: Early Work1,6,12–14
For the reasons summarised above, the hydrogenation of benzene in particular has commanded immense attention, and this has generated an enormous literature; it is therefore perhaps inevitable, although at first sight disappointing, that the results obtained are so diverse and contradictory. Keane and Patterson4 have noted that they are characterised by the following features.
(1)Rate maxima between 423 and 473 K.
(2)Product inhibition or easy desorption of cyclohexane.
(3)Competitive or non-competitive adsorption of the reactants.
(4)Temperature dependence of the order in hydrogen either from zero to 0.5 or from 0.5 to 3.
(5)Activation energies between 25 and 94 kJ mol−1.
And that is just with nickel catalysts; the cited paper gives some 20 references to support this summary. What are we to make of such a medley of results? We have to suppose that within the constraints imposed by the manner in which the reaction was conducted, the type of catalyst and the range of conditions explored, that each of the quantitative measurements is correct; only those such as (2) and (3) which depend on the interpretation of what was observed may possibly be in error. This situation is by no means unique, as we have seen, but is more obvious here simply because of the greater amount of work that has been performed. Catalysis is indeed like Cleopatra; a thing of ‘infinite variety’. How then are these conflicting conclusions to be reconciled? If we have faith in the scientific method we must conclude that somewhere there are logical explanations for these differences; and it is our task—not an easy one—to try to identify their causes. The best help comes from the more comprehensive kinetic studies, and it is these that will receive most attention.

HYDROGENATION OF THE AROMATIC RING |
441 |
One of the great problems with studying benzene hydrogenation is the pervasive presence of traces of thiophen, which is a notorious poison, and its removal to low levels is essential for obtaining reproducible results. A scientist was once asked if he took much trouble to purify his benzene. ‘No’, he replied; ‘Not much trouble: very much trouble’.
Early work on this family of reactions has several times been reviewed.1−3,12,15 That published by Hilton Smith in 1957 gives a particularly full account of work prior to that date, and will be of interest to historians of the science, as it describes inter alia several attempts by Russian scientists to establish a particle size effect and to confirm the validity of Balandin’s hypothesis that the fcc(111) plane would be particularly adept for the hydrogenation. The other reviews set the tone for what has been amply confirmed by later work, and the following generalisations can be offered. (1) Among the metals of Groups 8 to 10 the base metals (and palladium) are the least active. (2) Orders in hydrogen are larger than those of the aromatic, the former being often close to unity or even greater, and the latter generally between zero and 0.5; the aromatic molecule is therefore the more strongly adsorbed. (3) Activation energies lie mainly in the range 35–50 kJ mol−1, values over 55 kJ mol−1 being rare. (4) The rate is often observed to pass through a maximum as temperature is raised; while this is not unique to benzene hydrogenation, it has been much more often seen than with any other system. These observations provide the framework for a more detailed examination of the reactions’ characteristics.
10.2.2. Kinetics of Aromatic Ring Hydrogenation
The hydrogenation of aromatic hydrocarbons can be followed either in the vapour or the liquid phase or in solution, depending upon the molar mass and volatility. Much of the older qualitative work was performed in solution, where an additional dimension, namely, the pH, could be beneficially employed: such work does not however allow profound study of kinetics and mechanism, so it will not be dwelt on here. For the more active metals that can be used at moderate temperatures, the reaction ceases with formation of the alicyclic ring, but with the less active metals (Re, Tc) or metals in less active forms such as wire (Ir), the necessary higher temperatures cause deeper-seated changes, and alkanes containing six or fewer carbon atoms are among the products. These may arise from strongly adsorbed and dehydrogenated residues that at lower temperatures12,16 cause deactivation.
There are no recent comprehensive measurements of the relative activities of metals for benzene hydrogenation, and since most of the detailed work concerns only nickel, palladium and platinum a short summary of what is known about other metals is all that can be made. Quantitative measurements of seven of the metals of Groups 9 to 10 supported on silica were reported many years ago,1 and the low activity of the base metals has been confirmed by measurements on

442 |
CHAPTER 10 |
films.17 Rhenium in Group 7 also had low activity,16,18 but technetium is stated, in one of the very few papers mentioning the catalytic activity of this metal, to be as active as palladium.16 Catalysis by the metals of Group 7 is a very muchneglected area. Osmium had low activity,18 while tungsten as film was highly active;17 rhodium and iridium are comparable with platinum.1,18–20 All who have used palladium agree that it is only moderately active,4,5,16,19,21 sometimes being placed below nickel and iron in the hierarchy;17,22 in view of its outstanding activity for the hydrogenation of alkynes, this is somewhat surprising. Ru/C and Rh/C have been assigned highest activities for hydrogenating liquid benzene.2 Ring hydrogenations in solution catalysed by Adams platinum oxide proceed best under acidic conditions, which neutralise the retained sodium.23
The manner of performing the reaction appears to have some strange consequences. While most work has been carried out with continuous-flow reactors, the pulse-flow mode has sometimes been used,24,25 and this has been claimed to account of an observation26 that rate was a function of catalyst:support ratio in a physical mixture, an effect not observed in continuous flow.27,28 However as we shall see shortly there is now good evidence for support involvement, at least in some cases. The pulse-flow mode generates very much higher TOFs29 (by a factor of 103), presumably because the surface is thereby kept clean; it is however hard to imagine that only one part in 103 of the surface is active when continuous flow is used, although there is evidence to support such a conclusion.30
Table 10.1 shows some of the more recent kinetic measurements for benzene hydrogenation; extensive compilations of earlier results are to be found in references 1, 12, 15 and 29, but they do not alter or add to the picture significantly. The following points should be noted. (1) Values of apparent activation energy are remarkably consistent at 50 ± 10 kJ mol−1, although lower values are sometimes reported:16,31,32 they increase with benzene pressure over rhenium powder, and with hydrogen pressure over Ni/SiO2, but in this case the effect was removed by using rate constants rather than TOFs. Those for toluene and o-xylene are sometimes a little higher (64–80 kJ mol−1).4,29,33,34 (2) Orders of reaction are almost always expressed in the Power Rate Law approximation (Section 5.2), and only rarely have the reaction orders been interpreted using Langmuir-Hinshelwood formalism.22,26,35 (3) There is extensive information on the temperature-dependence of the reaction orders, which almost always increase with temperature (the table shows only maximum and minimum values): their significance will be considered further in connection with the inversion of the rate. Once again toluene and o-xylene behave almost exactly as benzene.4,33,36
Before proceeding to consider kinetic equations and implied reaction mechanisms, we may note some other pertinent features of these reactions. (A) Benzene hydrogenation was subject to the influence of the Strong Metal-Support Interaction (Section 3.35) when titania and vanadium sesquioxide were used as supports for rhodium, platinum and iridium;20,37,38 even Pt/SiO239 and Ni/SiO240 when heated

HYDROGENATION OF THE AROMATIC RING |
443 |
TABLE 10.1. Kinetics of the Hydrogenation of Benzene on Metals of Groups 8 and 11 r PH x PB y
Metal |
Form |
E /kJ mol−1 |
T /K |
x |
y |
Reference |
Co |
/SiO2 c |
24 |
338 |
1.0 |
0.2 |
31 |
|
|
50 |
452 |
1.7 |
0.5 |
|
Ni |
/SiO2 |
300 |
0.5b |
0.05 |
35 |
|
|
|
|
360 |
0.65 |
— |
|
Ni |
/SiO2 |
49 |
408 |
0.8 |
0 |
4,53 |
|
|
|
523 |
2.3 |
0.5 |
|
Ni |
/SiO2 |
52 |
298 |
0.5 |
0.1 |
60 |
|
|
|
473 |
2–3 |
0.3–0.5 |
|
Ni |
/Al2 O3 |
42 |
403 |
1.25 |
— |
25 |
Ni |
/K-Y zeolite |
59.5 |
403 |
0.7 |
0.02 |
122 |
|
|
|
523 |
2.3 |
0.5 |
|
Cu |
/SiO2 |
58 |
306 |
1.5 |
0.6 |
62 |
|
|
50 |
400 |
2.5 |
0.6 |
|
Pd |
variousa |
353 |
0.5 |
0 |
30,34 |
|
|
|
|
573 |
4 |
0.8 |
|
Pt |
variousa |
42–54 |
317 |
0.6 |
0.1 |
46 |
Ru |
/Al2 O3 |
50 |
313 |
1 |
0 |
|
Ru |
/SiO2 |
−26 |
303 |
1.2 |
0 |
19 |
|
|
|
400 |
2.0 |
0.2 |
61 |
a Includes supported catalysts and unsupported powders. b Order decreases with increasing PH
c Similar results for Co/A12 O3, Co/SiO2 -Al2 O3 and Co/C.
in hydrogen to a high enough temperature showed similar effects. (B) Inclusion of tungsten (as oxide) in Pt/Al2O341 and of molybdenum in Pt/SiO2 or Pt/Al2O342 led respectively to improved stability and greater activity. (C) In a study of the effect of sulfur and its compounds as poisons, it was found that hydrogen sulfide and sulfur dioxide are non-selective, as they affect hydrogenation and exchange equally, while sulfur formed by reaction between them is a selective poison, suppressing hydrogenation much more than exchange.43,44
There have been a number of attempts over the years to define the structure sensitivity of benzene hydrogenation. Extensive early work by Russian scientists, reviewed by Smith,2 covered cobalt, nickel, palladium and platinum on various supports, and alteration in particle size was supposedly achieved by changing the metal loading, in one case down to 0.03%. However when this work was conducted, X-ray diffraction was the only technique available for estimating particle size, although change in the intensity of the (111) reflection did sometimes produce evidence to support Balandin’s ideas, as noted above. Some of the more recent observations are summarised in Table 10.2. There is a general perception that the reaction is structure-insensitive, or almost so (Figure 10.1), but this conclusion may derive from the somewhat small range of dispersion covered, and the frequent use of
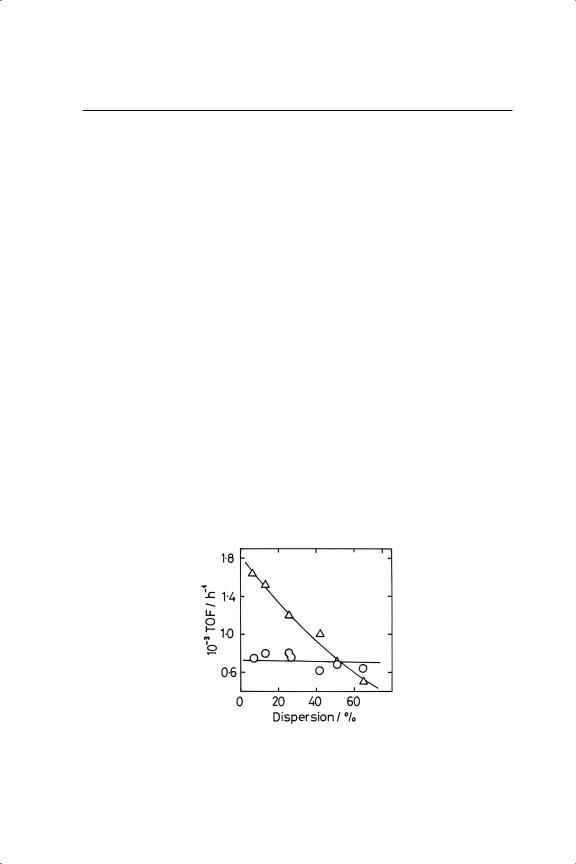
444 CHAPTER 10
TABLE 10.2. Structure-Sensitivity in the Hydrogenation of Benzene
Metal |
Form |
Size Range/Dispersion |
Conclusion |
Reference |
Ni |
/SiO2 |
0.5–5 nm |
Slight maximum at 1.3 nm |
123 |
Ru |
/SiO2 |
0.7–9.5 nm |
Maximum at 3.5 nm |
45 |
Rh |
/Al2 O3 |
10–150 nm |
TOF constant except at size <1 nm |
124 |
Pd |
/Al2 O3 |
10–150 nm |
TOF constant |
124,125 |
Pd |
/Al2 O3 |
12–100% |
TOF constant |
126 |
Pd |
/SiO2 |
4–48% |
TOF increases with dispersiona |
126 |
Pd |
/C |
2.5–21 nm |
Mild sensitivity below 4 nm |
127 |
Ir |
/γ -Al2 O3 |
0.5–3.3 nm |
TOF increases with sizec |
128 |
Pt |
/Al2 O3 |
7–65% |
TOF constant |
43 |
Pt |
/Al2 O3 |
7–65% |
Almost insensitiveb |
44 |
Pt |
/Al2 O3 |
4–89% |
See text |
27 |
Pt |
/SiO2 |
7–81% |
See text |
27 |
Pt |
/SiO2 |
4.5–64 nm |
TOF increases × 2 as size increases |
129 |
a This effect may be due to impurities in the support b See Figure 10.1
c Reactant was toluene.
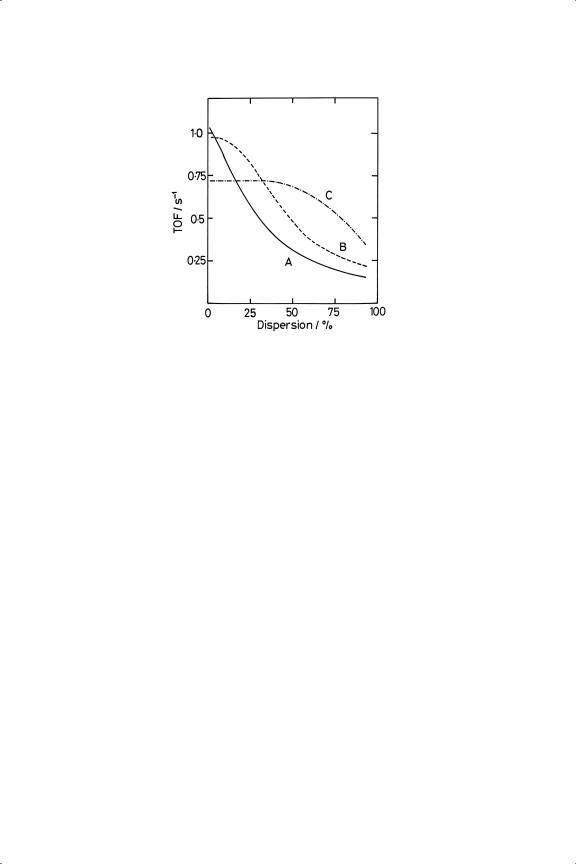
unnecessarily high reduction temperatures (>673 K), the need for which ‘seems to be part of the lore of hydrogenation catalysis’.27 In fact a careful study27 has shown that the situation is quite complex. With Pt/Al2O3, the TOF was only independent of dispersion between about 5 and 50% dispersion if it had been reduced at 723 K; at higher dispersions, and throughout if lower reduction temperatures were used, it decreased (see Figure 10.2) by as much as a factor of 10. There were differences too between Pt/Al2O3 and Pt/SiO2, especially when reduced at 723 K, where the latter’s TOF increased with dispersion. ‘Differences in the extent of surface reconstruction after the high temperature pre-treatment’ were thought to be responsible.
Figure 10.1. Turnover frequencies (h−1 ) for benzene hydrogenation (O) and exchange with deuterium ( )over Pt/Al2 O3 (T = 358 K; PB = 0.555 Torr; PD = 205 Torr).44
HYDROGENATION OF THE AROMATIC RING |
445 |
Figure 10.2. Turnover frequencies (s−1 ) for benzene hydrogenation over pre-oxidised Pt/Al2 O3 reduced at (A) 373 K; (B) 573 K; (C) 723 K (T = 353 K; PB = 68 Torr; PH = 692 Torr).27
No evidence was found of an effect of diluting Pt/Al2O3 with more support, although in other work a four-fold increase was reported. Ir/Al2O3 behaved as Pt/Al2O3 when reduced at low temperature,44 while with Ru/SiO2 the rate passed through a maximum as dispersion was altered.45 Extreme care has therefore to be taken before any generalisations about structure-sensitivity are advanced.
Very significant support effects have been seen in the work of Vannice’s group.30,34,46 TOFs for benzene hydrogenation on platinum catalysts were more than five times greater with acidic supports such as silica-alumina and titania, although activation energies and orders of reaction were unaffected. Similar results were obtained with toluene,29 and with palladium catalysts,21,34,47 although chlorination of Pt/Al2O3 of various dispersions had comparatively little effect.27 The enhancement was ascribed to the reaction of benzene adsorbed on acidic centres on the support with spiltover hydrogen: this will be referred to again when considering reaction mechanisms.
With metals active in hydrogenolysis (Ru, Re, Ni, Ir, see Chapters 13 and 14), the process of breaking C––C bonds overlaps with that of hydrogenation, and because its activation energy is higher it grows in importance as temperature is raised, and finally becomes dominant. Thus with ruthenium above 405–420 K, technetium above about 440 K and rhenium above about 475 K, the hydrogenolysis rate was easily measurable,48 activation energies being respectively 125, 121 and 134 kJ mol−1. The process usually goes all the way to methane, although occasionally intermediates have been seen (e.g. with cobalt catalysts48). In most cases,

446 |
CHAPTER 10 |
however, the occurrence of hydrogenolysis is either not apparent in the range of temperature used, or is ignored.
10.2.3. Rate Expressions and Reaction Mechanisms
In kinetic studies of the hydrogenation of aromatic hydrocarbons, the dependence of rate upon reactant pressures has usually been expressed in Power Rate Law formulations, that is, by orders of reaction that are simple exponents of the pressures. These as we have seen (Section 5.2) are at best approximations to more fundamental expressions based on concentrations of adsorbed species,4,5,14 although they may well represent results over the limited range in which measurements were made. The Langmuir-Hinshelwood formalism has however sometimes been used, and heats of adsorption of the reactants in their ‘reactive’ states derived from the temperature-dependence of their adsorption coefficients.4,5,22,30,35
The next step is to develop a quantitative framework based on an assumed mechanism, and to test this against the experimental results. A particular feature is the dependence of the orders, especially those of hydrogen, upon temperature (Table 10.1). Aspects of the perceived mechanisms, which will now be briefly reviewed, usually ignore any precise description of the reactant hydrocarbon or derived species: these matters will be covered in the section dealing with the exchange reaction.
One particularly vexed question is whether the reaction occurs only on the metal particles in supported metal catalysts. Clearly, reaction can occur on them, as unsupported metals are effective, but TOFs are lower than on supported metals. This may be in part because metal sites may be deactivated by ‘carbonaceous deposits’, especially at higher temperatures.30 Vannice and his associates34,46−48 have interpreted the larger TOFs found with acidic supports as evidence for a contribution from some spillover catalysis, i.e. reaction of hydrogen atoms moving from the metal to aromatic molecules adsorbed at Brønsted acid centres on the support: but, since the enhancement (that is, the TOF on an acidic support compared to that on a neutral support) can be as much as five-fold, we must conclude that up to 80% of the reaction proceeds by the spillover route—which is no mean contribution. Other workers appear to ignore this possibility,4,5 which, since neutral or weakly acidic supports are commonly used, may be a reasonable thing to do. A distinction must be drawn between spillover of hydrogen from metal to the support granule on which the metal sits, which is easy, and spillover to another granule of support admixed with the catalyst, which is more difficult (Section 3.34). With Pt/C, however, the latter is possible, as the rate increased to a limiting value as the catalyst was diluted with support.49
A further vexed question is whether the reaction proceeds through hydrogen molecules arriving from the gas phase or through chemisorbed hydrogen atoms. Most workers opt for the latter, and, when the temperature-variation of their